
Formulation, Characterisation and Evaluation of the Antihypertensive Peptides, Isoleucine-Proline-Proline and Leucine-Lysine-Proline in Chitosan Nanoparticles Coated with Zein for Oral Drug Delivery
 ,
,  ,
,  ,
,  and
and
Abstract
:
1. Introduction
2. Results
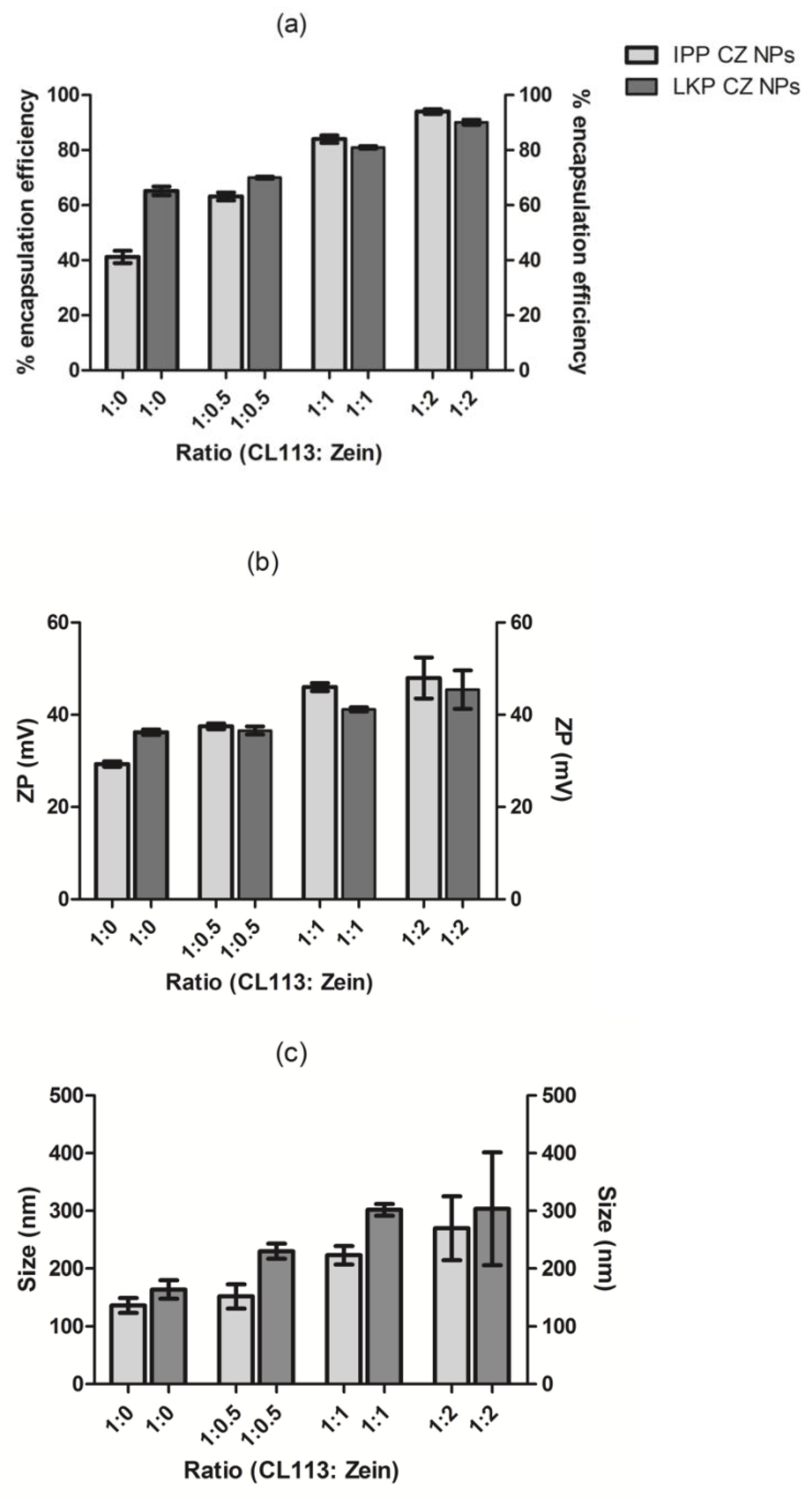
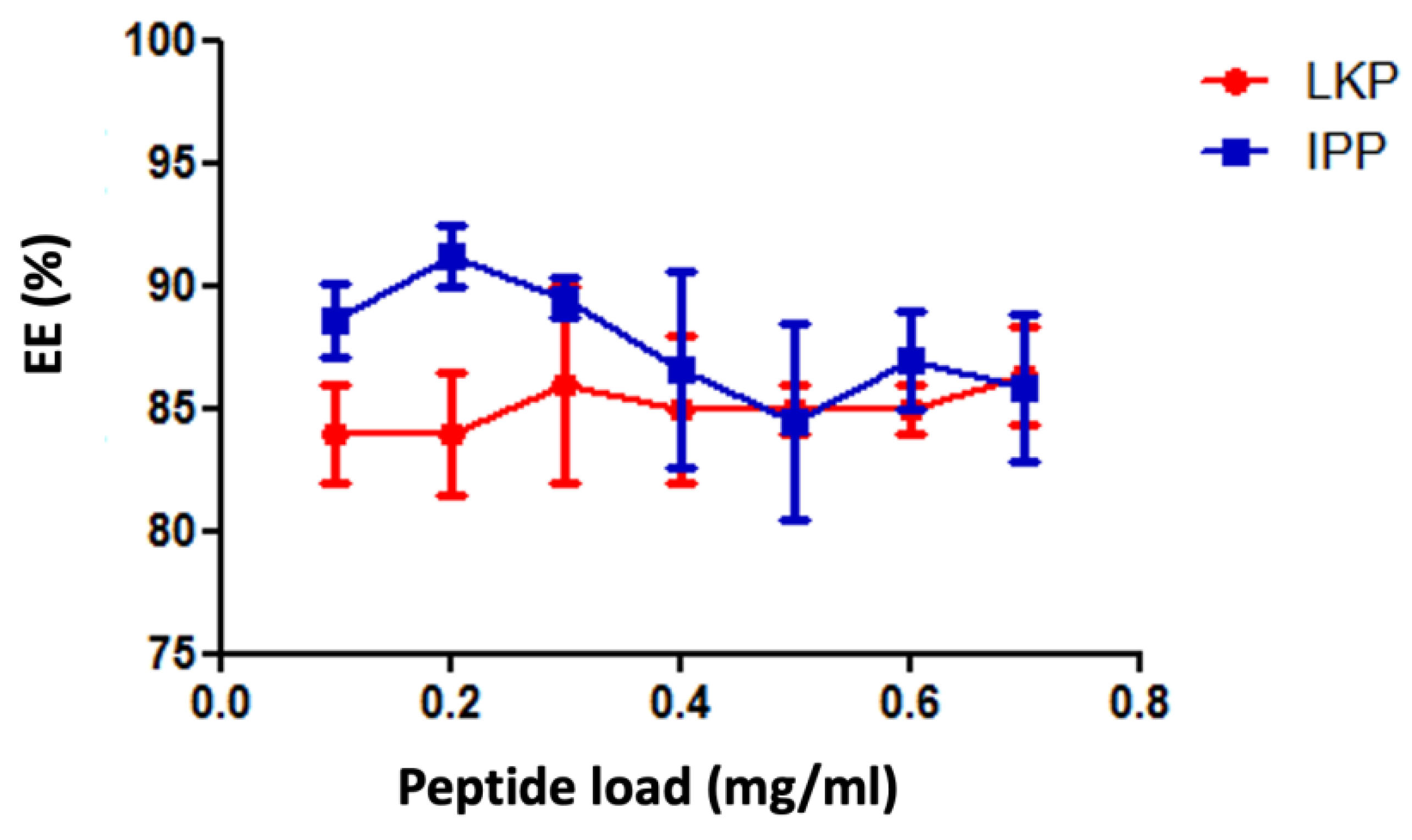
2.1. Physicochemical Characterisation of Peptide Loaded Chitosan (CL113 NP) and Chitosan Zein Nanoparticles (CZ NP)
