
Unveiling Mycoviromes Using Fungal Transcriptomes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
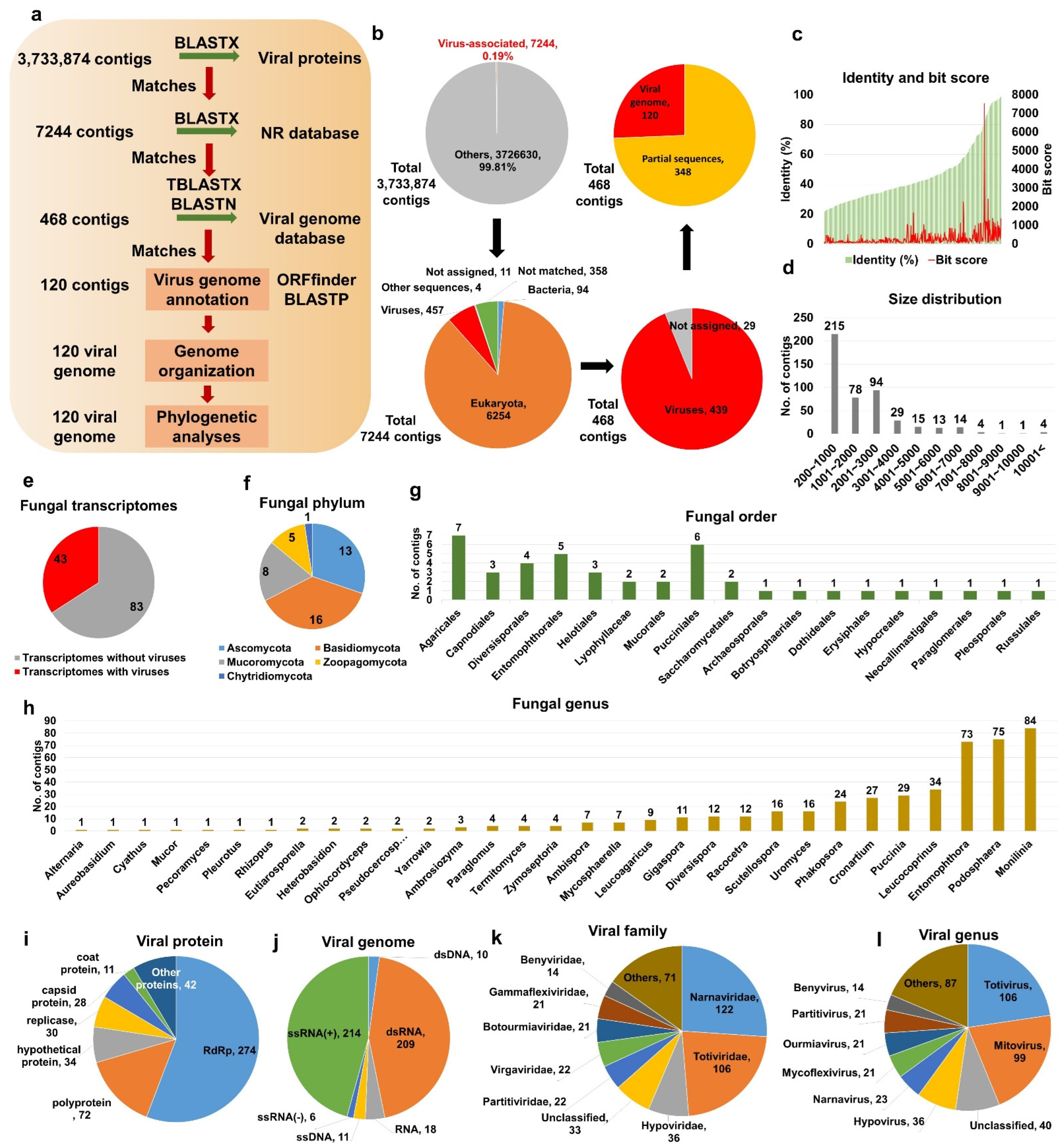
2.1. Identification of Virus-Associated Contigs from Fungal Transcriptomes
2.2. Viral Genome Assembly and Phylogeny of Novel Mycoviruses
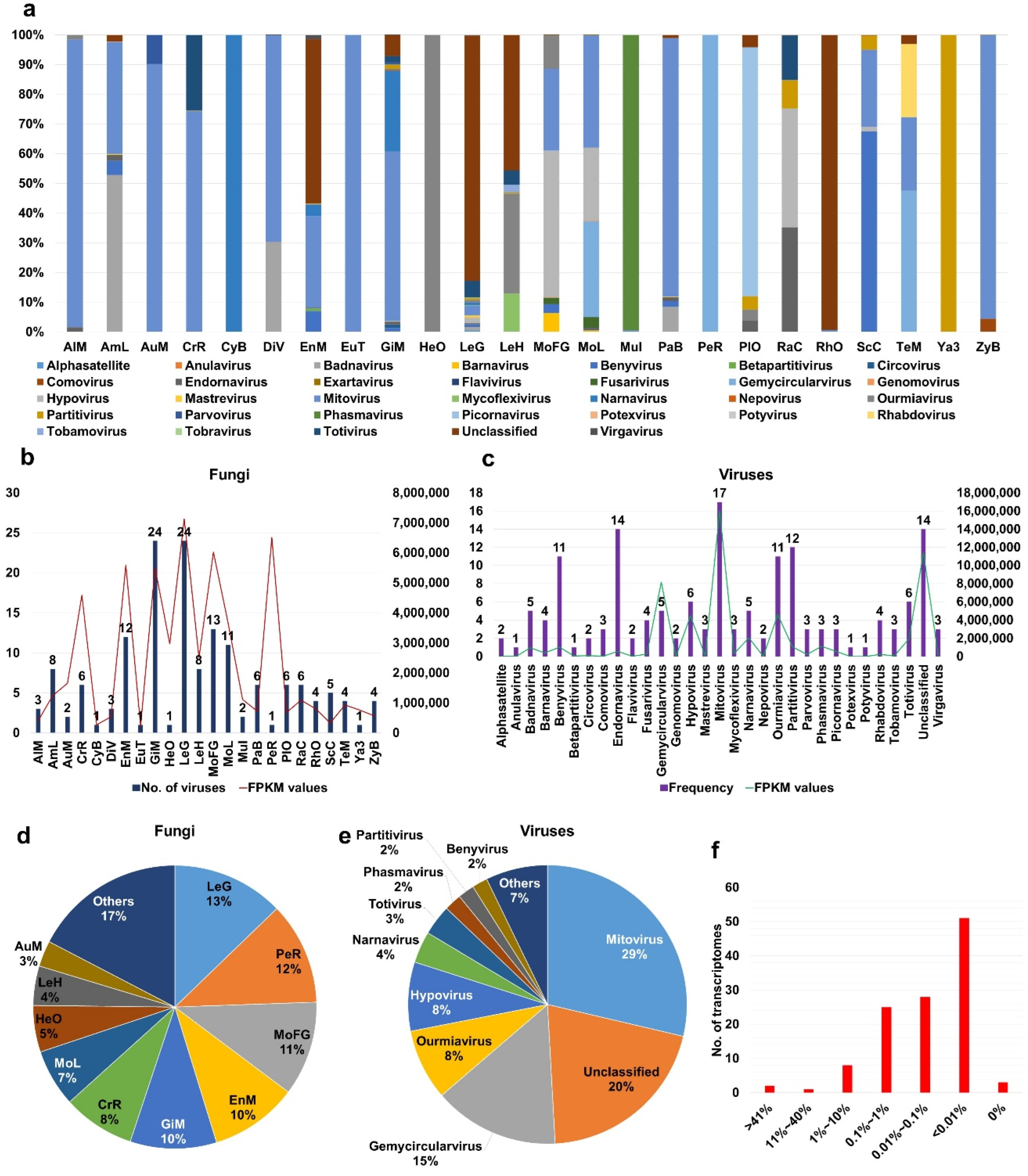
2.3. Viral Abundance in Different Fungal Transcriptomes
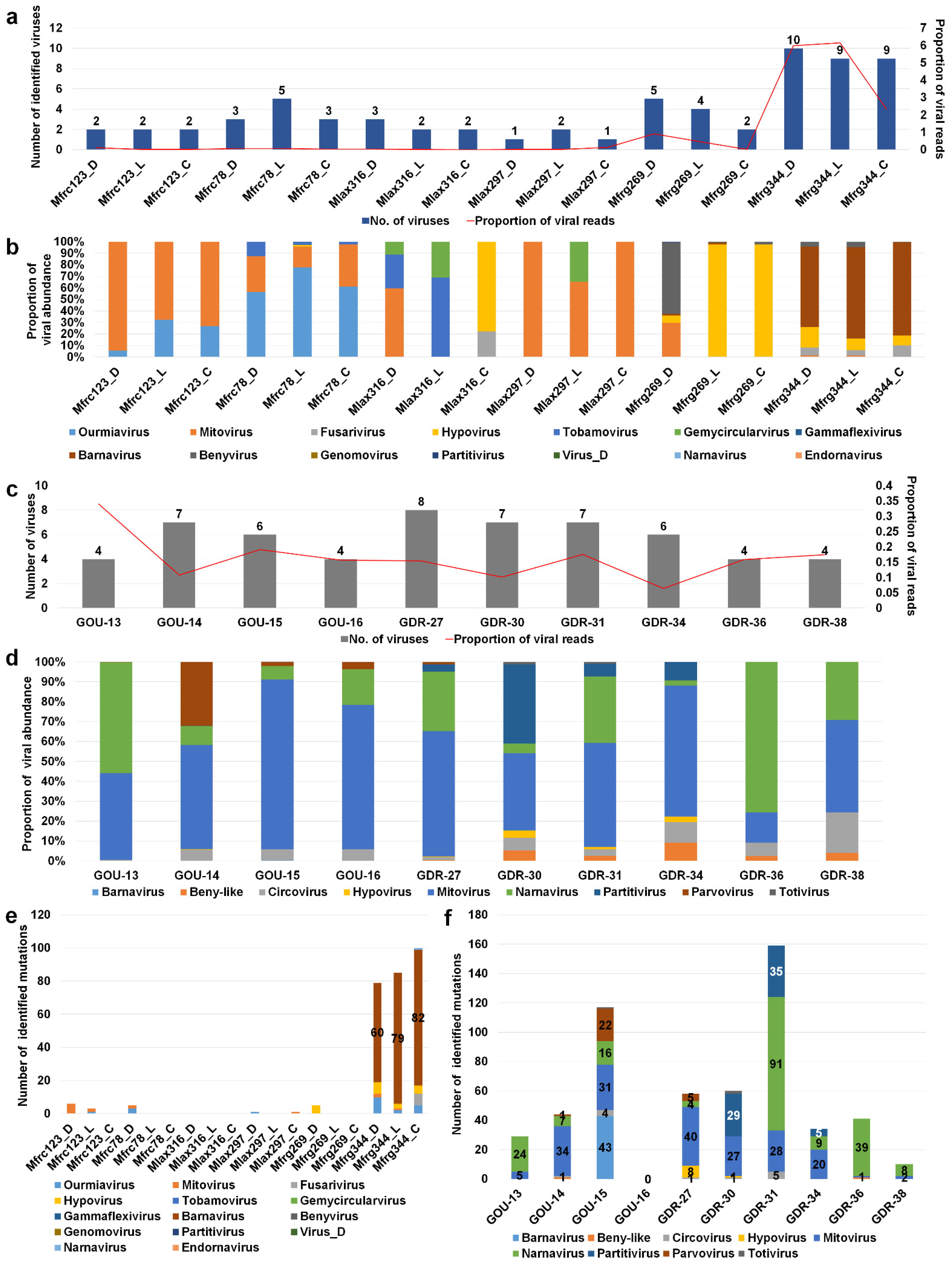
2.4. Analyses of Diverse Monilinia and Gigaspora Viromes
3. Discussion
4. Materials and Methods
4.1. Download of Fungal Transcriptomes
4.2. Construction of Viral Genome and Protein Database
4.3. Identification of Virus-Associated Contigs
4.4. Viral Genome Annotation
4.5. Phylogenetic Tree Construction
4.6. Estimation of Viral Abundance in Fungal Transcriptomes
4.7. Fungal Transcriptome Assembly and Calculation of Viral Mutations
4.8. Identification of Virus Mutations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lefkowitz, E.J.; Dempsey, D.M.; Hendrickson, R.C.; Orton, R.J.; Siddell, S.G.; Smith, D.B. Virus taxonomy: The database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Res. 2017, 46, D708–D717. [Google Scholar] [CrossRef]
- Monier, A.; Claverie, J.-M.; Ogata, H. Taxonomic distribution of large DNA viruses in the sea. Genome Biol. 2008, 9, R106. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.-D.; Tian, J.-H.; Chen, L.-J.; Chen, X.; Li, C.-X.; Qin, X.-C.; Li, J.; Cao, J.-P.; Eden, J.-S. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.-D.; Chen, X.; Tian, J.-H.; Chen, L.-J.; Li, K.; Wang, W.; Eden, J.-S.; Shen, J.-J.; Liu, L. The evolutionary history of vertebrate RNA viruses. Nature 2018, 556, 197. [Google Scholar] [CrossRef]
