

Allyl-, Butyl- and Phenylethyl-Isothiocyanate Modulate Akt–mTOR and Cyclin–CDK Signaling in Gemcitabine- and Cisplatin-Resistant Bladder Cancer Cell Lines


 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
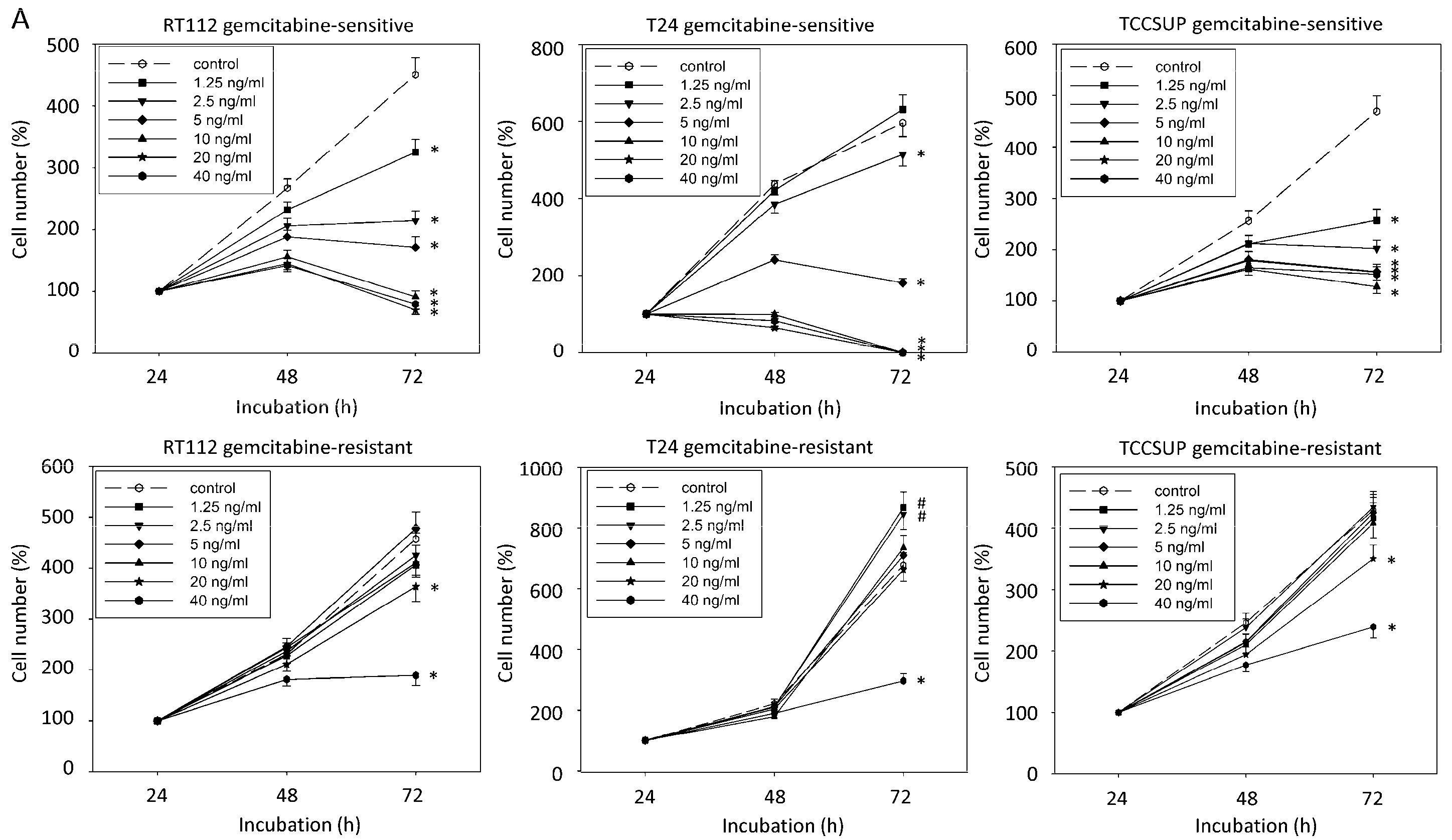
2.1. Resistance Induction
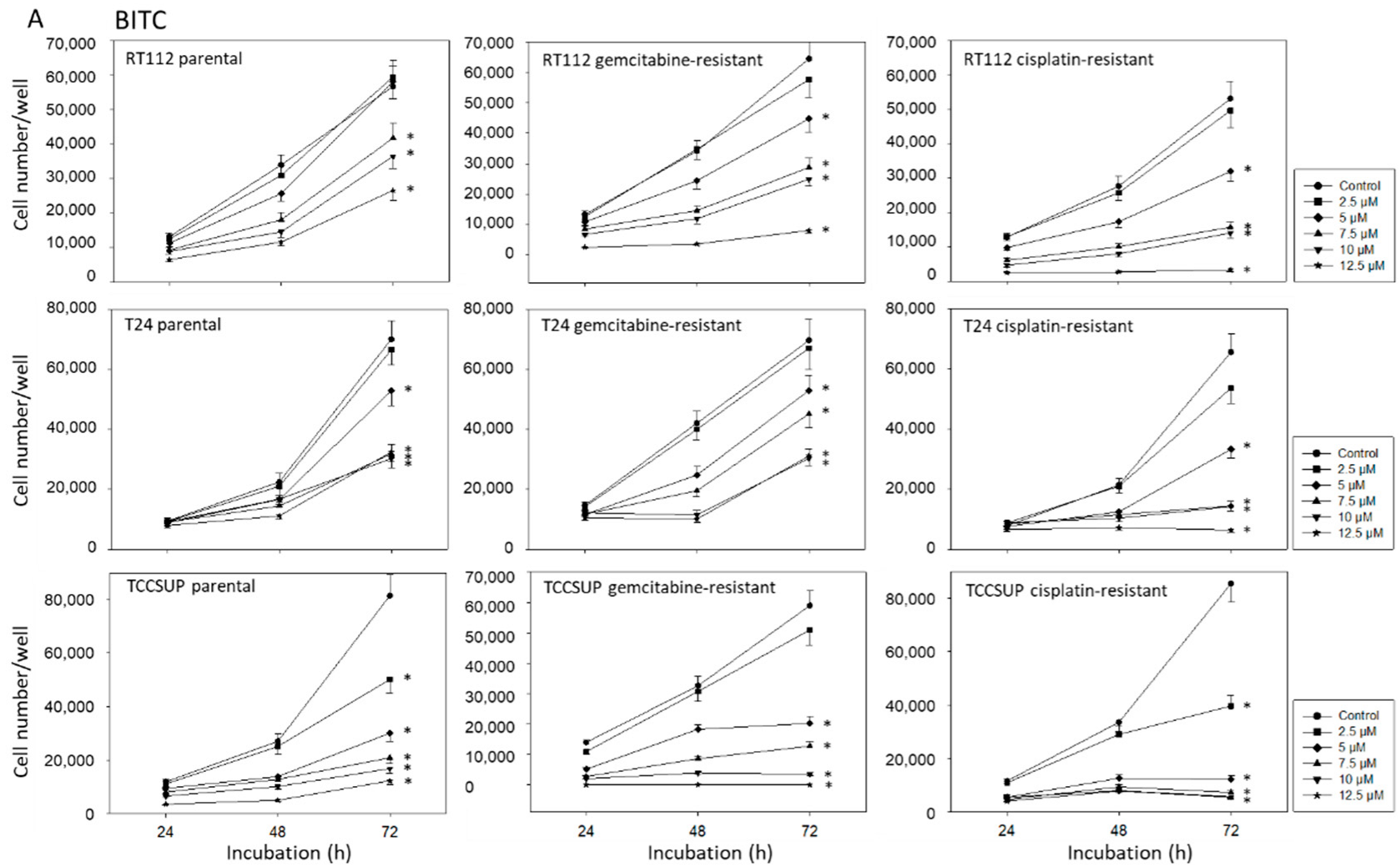
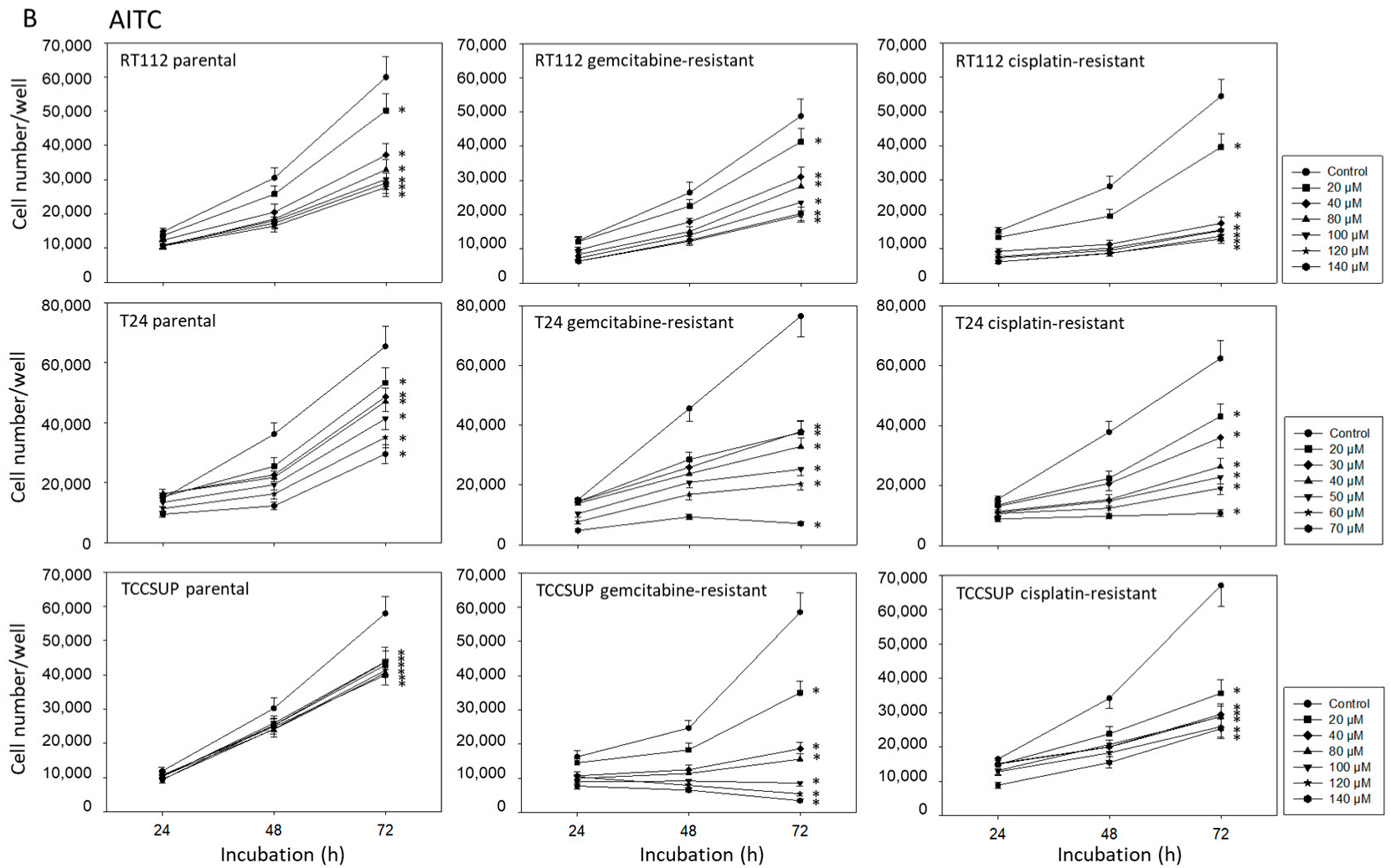
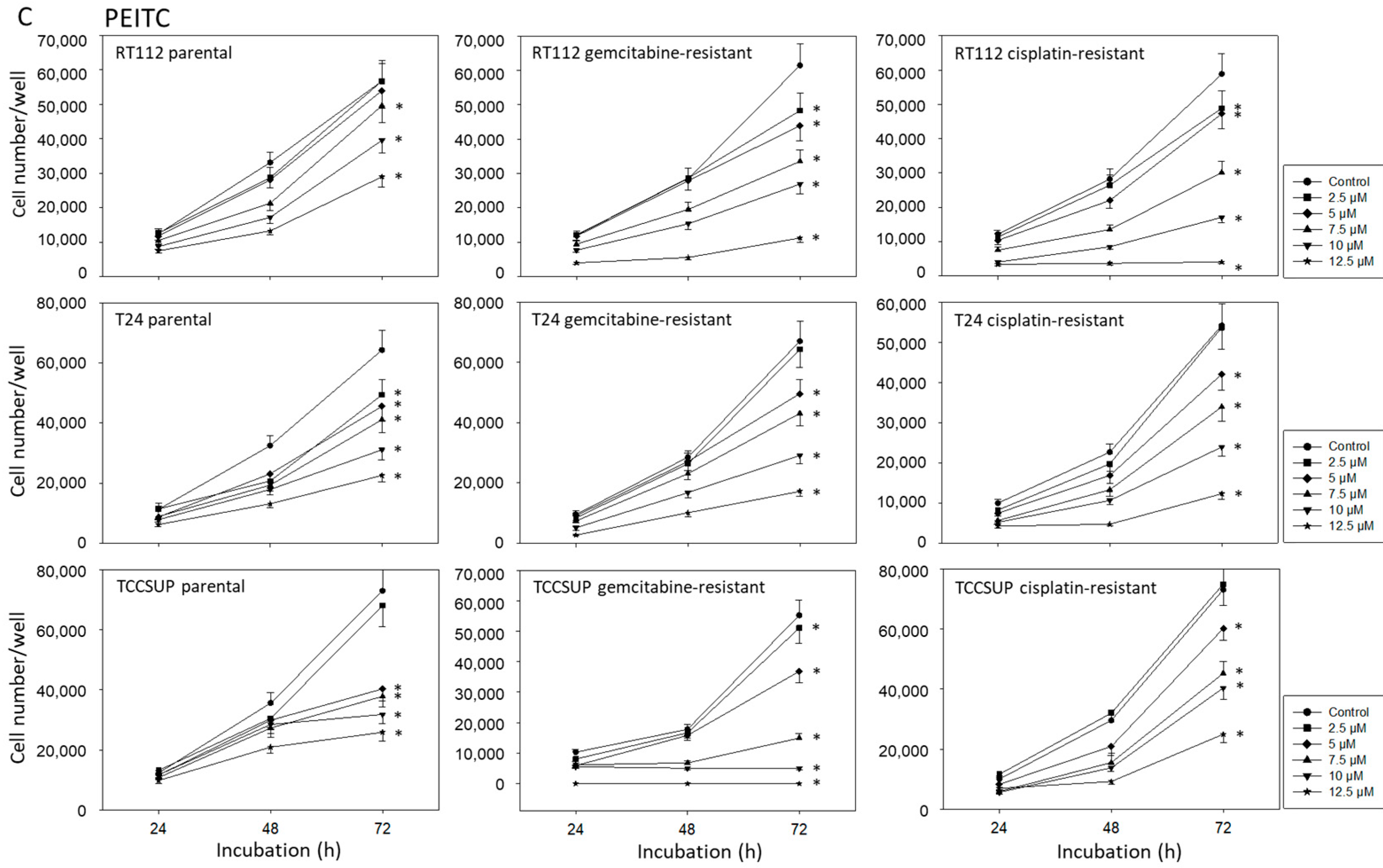
2.2. BITC, AITC, and PEITC Block Growth of Drug-Resistant Tumor Cells
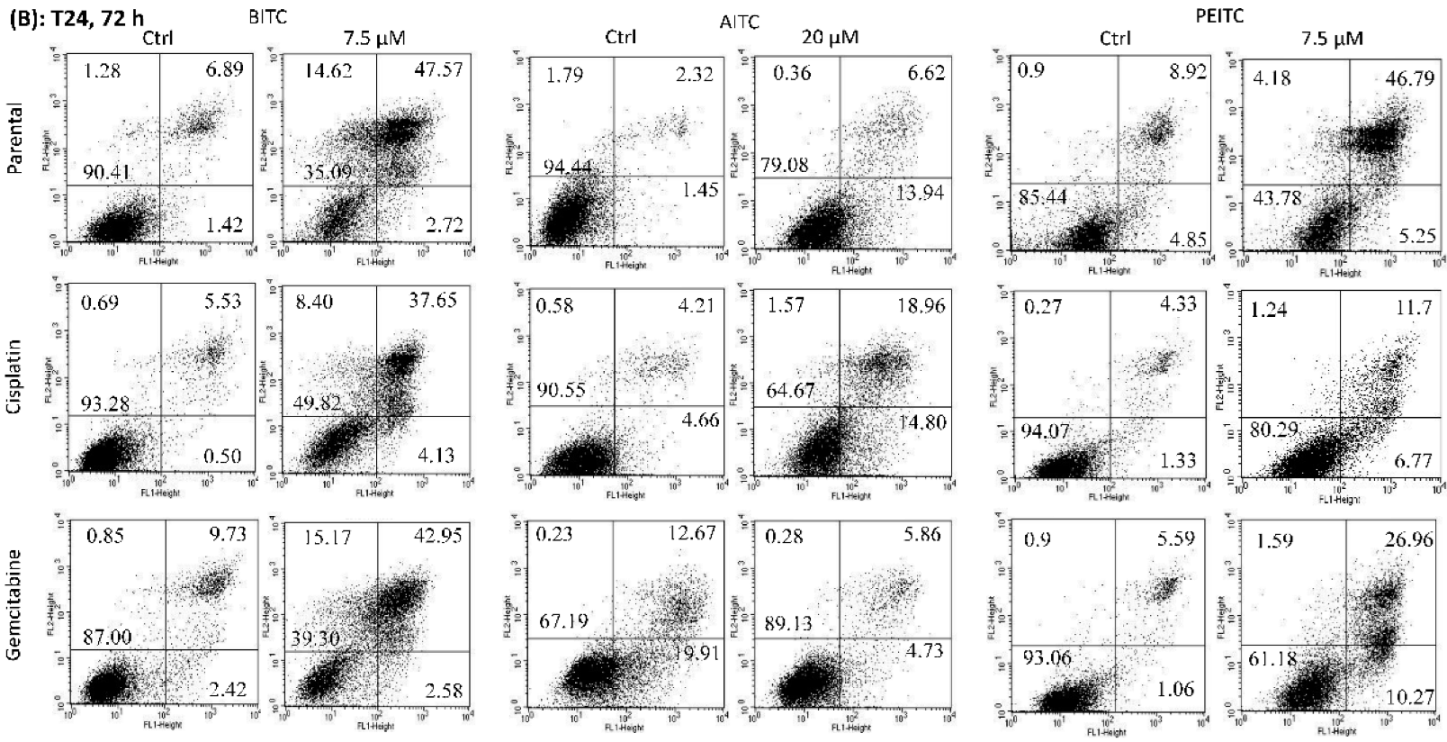
2.3. Apoptosis Induction by BITC, AITC, and PEITC
2.4. Suppression of Clonogenic Tumor Growth
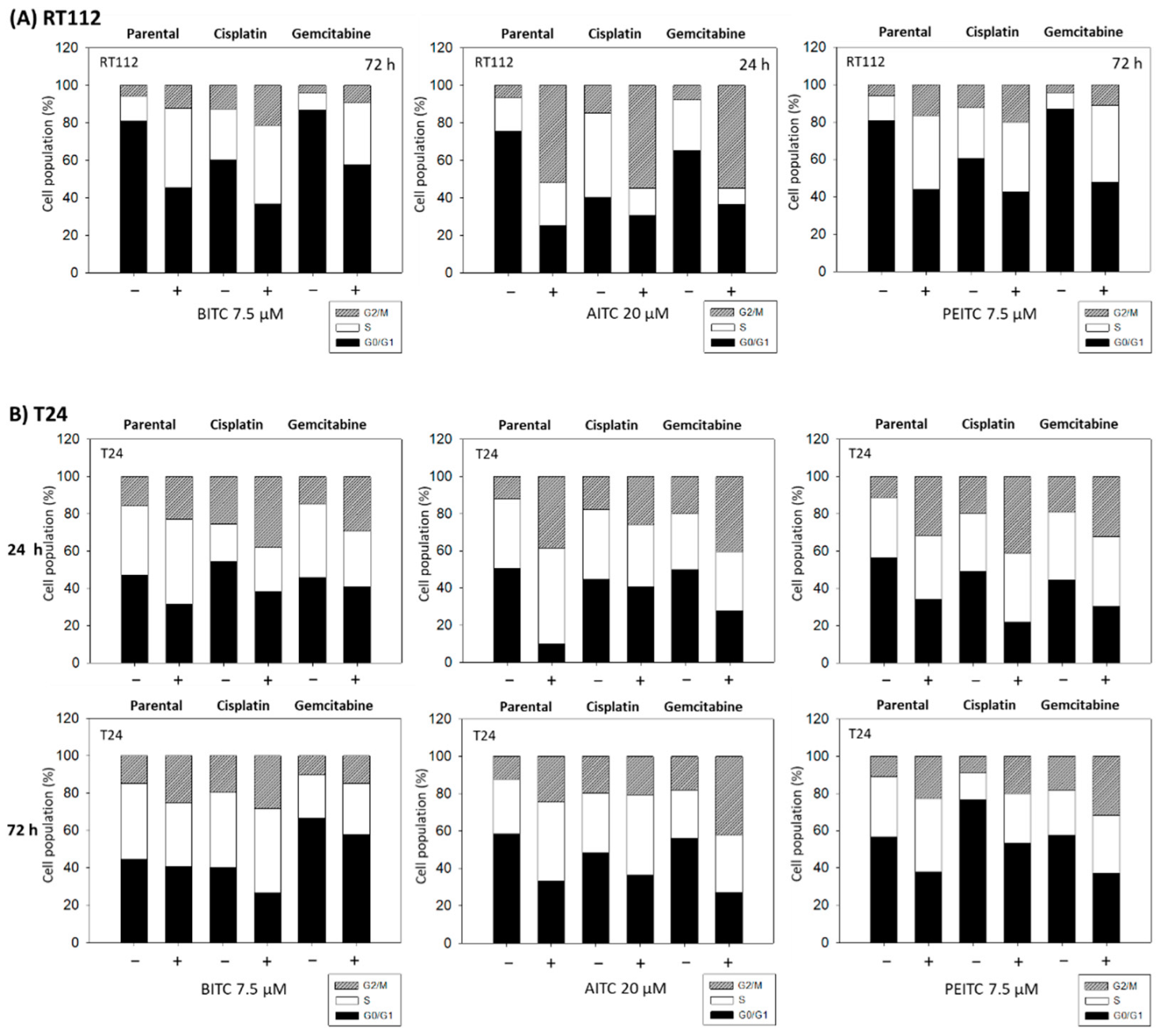
2.5. Cell Cycle Arrest by BITC, AITC, and PEITC
2.6. Alteration of Cell Cycle Protein Expression
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Resistance Induction
4.3. Cell Growth
4.4. Apoptosis
4.5. Clonogenic Growth
4.6. Cell Cycle Analysis
4.7. Western Blot Analysis
4.8. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Witjes, J.A.; Bruins, H.M.; Cathomas, R.; Compérat, E.M.; Cowan, N.C.; Gakis, G.; Hernández, V.; Linares Espinós, E.; Lorch, A.; Neuzillet, Y.; et al. European Association of Urology Guidelines on Muscle-invasive and Metastatic Bladder Cancer: Summary of the 2020 Guidelines. Eur. Urol. 2021, 79, 82–104. [Google Scholar] [CrossRef] [PubMed]
- Carril-Ajuria, L.; Martin-Soberón, M.C.; de Velasco, G.; Agarwal, N.; Castellano, D. Immunotherapy maintenance therapy for advanced urothelial carcinoma (aUC): A comprehensive review. J. Cancer Res. Clin. Oncol. 2022, 148, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Mancini, M.; Righetto, M.; Noessner, E. Checkpoint Inhibition in Bladder Cancer: Clinical Expectations, Current Evidence, and Proposal of Future Strategies Based on a Tumor-Specific Immunobiological Approach. Cancers 2021, 13, 6016. [Google Scholar] [CrossRef]