2.2. Chemical Characterisation of CZ NPs Using FTIR Spectroscopy
2.3. ACE Inhibition of CZ NP
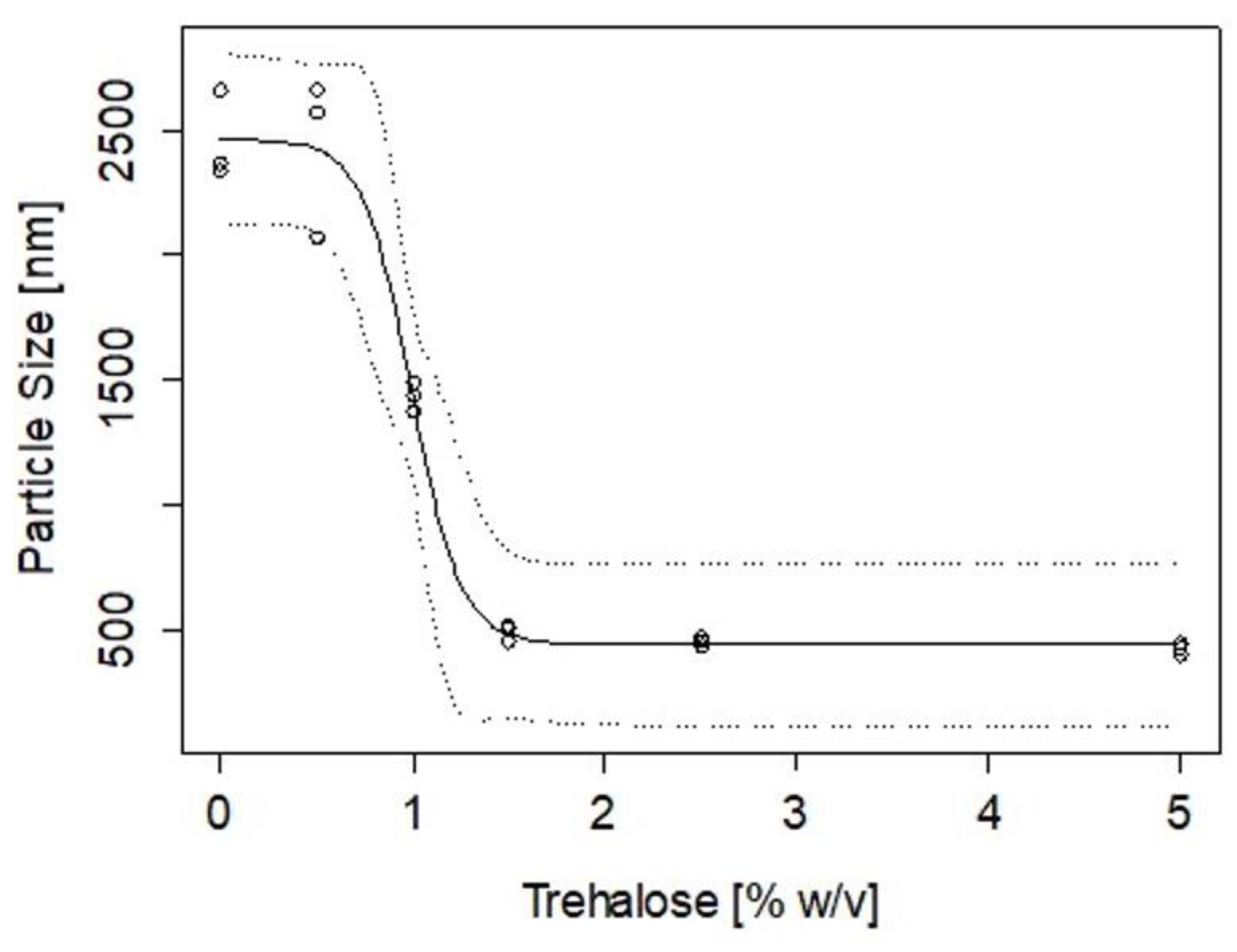
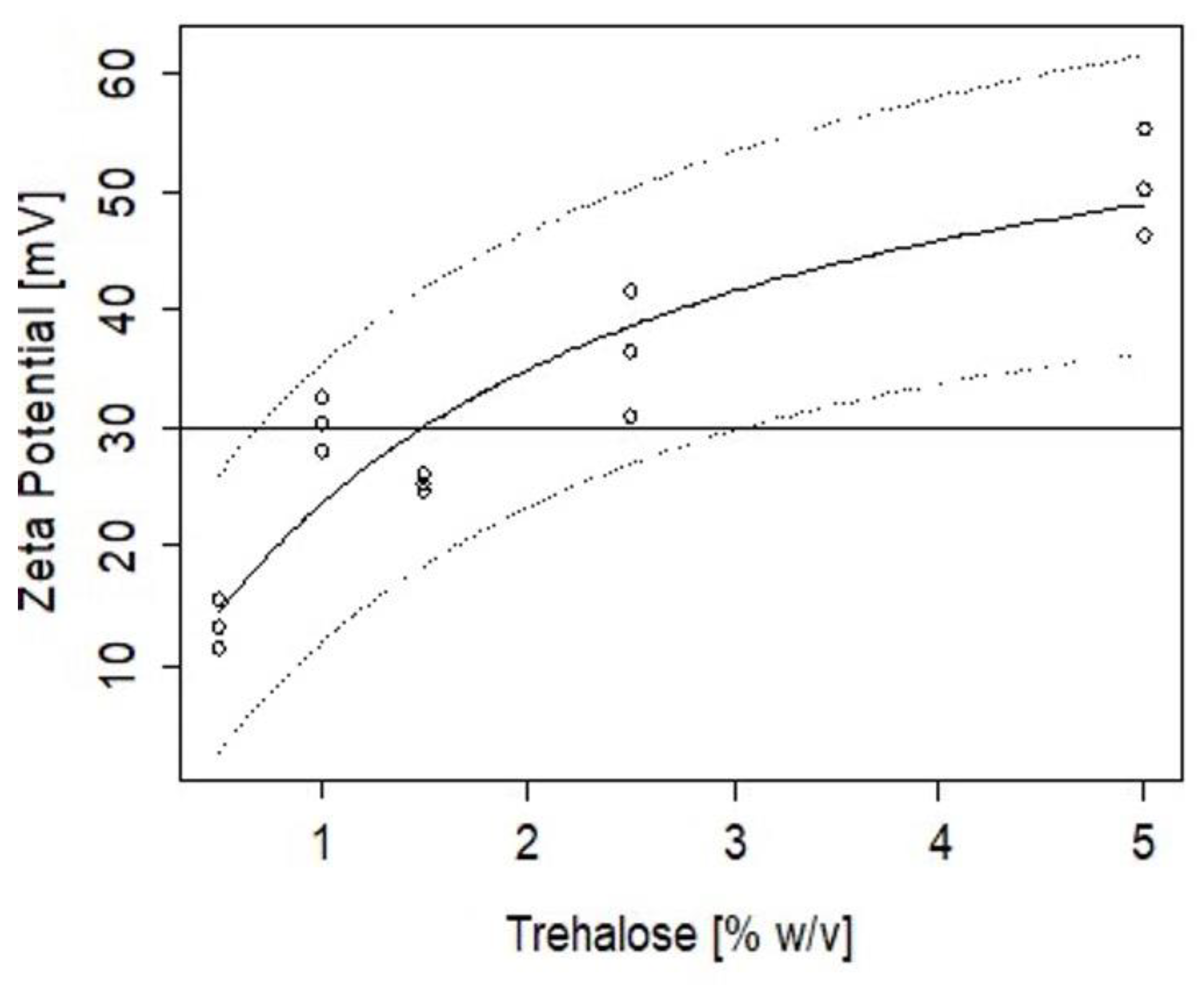
2.4. Stability of CZ NP during Lyophilisation
2.5. Cytotoxicity Assessment of Peptide CZ NP
2.6. In Vitro Controlled Release Studies of CZ NP
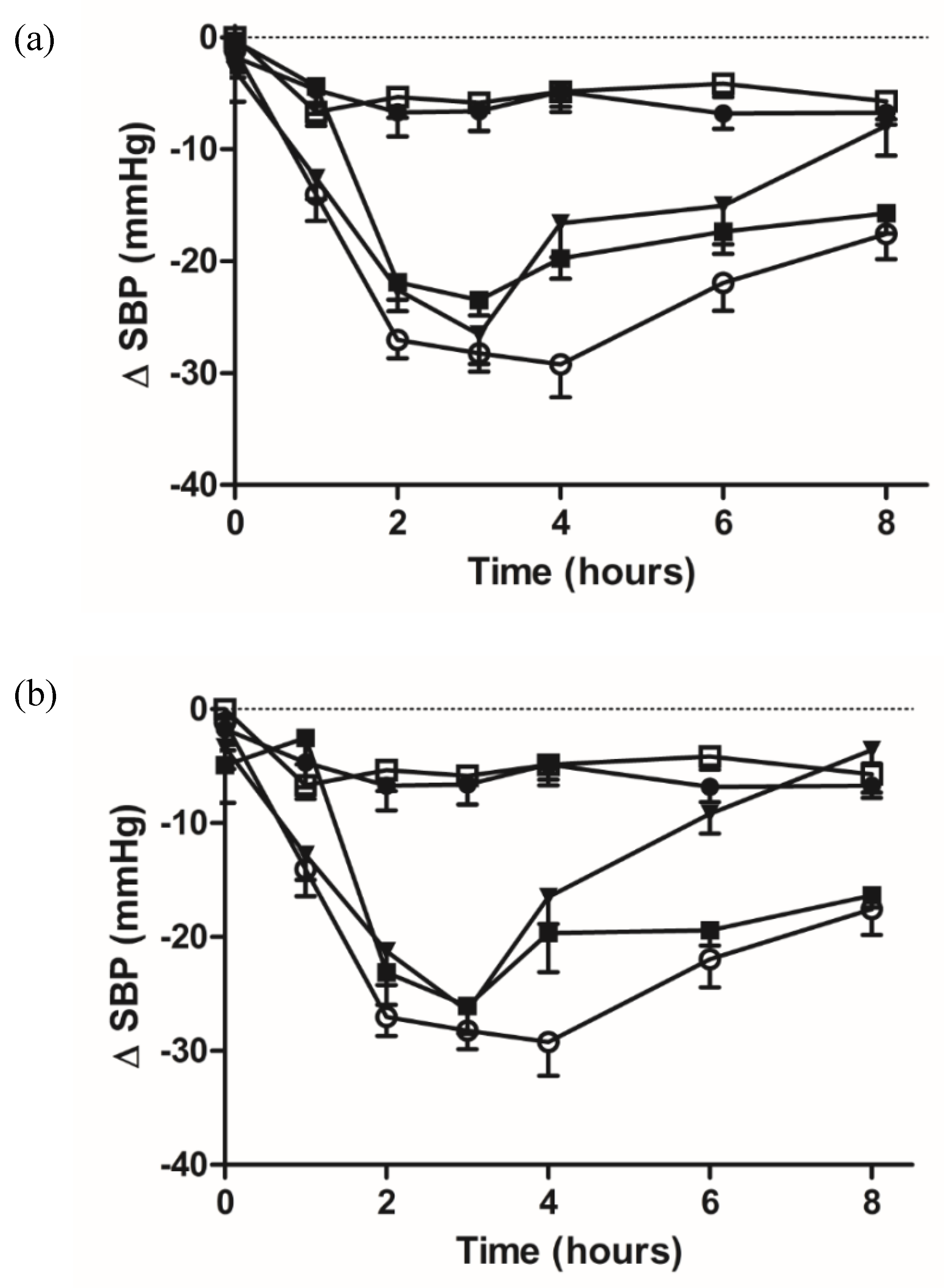