- Mushegian, A.; Shipunov, A.; Elena, S.F. Changes in the composition of the RNA virome mark evolutionary transitions in green plants. BMC Biol. 2016, 14, 68. [Google Scholar] [CrossRef]
- Shkoporov, A.N.; Hill, C. Bacteriophages of the human gut: The “known unknown” of the microbiome. Cell Host Microbe 2019, 25, 195–209. [Google Scholar] [CrossRef]
- Prussin, A.J.; Marr, L.C.; Bibby, K.J. Challenges of studying viral aerosol metagenomics and communities in comparison with bacterial and fungal aerosols. FEMS Microbiol. Lett. 2014, 357, 1–9. [Google Scholar] [CrossRef]
- Kumar, N.; Barua, S.; Riyesh, T.; Chaubey, K.K.; Rawat, K.D.; Khandelwal, N.; Mishra, A.K.; Sharma, N.; Chandel, S.S.; Sharma, S. Complexities in isolation and purification of multiple viruses from mixed viral infections: Viral interference, persistence and exclusion. PLoS ONE 2016, 11, e0156110. [Google Scholar]
- Nowrousian, M. Next-generation sequencing techniques for eukaryotic microorganisms: Sequencing-based solutions to biological problems. Eukaryot. Cell 2010, 9, 1300–1310. [Google Scholar] [CrossRef]
- Zhang, Y.-Z.; Shi, M.; Holmes, E.C. Using metagenomics to characterize an expanding virosphere. Cell 2018, 172, 1168–1172. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.; Tzanetakis, I.E. Development of a virus detection and discovery pipeline using next generation sequencing. Virology 2014, 471, 54–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, A.; Sekizuka, T.; Kuroda, M. VirusTAP: Viral genome-targeted assembly pipeline. Front. Microbiol. 2016, 7, 32. [Google Scholar] [CrossRef] [PubMed]