- Bellmunt, J.; de Wit, R.; Vaughn, D.J.; Fradet, Y.; Lee, J.L.; Fong, L.; Vogelzang, N.J.; Climent, M.A.; Petrylak, D.P.; Choueiri, T.K.; et al. KEYNOTE-045 Investigators. Pembrolizumab as Second-Line Therapy for Advanced Urothelial Carcinoma. N. Engl. J. Med. 2017, 376, 1015–1026. [Google Scholar] [CrossRef]
- Buckner, C.A.; Lafrenie, R.M.; Dénommée, J.A.; Caswell, J.M.; Want, D.A. Complementary and alternative medicine use in patients before and after a cancer diagnosis. Curr Oncol. 2018, 25, e275–e281. [Google Scholar] [CrossRef]
- Marijanović, I.; Bevanda Glibo, D.; Lasić, T.; Kraljević, M.; Buhovac, T.; Cerić, T.; Sokolović, E. The Use of Complementary and Alternative Medicine among Cancer Patients Undergoing Conventional Treatment: A Single-Centre Experience. Psychiatr. Danub. 2021, 33, 304–307. [Google Scholar]
- Jędrzejewska, A.; Ślusarska, B.J.; Szadowska-Szlachetka, Z.; Rudnicka-Drożak, E.; Panasiuk, L. Use of complementary and alternative medicine in patients with cancer and their relationship with health behaviours—Cross-sectional study. Ann. Agric. Environ. Med. 2021, 28, 475–482. [Google Scholar] [CrossRef]
- Park, T.; Lee, S. Clinical Experiences of Korean Medicine Treatment against Urinary Bladder Cancer in General Practice. Evid.-Based Complement. Altern. Med. 2016, 2016, 3759069. [Google Scholar] [CrossRef]
- Zhou, S.; Fan, X.; Du, X.; Liu, S.; Sun, H.; Zhang, Y. Effect of Traditional Chinese Medicine Combined with Bladder Perfusion with Hydroxycamptothecin on Color Ultrasound and Clinical Efficacy in Patients with Bladder Cancer Surgery. Evid.-Based Complement. Altern. Med. 2021, 2021, 7178414. [Google Scholar] [CrossRef]