2.7. Pharmacodynamic Studies of Oral Administered Zein/Chitosan Loaded Nanoparticles Using the SHR Model
3. Discussion
4. Materials and Methods
4.1. Materials
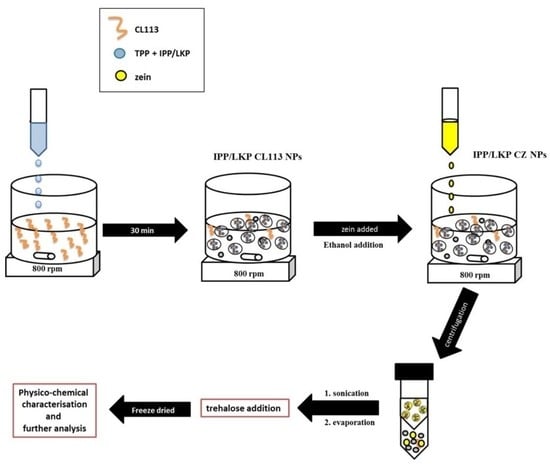
4.2. Formulation of Peptide Loaded Chitosan Nanoparticles
4.3. Coating of Peptide Loaded Chitosan Nanoparticles with Zein
4.4. Physicochemical Characterisation of CZ NP
4.5. Determination of Encapsulation Efficiency and Loading Capacity
4.6. ACE Inhibition Assessment of CZ NP
4.7. Stability of CZ NP after Freeze Drying
4.8. Cytotoxicity Assessment
4.9. In Vitro Dissolution Studies
4.10. Release Kinetics
4.11. In Vivo Pharmacodynamic Studies
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- World Health Organization. Hypertension. 2019. Available online: https://www.who.int/news-room/fact-sheets/detail/hypertension (accessed on 10 March 2020).