- Prosperi, M.C.; Prosperi, L.; Bruselles, A.; Abbate, I.; Rozera, G.; Vincenti, D.; Solmone, M.C.; Capobianchi, M.R.; Ulivi, G. Combinatorial analysis and algorithms for quasispecies reconstruction using next-generation sequencing. BMC Bioinform. 2011, 12, 5–13. [Google Scholar] [CrossRef]
- Watson, S.J.; Welkers, M.R.; Depledge, D.P.; Coulter, E.; Breuer, J.M.; de Jong, M.D.; Kellam, P. Viral population analysis and minority-variant detection using short read next-generation sequencing. Philos. Trans. R. Soc. B 2013, 368, 20120205. [Google Scholar] [CrossRef] [PubMed]
- Paez-Espino, D.; Eloe-Fadrosh, E.A.; Pavlopoulos, G.A.; Thomas, A.D.; Huntemann, M.; Mikhailova, N.; Rubin, E.; Ivanova, N.N.; Kyrpides, N.C. Uncovering Earth’s virome. Nature 2016, 536, 425. [Google Scholar] [CrossRef]
- Nuss, D.L. Hypovirulence: Mycoviruses at the fungal–plant interface. Nat. Rev. Microbiol. 2005, 3, 632. [Google Scholar] [CrossRef]
- Liu, L.; Xie, J.; Cheng, J.; Fu, Y.; Li, G.; Yi, X.; Jiang, D. Fungal negative-stranded RNA virus that is related to bornaviruses and nyaviruses. Proc. Nat. Acad. Sci. USA 2014, 111, 12205–12210. [Google Scholar] [CrossRef]
- Yu, X.; Li, B.; Fu, Y.; Jiang, D.; Ghabrial, S.A.; Li, G.; Peng, Y.; Xie, J.; Cheng, J.; Huang, J. A geminivirus-related DNA mycovirus that confers hypovirulence to a plant pathogenic fungus. Proc. Nat. Acad. Sci. USA 2010, 107, 8387–8392. [Google Scholar] [CrossRef]