- Juengel, E.; Erb, H.H.H.; Haferkamp, A.; Rutz, J.; Chun, F.K.; Blaheta, R.A. Relevance of the natural HDAC inhibitor sulforaphane as a chemopreventive agent in urologic tumors. Cancer Lett. 2018, 435, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Mastuo, T.; Miyata, Y.; Yuno, T.; Mukae, Y.; Otsubo, A.; Mitsunari, K.; Ohba, K.; Sakai, H. Molecular Mechanisms of the Anti-Cancer Effects of Isothiocyanates from Cruciferous Vegetables in Bladder Cancer. Molecules 2020, 25, 575. [Google Scholar] [CrossRef] [PubMed]
- Yeary, K.H.K.; Clark, N.; Saad-Harfouche, F.; Erwin, D.; Kuliszewski, M.G.; Li, Q.; McCann, S.E.; Yu, H.; Lincourt, C.; Zoellner, J.; et al. Cruciferous Vegetable Intervention to Reduce the Risk of Cancer Recurrence in Non-Muscle-Invasive Bladder Cancer Survivors: Development Using a Systematic Process. JMIR Cancer 2022, 8, e32291. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.F.; Tsai, T.F.; Lin, Y.C.; Chen, H.E.; Chou, K.Y.; Hwang, T.I. Benzyl isothiocyanate suppresses IGF1R, FGFR3 and mTOR expression by upregulation of miR-99a-5p in human bladder cancer cells. Int. J. Oncol. 2019, 54, 2106–2116. [Google Scholar] [CrossRef]
- Tang, L.; Zhang, Y. Mitochondria are the primary target in isothiocyanate-induced apoptosis in human bladder cancer cells. Mol. Cancer Ther. 2005, 4, 1250–1259. [Google Scholar] [CrossRef]
- Tang, K.; Lin, Y.; Li, L.M. The role of phenethyl isothiocyanate on bladder cancer ADM resistance reversal and its molecular mechanism. Anat. Rec. 2013, 296, 899–906. [Google Scholar] [CrossRef]
- Sávio, A.L.; da Silva, G.N.; Salvadori, D.M. Inhibition of bladder cancer cell proliferation by allyl isothiocyanate (mustard essential oil). Mutat. Res. 2015, 771, 29–35. [Google Scholar] [CrossRef]