- Al Ghorani, H.; Götzinger, F.; Böhm, M.; Mahfoud, F. Arterial hypertension—Clinical trials update 2021. Nutr. Metab. Cardiovasc. Dis. 2022, 32, 21–31. [Google Scholar]
- Ott, C.; Schmieder, R.E. Diagnosis and treatment of arterial hypertension 2021. Kidney Int. 2021, 101, 36–46. [Google Scholar] [CrossRef] [PubMed]
- April-Sanders, A.K.; Golestaneh, L.; Zhang, L.; Swett, K.; Meissner, P.; Rodriguez, C.J. Hypertension Treatment and Control in a New York City Health Care System. J. Am. Heart Assoc. 2022, 11, e026077. [Google Scholar] [CrossRef]
- Slomski, A. Polypill for Initial Hypertension Treatment Is Promising. JAMA 2021, 326, 1572. [Google Scholar] [CrossRef]
- Plosker, G.L.; McTavish, D. Captopril. A review of its pharmacology and therapeutic efficacy after myocardial infarction and in ischaemic heart disease. Drugs Aging 1995, 7, 226–253. [Google Scholar] [CrossRef] [PubMed]
- Migdalof, B.H.; Antonaccio, M.J.; Mc Kinstry, D.N.; Singhvi, S.M.; Lan, S.-J.; Egli, P.; Kripalani, K.J. Captopril: Pharmacology, Metabolism, and Disposition. Drug Metab. Rev. 1984, 15, 841–869. [Google Scholar] [CrossRef] [PubMed]
- Isles, C.; Hodsman, G.; Robertson, J. Side-Effects of Captopril. Lancet 1983, 321, 355. [Google Scholar] [CrossRef]
- Abeer, M.M.; Trajkovic, S.; Brayden, D.J. Measuring the oral bioavailability of protein hydrolysates derived from food sources: A critical review of current bioassays. Biomed. Pharmacother. 2021, 144, 112275. [Google Scholar] [CrossRef]
- Turpeinen, A.M.; Ikonen, M.; Kivimäki, A.S.; Kautiainen, H.; Vapaatalo, H.; Korpela, R. A spread containing bioactive milk peptides Ile–Pro–Pro and Val–Pro–Pro, and plant sterols has antihypertensive and cholesterol-lowering effects. Food Funct. 2012, 3, 621–627. [Google Scholar] [CrossRef]
- Nakamura, Y.; Yamamoto, N.; Sakai, K.; Takano, T. Antihypertensive Effect of Sour Milk and Peptides Isolated from It That are Inhibitors to Angiotensin I-Converting Enzyme. J. Dairy Sci. 1995, 78, 1253–1257. [Google Scholar] [CrossRef]
- Brayden, D.; Hill, T.; Fairlie, D.; Maher, S.; Mrsny, R. Systemic delivery of peptides by the oral route: Formulation and medicinal chemistry approaches. Adv. Drug Deliv. Rev. 2020, 157, 2–36. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, J.P.; Heade, J.; Ryan, S.M.; Brayden, D.J. Stability, toxicity and intestinal permeation enhancement of two food-derived antihypertensive tripeptides, Ile-Pro-Pro and Leu-Lys-Pro. Peptides 2015, 71, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.; ElSayed, M.E. Progress and limitations of oral peptide delivery as a potentially transformative therapy. Expert Opin. Drug Deliv. 2022, 19, 163–178. [Google Scholar] [CrossRef] [PubMed]
- Frigaard, J.; Jensen, J.L.; Galtung, H.K.; Hiorth, M. The Potential of Chitosan in Nanomedicine: An Overview of the Cytotoxicity of Chitosan Based Nanoparticles. Front. Pharmacol. 2022, 13, 880377. [Google Scholar] [CrossRef] [PubMed]
- Pathomthongtaweechai, N.; Muanprasat, C. Potential Applications of Chitosan-Based Nanomaterials to Surpass the Gastrointestinal Physiological Obstacles and Enhance the Intestinal Drug Absorption. Pharmaceutics 2021, 13, 887. [Google Scholar] [CrossRef]
- Li, S.; Zhang, H.; Chen, K.; Jin, M.; Vu, S.H.; Jung, S.; He, N.; Zheng, Z.; Lee, M.-S. Application of chitosan/alginate nanoparticle in oral drug delivery systems: Prospects and challenges. Drug Deliv. 2022, 29, 1142–1149. [Google Scholar] [CrossRef]
- Amin, M.K.; Boateng, J.S. Enhancing Stability and Mucoadhesive Properties of Chitosan Nanoparticles by Surface Modification with Sodium Alginate and Polyethylene Glycol for Potential Oral Mucosa Vaccine Delivery. Mar. Drugs 2022, 20, 156. [Google Scholar] [CrossRef]
- Garcia-Fuentes, M.; Alonso, M.J. Chitosan-based drug nanocarriers: Where do we stand? J. Control. Release 2012, 161, 496–504. [Google Scholar] [CrossRef]
- Bayat, A.; Larijani, B.; Ahmadian, S.; Junginger, H.E.; Rafiee-Tehrani, M. Preparation and characterization of insulin nanoparticles using chitosan and its quaternized derivatives. Nanomed. Nanotechnol. Biol. Med. 2008, 4, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Yun, Y.; Cho, Y.W.; Park, K. Nanoparticles for oral delivery: Targeted nanoparticles with peptidic ligands for oral protein delivery. Adv. Drug Deliv. Rev. 2013, 65, 822–832. [Google Scholar] [CrossRef]