- Son, M.; Yu, J.; Kim, K.-H. Five questions about mycoviruses. PLoS Pathog. 2015, 11, e1005172. [Google Scholar] [CrossRef]
- Marzano, S.-Y.L.; Nelson, B.D.; Ajayi-Oyetunde, O.; Bradley, C.A.; Hughes, T.J.; Hartman, G.L.; Eastburn, D.M.; Domier, L.L. Identification of diverse mycoviruses through metatranscriptomics characterization of the viromes of five major fungal plant pathogens. J. Virol. 2016, 90, 6846–6863. [Google Scholar] [CrossRef]
- Gilbert, K.B.; Holcomb, E.E.; Allscheid, R.L.; Carrington, J.C. Hiding in plain sight: New virus genomes discovered via a systematic analysis of fungal public transcriptomes. PLoS ONE 2019, 14, e0219207. [Google Scholar] [CrossRef] [Green Version]
- Thapa, V.; Roossinck, M.J. Determinants of Coinfection in the Mycoviruses. Front. Cell. Infect. Microbiol. 2019, 9, 169. [Google Scholar] [CrossRef] [PubMed]
- Jo, Y.; Choi, H.; Cho, J.K.; Yoon, J.-Y.; Choi, S.-K.; Cho, W.K. In silico approach to reveal viral populations in grapevine cultivar Tannat using transcriptome data. Sci. Rep. 2015, 5, 15841. [Google Scholar] [CrossRef] [PubMed]
- Cordey, S.; Laubscher, F.; Hartley, M.-A.; Junier, T.; Pérez-Rodriguez, F.J.; Keitel, K.; Vieille, G.; Samaka, J.; Mlaganile, T.; Kagoro, F. Detection of dicistroviruses RNA in blood of febrile Tanzanian children. Emerg. Microbes Infect. 2019, 8, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Angelini, R.M.D.M.; Abate, D.; Rotolo, C.; Gerin, D.; Pollastro, S.; Faretra, F. De novo assembly and comparative transcriptome analysis of Monilinia fructicola, Monilinia laxa and Monilinia fructigena, the causal agents of brown rot on stone fruits. BMC Genom. 2018, 19, 436. [Google Scholar]