- Savio, A.L.; da Silva, G.N.; de Camargo, E.A.; Salvadori, D.M. Cell cycle kinetics, apoptosis rates, DNA damage and TP53 gene expression in bladder cancer cells treated with allyl isothiocyanate (mustard essential oil). Mutat. Res. 2014, 762, 40–46. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Li, Y.; Geng, F.; Munday, R.; Zhang, Y. The principal urinary metabolite of allyl isothiocyanate, N-acetyl-S-(N-allylthiocarbamoyl)cysteine, inhibits the growth and muscle invasion of bladder cancer. Carcinogenesis 2012, 33, 394–398. [Google Scholar] [CrossRef]
- Lee, C.F.; Chiang, N.N.; Lu, Y.H.; Huang, Y.S.; Yang, J.S.; Tsai, S.C.; Lu, C.C.; Chen, F.A. Benzyl isothiocyanate (BITC) triggers mitochondria-mediated apoptotic machinery in human cisplatin-resistant oral cancer CAR cells. Biomedicine 2018, 8, 15. [Google Scholar] [CrossRef]
- Chang, P.Y.; Tsai, F.J.; Bau, D.T.; Hsu, Y.M.; Yang, J.S.; Tu, M.G.; Chiang, S.L. Potential effects of allyl isothiocyanate on inhibiting cellular proliferation and inducing apoptotic pathway in human cisplatin-resistant oral cancer cells. J. Formos. Med. Assoc. 2021, 120, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, E.; Sengupta, D.; Kumar, R.; Dehury, B.; Das, S.; Roy, M.; Mukherjee, S. Phenethylisothiocyanate Potentiates Platinum Therapy by Reversing Cisplatin Resistance in Cervical Cancer. Front. Pharmacol. 2022, 13, 803114. [Google Scholar] [CrossRef]
- Yu, T.T.; Chang, M.Y.; Hsieh, Y.J.; Chang, C.J. Suppression of multiple processes relevant to cancer progression by benzyl isothiocyanate may result from the inhibition of Aurora A kinase activity. Food Funct. 2020, 11, 9010–9019. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.L.; Huang, A.C.; Yang, J.S.; Liao, C.L.; Lu, H.F.; Chou, S.T.; Ma, C.Y.; Hsia, T.C.; Ko, Y.C.; Chung, J.G. Benzyl isothiocyanate (BITC) and phenethyl isothiocyanate (PEITC)-mediated generation of reactive oxygen species causes cell cycle arrest and induces apoptosis via activation of caspase-3, mitochondria dysfunction and nitric oxide (NO) in human osteogenic sarcoma U-2 OS cells. J. Orthop. Res. 2011, 29, 1199–1209. [Google Scholar] [PubMed]
- Chiang, J.H.; Tsai, F.J.; Hsu, Y.M.; Yin, M.C.; Chiu, H.Y.; Yang, J.S. Sensitivity of allyl isothiocyanate to induce apoptosis via ER stress and the mitochondrial pathway upon ROS production in colorectal adenocarcinoma cells. Oncol. Rep. 2020, 44, 1415–1424. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Lim, D.Y.; Kwon, G.T.; Kim, J.H.; Huang, Z.; Song, H.; Oh, Y.S.; Kang, Y.H.; Lee, K.W.; Dong, Z.; et al. Benzyl Isothiocyanate Inhibits Prostate Cancer Development in the Transgenic Adenocarcinoma Mouse Prostate (TRAMP) Model, Which Is Associated with the Induction of Cell Cycle G1 Arrest. Int. J. Mol. Sci. 2016, 17, 264. [Google Scholar] [CrossRef]
- Shang, H.S.; Shih, Y.L.; Lu, T.J.; Lee, C.H.; Hsueh, S.C.; Chou, Y.C.; Lu, H.F.; Liao, N.C.; Chung, J.G. Benzyl isothiocyanate (BITC) induces apoptosis of GBM 8401 human brain glioblastoma multiforms cells via activation of caspase-8/Bid and the reactive oxygen species-dependent mitochondrial pathway. Environ. Toxicol. 2016, 31, 1751–1760. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014, 507, 315–322. [Google Scholar] [CrossRef]
- Han, C.; Chen, S.; Ma, H.; Wen, X.; Wang, Z.; Xu, Y.; Jin, X.; Yu, X.; Wang, M. RPN2 Predicts Poor Prognosis and Promotes Bladder Cancer Growth and Metastasis via the PI3K-Akt Pathway. OncoTargets Ther. 2021, 14, 1643–1657. [Google Scholar] [CrossRef]