- Ramasamy, T.; Tran, T.H.; Cho, H.J.; Kim, J.H.; Kim, Y.I.; Jeon, J.Y.; Choi, H.-G.; Yong, C.S.; Kim, J.O. Chitosan-Based Polyelectrolyte Complexes as Potential Nanoparticulate Carriers: Physicochemical and Biological Characterization. Pharm. Res. 2014, 31, 1302–1314. [Google Scholar] [CrossRef]
- Amidi, M.; Mastrobattista, E.; Jiskoot, W.; Hennink, W.E. Chitosan-based delivery systems for protein therapeutics and antigens. Adv. Drug Deliv. Rev. 2010, 62, 59–82. [Google Scholar] [CrossRef]
- Sarmento, B.; Ferreira, D.; Jorgensen, L.; van de Weert, M. Probing insulin’s secondary structure after entrapment into alginate/chitosan nanoparticles. Eur. J. Pharm. Biopharm. 2007, 65, 10–17. [Google Scholar] [CrossRef]
- Garrait, G.; Beyssac, E.; Subirade, M. Development of a novel drug delivery system: Chitosan nanoparticles entrapped in alginate microparticles. J. Microencapsul. 2014, 31, 363–372. [Google Scholar] [CrossRef]
- Surendran, V.; Palei, N.N. Formulation and Characterization of Rutin Loaded Chitosan-alginate Nanoparticles: Antidiabetic and Cytotoxicity Studies. Curr. Drug Deliv. 2022, 19, 379–394. [Google Scholar] [CrossRef]
- Sarmento, B.; Ribeiro, A.; Veiga, F.; Ferreira, D. Development and characterization of new insulin containing polysaccharide nanoparticles. Colloids Surf. B Biointerfaces 2006, 53, 193–202. [Google Scholar] [CrossRef]
- Wang, F.; Li, J.; Tang, X.; Huang, K.; Chen, L. Polyelectrolyte three layer nanoparticles of chitosan/dextran sulfate/chitosan for dual drug delivery. Colloids Surf. B Biointerfaces 2020, 190, 110925. [Google Scholar] [CrossRef]
- Ryan, S.M.; McMorrow, J.; Umerska, A.; Patel, H.B.; Kornerup, K.N.; Tajber, L.; Murphy, E.P.; Perretti, M.; Corrigan, O.I.; Brayden, D.J. An intra-articular salmon calcitonin-based nanocomplex reduces experimental inflammatory arthritis. J. Control. Release 2013, 167, 120–129. [Google Scholar] [CrossRef]
- Silva, B.; Gonçalves, L.M.; Braz, B.S.; Delgado, E. Chitosan and Hyaluronic Acid Nanoparticles as Vehicles of Epoetin Beta for Subconjunctival Ocular Delivery. Mar. Drugs 2022, 20, 151. [Google Scholar] [CrossRef]
- Vozza, G.; Khalid, M.; Byrne, H.J.; Ryan, S.M.; Frias, J.M. Nutraceutical formulation, characterisation, and in-vitro evaluation of methylselenocysteine and selenocystine using food derived chitosan:zein nanoparticles. Food Res. Int. 2019, 120, 295–304. [Google Scholar] [CrossRef]
- Loureiro, J.; Miguel, S.P.; Seabra, I.J.; Ribeiro, M.P.; Coutinho, P. Single-Step Self-Assembly of Zein–Honey–Chitosan Nanoparticles for Hydrophilic Drug Incorporation by Flash Nanoprecipitation. Pharmaceutics 2022, 14, 920. [Google Scholar] [CrossRef] [PubMed]
- Paliwal, R.; Palakurthi, S. Zein in controlled drug delivery and tissue engineering. J. Control. Release 2014, 189, 108–122. [Google Scholar] [CrossRef]
- Yuan, Y.; Ma, M.; Wang, D.; Xu, Y. A review of factors affecting the stability of zein-based nanoparticles loaded with bioactive compounds: From construction to application. Crit. Rev. Food Sci. Nutr. 2022, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cui, L.; Che, X.; Zhang, H.; Shi, N.; Li, C.; Chen, Y.; Kong, W. Zein-based films and their usage for controlled delivery: Origin, classes and current landscape. J. Control. Release 2015, 206, 206–219. [Google Scholar] [CrossRef] [PubMed]
- Reboredo, C.; González-Navarro, C.J.; Martínez-López, A.L.; Martínez-Ohárriz, C.; Sarmento, B.; Irache, J.M. Zein-Based Nanoparticles as Oral Carriers for Insulin Delivery. Pharmaceutics 2021, 14, 39. [Google Scholar] [CrossRef]
- Müller, V.; Piai, J.F.; Fajardo, A.R.; Fávaro, S.L.; Rubira, A.; Muniz, E. Preparation and Characterization of Zein and Zein-Chitosan Microspheres with Great Prospective of Application in Controlled Drug Release. J. Nanomater. 2011, 2011, 928728. [Google Scholar] [CrossRef]
- Kretschmer, B. Infrared spectroscopy and optical rotatory dispersion of zein, wheat gluten and gliadin. J. Phys. Chem. 1957, 61, 1627–1631. [Google Scholar] [CrossRef]
- Zou, T.; Li, Z.; Percival, S.S.; Bonard, S.; Gu, L. Fabrication, characterization, and cytotoxicity evaluation of cranberry procyanidins-zein nanoparticles. Food Hydrocoll. 2012, 27, 293–300. [Google Scholar] [CrossRef]
- Sessa, D.J.; Mohamed, A.; Byars, J.A. Chemistry and Physical Properties of Melt-Processed and Solution-Cross-Linked Corn Zein. J. Agric. Food Chem. 2008, 56, 7067–7075. [Google Scholar] [CrossRef]