- Salvioli, A.; Ghignone, S.; Novero, M.; Navazio, L.; Bagnaresi, P.; Bonfante, P. Symbiosis with an endobacterium increases the fitness of a mycorrhizal fungus, raising its bioenergetic potential. ISME J. 2016, 10, 130. [Google Scholar] [CrossRef]
- Grybchuk, D.; Kostygov, A.Y.; Macedo, D.H.; Votýpka, J.; Lukeš, J.; Yurchenko, V. RNA Viruses in Blechomonas (Trypanosomatidae) and Evolution of Leishmaniavirus. mBio 2018, 9, e01932-18. [Google Scholar] [CrossRef]
- Niagro, F.; Forsthoefel, A.; Lawther, R.; Kamalanathan, L.; Ritchie, B.; Latimer, K.; Lukert, P. Beak and feather disease virus and porcine circovirus genomes: Intermediates between the geminiviruses and plant circoviruses. Arch. Virol. 1998, 143, 1723–1744. [Google Scholar] [CrossRef]
- Andika, I.B.; Wei, S.; Cao, C.; Salaipeth, L.; Kondo, H.; Sun, L. Phytopathogenic fungus hosts a plant virus: A naturally occurring cross-kingdom viral infection. Proc. Nat. Acad. Sci. USA 2017, 114, 12267–12272. [Google Scholar] [CrossRef]
- Nerva, L.; Varese, G.; Falk, B.; Turina, M. Mycoviruses of an endophytic fungus can replicate in plant cells: Evolutionary implications. Sci. Rep. 2017, 7, 1908. [Google Scholar] [CrossRef]