- Chi, M.; Liu, J.; Mei, C.; Shi, Y.; Liu, N.; Jiang, X.; Liu, C.; Xue, N.; Hong, H.; Xie, J.; et al. TEAD4 functions as a prognostic biomarker and triggers EMT via PI3K/AKT pathway in bladder cancer. J. Exp. Clin. Cancer Res. 2022, 41, 175. [Google Scholar] [CrossRef]
- Chang, Y.H.; Tam, H.L.; Lu, M.C.; Huang, H.S. Gemcitabine-induced Gli-dependent activation of hedgehog pathway resists to the treatment of urothelial carcinoma cells. PLoS ONE 2021, 16, e0254011. [Google Scholar]
- Xiong, Y.; Ju, L.; Yuan, L.; Chen, L.; Wang, G.; Xu, H.; Peng, T.; Luo, Y.; Xiao, Y.; Wang, X. KNSTRN promotes tumorigenesis and gemcitabine resistance by activating AKT in bladder cancer. Oncogene 2021, 40, 1595–1608. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Su, H.; Ruan, C.; Li, X. Keratin 17 knockdown suppressed malignancy and cisplatin tolerance of bladder cancer cells, as well as the activation of AKT and ERK pathway. Folia Histochem. Cytobiol. 2021, 59, 40–48. [Google Scholar] [CrossRef]
- Shigeta, K.; Hasegawa, M.; Kikuchi, E.; Yasumizu, Y.; Kosaka, T.; Mizuno, R.; Mikami, S.; Miyajima, A.; Kufe, D.; Oya, M. Role of the MUC1-C oncoprotein in the acquisition of cisplatin resistance by urothelial carcinoma. Cancer Sci. 2020, 111, 3639–3652. [Google Scholar] [CrossRef] [PubMed]
- McPherson, V.; Reardon, B.; Bhayankara, A.; Scott, S.N.; Boyd, M.E.; Garcia-Grossman, I.R.; Regazzi, A.M.; McCoy, A.S.; Kim, P.H.; Al-Ahmadie, H.; et al. A phase 2 trial of buparlisib in patients with platinum-resistant metastatic urothelial carcinoma. Cancer 2020, 126, 4532–4544. [Google Scholar] [CrossRef] [PubMed]
- Janczewski, Ł. Sulforaphane and Its Bifunctional Analogs: Synthesis and Biological Activity. Molecules 2022, 27, 1750. [Google Scholar] [CrossRef]