- Luo, Y.; Teng, Z.; Wang, Q. Development of Zein Nanoparticles Coated with Carboxymethyl Chitosan for Encapsulation and Controlled Release of Vitamin D3. J. Agric. Food Chem. 2012, 60, 836–843. [Google Scholar] [CrossRef]
- He, Z.; Santos, J.L.; Tian, H.; Huang, H.; Hu, Y.; Liu, L.; Leong, K.W.; Chen, Y.; Mao, H.-Q. Scalable fabrication of size-controlled chitosan nanoparticles for oral delivery of insulin. Biomaterials 2017, 130, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Date, A.A.; Hanes, J.; Ensign, L.M. Nanoparticles for oral delivery: Design, evaluation and state-of-the-art. J. Control. Release 2016, 240, 504–526. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Hafez, S.M.; Hathout, R.M.; Sammour, O.A. Towards better modeling of chitosan nanoparticles production: Screening different factors and comparing two experimental designs. Int. J. Biol. Macromol. 2014, 64, 334–340. [Google Scholar] [CrossRef]
- Dubey, S.K.; Parab, S.; Dabholkar, N.; Agrawal, M.; Singhvi, G.; Alexander, A.; Bapat, R.A.; Kesharwani, P. Oral peptide delivery: Challenges and the way ahead. Drug Discov. Today 2021, 26, 931–950. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Goand, U.K.; Husain, A.; Katekar, R.A.; Garg, R.; Gayen, J.R. Challenges of peptide and protein drug delivery by oral route: Current strategies to improve the bioavailability. Drug Dev. Res. 2021, 82, 927–944. [Google Scholar] [CrossRef]
- Gleeson, J.; Brayden, D.; Ryan, S.M. Evaluation of PepT1 transport of food-derived antihypertensive peptides, Ile-Pro-Pro and Leu-Lys-Pro using in vitro, ex vivo and in vivo transport models. Eur. J. Pharm. Biopharm. 2017, 115, 276–284. [Google Scholar] [CrossRef]
- Karthikeyan, K.; Vijayalakshmi, E.; Korrapati, P.S. Selective Interactions of Zein Microspheres with Different Class of Drugs: An In Vitro and In Silico Analysis. AAPS PharmSciTech 2014, 15, 1172–1180. [Google Scholar] [CrossRef]
- Lai, L.; Guo, H. Preparation of new 5-fluorouracil-loaded zein nanoparticles for liver targeting. Int. J. Pharm. 2011, 404, 317–323. [Google Scholar] [CrossRef]
- Thapa, R.K.; Nguyen, H.T.; Jeong, J.-H.; Shin, B.S.; Ku, S.K.; Choi, H.-G.; Yong, C.S.; Kim, J.O. Synergistic anticancer activity of combined histone deacetylase and proteasomal inhibitor-loaded zein nanoparticles in metastatic prostate cancers. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 885–896. [Google Scholar] [CrossRef]
- Zhang, L.; Li, K.; Yu, D.; Regenstein, J.M.; Dong, J.; Chen, W.; Xia, W. Chitosan/zein bilayer films with one-way water barrier characteristic: Physical, structural and thermal properties. Int. J. Biol. Macromol. 2022, 200, 378–387. [Google Scholar] [CrossRef]
- Zhang, Y.; Cui, L.; Li, F.; Shi, N.; Li, C.; Yu, X.; Chen, Y.; Kong, W. Design, fabrication and biomedical applications of zein-based nano/micro-carrier systems. Int. J. Pharm. 2016, 513, 191–210. [Google Scholar] [CrossRef]
- Regier, M.C.; Taylor, J.D.; Borcyk, T.; Yang, Y.; Pannier, A.K. Fabrication and characterization of DNA-loaded zein nanospheres. J. Nanobiotechnol. 2012, 10, 44. [Google Scholar] [CrossRef] [PubMed]
- Erickson, D.P.; Dunbar, M.; Hamed, E.; Ozturk, O.K.; Campanella, O.H.; Keten, S.; Hamaker, B.R. Atomistic Modeling of Peptide Aggregation and β-Sheet Structuring in Corn Zein for Viscoelasticity. Biomacromolecules 2021, 22, 1856–1866. [Google Scholar] [CrossRef] [PubMed]
- Debnath, K.; Pradhan, N.; Singh, B.K.; Jana, N.R.; Jana, N.R. Poly(trehalose) Nanoparticles Prevent Amyloid Aggregation and Suppress Polyglutamine Aggregation in a Huntington’s Disease Model Mouse. ACS Appl. Mater. Interfaces 2017, 9, 24126–24139. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lee, J.; Mansfield, K.M.; Ko, J.H.; Sallam, S.; Wesdemiotis, C.; Maynard, H.D. Trehalose Glycopolymer Enhances Both Solution Stability and Pharmacokinetics of a Therapeutic Protein. Bioconj. Chem. 2017, 28, 836–845. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.R.; Velikov, K.P. Zein as a source of functional colloidal nano- and microstructures. Curr. Opin. Colloid Interface Sci. 2014, 19, 450–458. [Google Scholar] [CrossRef]
- Podaralla, S.; Perumal, O. Influence of Formulation Factors on the Preparation of Zein Nanoparticles. AAPS PharmSciTech 2012, 13, 919–927. [Google Scholar] [CrossRef]