- Marzec, M.; Muszynska, A.; Gruszka, D. The role of strigolactones in nutrient-stress responses in plants. Int. J. Mol. Sci. 2013, 14, 9286–9304. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xie, J.; Fu, Y.; Cheng, J.; Qu, Z.; Zhao, Z.; Cheng, S.; Chen, T.; Li, B.; Wang, Q. A 2-kb mycovirus converts a pathogenic fungus into a beneficial endophyte for Brassica protection and yield enhancement. Mol. Plant 2020, 13, 1420–1433. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Doerks, T.; Bork, P. SMART 7: Recent updates to the protein domain annotation resource. Nucleic Acids Res. 2011, 40, D302–D305. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2014, 32, 268–274. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2017, 35, 518–522. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Kodama, Y.; Shumway, M.; Leinonen, R. The Sequence Read Archive: Explosive growth of sequencing data. Nucleic Acids Res. 2011, 40, D54–D56. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jo, Y.; Choi, H.; Chu, H.; Cho, W.K. Unveiling Mycoviromes Using Fungal Transcriptomes. Int. J. Mol. Sci. 2022, 23, 10926. https://doi.org/10.3390/ijms231810926
Jo Y, Choi H, Chu H, Cho WK. Unveiling Mycoviromes Using Fungal Transcriptomes. International Journal of Molecular Sciences. 2022; 23(18):10926. https://doi.org/10.3390/ijms231810926
Chicago/Turabian StyleJo, Yeonhwa, Hoseong Choi, Hyosub Chu, and Won Kyong Cho. 2022. "Unveiling Mycoviromes Using Fungal Transcriptomes" International Journal of Molecular Sciences 23, no. 18: 10926. https://doi.org/10.3390/ijms231810926
APA StyleJo, Y., Choi, H., Chu, H., & Cho, W. K. (2022). Unveiling Mycoviromes Using Fungal Transcriptomes. International Journal of Molecular Sciences, 23(18), 10926. https://doi.org/10.3390/ijms231810926