- Makarević, J.; Rutz, J.; Juengel, E.; Maxeiner, S.; Tsaur, I.; Chun, F.K.; Bereiter-Hahn, J.; Blaheta, R.A. Influence of the HDAC Inhibitor Valproic Acid on the Growth and Proliferation of Temsirolimus-Resistant Prostate Cancer Cells In Vitro. Cancers 2019, 11, 566. [Google Scholar] [CrossRef]
- Makarević, J.; Tawanaie, N.; Juengel, E.; Reiter, M.; Mani, J.; Tsaur, I.; Bartsch, G.; Haferkamp, A.; Blaheta, R.A. Cross-communication between histone H3 and H4 acetylation and Akt-mTOR signalling in prostate cancer cells. J. Cell. Mol. Med. 2014, 18, 1460–1466. [Google Scholar] [CrossRef]
- Yokoi, K.; Kobayashi, A.; Motoyama, H.; Kitazawa, M.; Shimizu, A.; Notake, T.; Yokoyama, T.; Matsumura, T.; Takeoka, M.; Miyagawa, S.I. Survival pathway of cholangiocarcinoma via AKT/mTOR signaling to escape RAF/MEK/ERK pathway inhibition by sorafenib. Oncol. Rep. 2018, 39, 843–850. [Google Scholar]
- Huan, J.; Grivas, P.; Birch, J.; Hansel, D.E. Emerging Roles for Mammalian Target of Rapamycin (mTOR) Complexes in Bladder Cancer Progression and Therapy. Cancers 2022, 14, 1555. [Google Scholar] [CrossRef]
- Li, X.; He, S.; Tian, Y.; Weiss, R.M.; Martin, D.T. Synergistic inhibition of GP130 and ERK signaling blocks chemoresistant bladder cancer cell growth. Cell. Signal. 2019, 63, 109381. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.H.; You, S.; Oh, J.W.; Yoon, J.; Yeon, A.; Shahid, M.; Cho, E.; Sairam, V.; Park, T.D.; Kim, K.P.; et al. Integrated proteomic and phosphoproteomic analyses of cisplatin-sensitive and resistant bladder cancer cells reveal CDK2 network as a key therapeutic target. Cancer Lett. 2018, 437, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xie, W.; Gu, P.; Cai, Q.; Wang, B.; Xie, Y.; Dong, W.; He, W.; Zhong, G.; Lin, T.; et al. Upregulated WDR5 promotes proliferation, self-renewal and chemoresistance in bladder cancer via mediating H3K4 trimethylation. Sci. Rep. 2015, 5, 8293. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rutz, J.; Maxeiner, S.; Grein, T.; Sonnenburg, M.; Khadir, S.E.; Makhatelashvili, N.; Mann, J.; Xie, H.; Cinatl, J.; Thomas, A.; et al. Allyl-, Butyl- and Phenylethyl-Isothiocyanate Modulate Akt–mTOR and Cyclin–CDK Signaling in Gemcitabine- and Cisplatin-Resistant Bladder Cancer Cell Lines. Int. J. Mol. Sci. 2022, 23, 10996. https://doi.org/10.3390/ijms231910996
Rutz J, Maxeiner S, Grein T, Sonnenburg M, Khadir SE, Makhatelashvili N, Mann J, Xie H, Cinatl J, Thomas A, et al. Allyl-, Butyl- and Phenylethyl-Isothiocyanate Modulate Akt–mTOR and Cyclin–CDK Signaling in Gemcitabine- and Cisplatin-Resistant Bladder Cancer Cell Lines. International Journal of Molecular Sciences. 2022; 23(19):10996. https://doi.org/10.3390/ijms231910996
Chicago/Turabian StyleRutz, Jochen, Sebastian Maxeiner, Timothy Grein, Marlon Sonnenburg, Salma El Khadir, Nino Makhatelashvili, Johanna Mann, Hui Xie, Jindrich Cinatl, Anita Thomas, and et al. 2022. "Allyl-, Butyl- and Phenylethyl-Isothiocyanate Modulate Akt–mTOR and Cyclin–CDK Signaling in Gemcitabine- and Cisplatin-Resistant Bladder Cancer Cell Lines" International Journal of Molecular Sciences 23, no. 19: 10996. https://doi.org/10.3390/ijms231910996
APA StyleRutz, J., Maxeiner, S., Grein, T., Sonnenburg, M., Khadir, S. E., Makhatelashvili, N., Mann, J., Xie, H., Cinatl, J., Thomas, A., Chun, F. K.-H., Haferkamp, A., Blaheta, R. A., & Tsaur, I. (2022). Allyl-, Butyl- and Phenylethyl-Isothiocyanate Modulate Akt–mTOR and Cyclin–CDK Signaling in Gemcitabine- and Cisplatin-Resistant Bladder Cancer Cell Lines. International Journal of Molecular Sciences, 23(19), 10996. https://doi.org/10.3390/ijms231910996