- Fujita, H.; Yoshikawa, M. LKPNM: A prodrug-type ACE-inhibitory peptide derived from fish protein. Immunopharmacology 1999, 44, 123–127. [Google Scholar] [CrossRef]
- Valjakka, J.; Siltari, A.; Kukkurainen, S.; Vapaatalo, H.; Viitanen, R. Does the cis/trans configuration of peptide bonds in bioactive tripeptides play a role in ACE-1 enzyme inhibition? Biol. Targets Ther. 2014, 8, 59–65. [Google Scholar] [CrossRef]
- van Platerink, C.J.; Janssen, H.-G.M.; Haverkamp, J. Development of an at-line method for the identification of angiotensin-I inhibiting peptides in protein hydrolysates. J. Chromatogr. B 2007, 846, 147–154. [Google Scholar] [CrossRef]
- Maeno, M.; Mizuno, S.; Mennear, J.H.; Bernard, B.K. Studies of the Toxicological Potential of Tripeptides (L-Valyl-L-prolyl-L-proline and L-lsoleucyl-L-prolyl-L-proline): VIII. Assessment of Cytotoxicity and Clastogenicity of Tripeptides-Containing Casein Hydrolysate and Lactobacillus helveticus-fermented Milk Powders in Chinese Hamster Lung Cells. Int. J. Toxicol. 2005, 24, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.-S.; Dong, J.; Lin, Z.-X.; Yang, B.; Wang, J.-Y. Comparison of cytocompatibility of zein film with other biomaterials and its degradability in vitro. Biopolymers 2005, 78, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Casey, A.; Herzog, E.; Davoren, M.; Lyng, F.; Byrne, H.; Chambers, G. Spectroscopic analysis confirms the interactions between single walled carbon nanotubes and various dyes commonly used to assess cytotoxicity. Carbon 2007, 45, 1425–1432. [Google Scholar] [CrossRef]
- Penalva, R.; Esparza, I.; Larraneta, E.; González-Navarro, C.J.; Gamazo, C.; Irache, J.M. Zein-Based Nanoparticles Improve the Oral Bioavailability of Resveratrol and Its Anti-inflammatory Effects in a Mouse Model of Endotoxic Shock. J. Agric. Food Chem. 2015, 63, 5603–5611. [Google Scholar] [CrossRef] [PubMed]
- Inchaurraga, L.; Martínez-López, A.L.; Martin-Arbella, N.; Irache, J.M. Zein-based nanoparticles for the oral delivery of insulin. Drug Deliv. Transl. Res. 2020, 10, 1601–1611. [Google Scholar] [CrossRef] [PubMed]
- Calvo, P.; Remu, C.; Pez, N.-L.; Vila-Jato, J.L. and Alonso, M.J. Novel Hydrophilic Chitosan–Polyethylene Oxide Nanoparticles as Protein Carriers. J. Appl. Polym. Sci. 1997, 63, 125–132. [Google Scholar] [CrossRef]
- Vimal, S.; Majeed, S.A.; Taju, G.; Nambi, K.; Raj, N.S.; Madan, N.; Farook, M.; Rajkumar, T.; Gopinath, D.; Hameed, A.S. RETRACTED: Chitosan tripolyphosphate (CS/TPP) nanoparticles: Preparation, characterization and application for gene delivery in shrimp. Acta Trop. 2013, 128, 486–493. [Google Scholar] [CrossRef] [PubMed]
- Al-Qadi, S.; Grenha, A.; Remuñán-López, C. Microspheres loaded with polysaccharide nanoparticles for pulmonary delivery: Preparation, structure and surface analysis. Carbohydr. Polym. 2011, 86, 25–34. [Google Scholar] [CrossRef]
- Abdelwahed, W.; Degobert, G.; Stainmesse, S.; Fessi, H. Freeze-drying of nanoparticles: Formulation, process and storage considerations. Adv. Drug Deliv. Rev. 2006, 58, 1688–1713. [Google Scholar] [CrossRef]
- Szymańska, E.; Winnicka, K. Stability of Chitosan—A Challenge for Pharmaceutical and Biomedical Applications. Mar. Drugs 2015, 13, 1819–1846. [Google Scholar] [CrossRef]
- RC Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022. [Google Scholar]
- Bagre, A.P.; Jain, K.; Jain, N.K. Alginate coated chitosan core shell nanoparticles for oral delivery of enoxaparin: In vitro and in vivo assessment. Int. J. Pharm. 2013, 456, 31–40. [Google Scholar] [CrossRef] [PubMed]
- British Pharmacopoeia. Dissolution for Delayed Release of Solid Dosage Forms (Method B); TSO: London, UK, 2016. [Google Scholar]
- Siepmann, J.; Peppas, N.A. Higuchi equation: Derivation, applications, use and misuse. Int. J. Pharm. 2011, 418, 6–12. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Peptide Load (µg/mL) | CL113 (mg/mL) | TPP (mg/mL) | Ratio (CL113:TPP) | Particle Size (nm) | ZP (mV) | EE (%) | LC (%) |
|---|---|---|---|---|---|---|---|---|
| LKP | 100 | 1.58 | 0.27 | 6:1 | 166 ± 14 | 32 ± 2 | 65 ± 3 | 3 ± 0.5 |
| IPP | 100 | 1.45 | 0.40 | 4:1 | 136 ± 23 | 29 ± 2 | 41 ± 5 | 2 ± 0.8 |
| Peptide | Peptide Load (µg/mL) | CL113 (mg/mL) | TPP (mg/mL) | Zein (mg/mL) | Particle Size (nm) | ZP (mV) | EE (%) | LC (%) |
|---|---|---|---|---|---|---|---|---|
| LKP | 100 | 1.58 | 0.27 | 1.58 | 301 ± 16 | 41 ± 1 | 80 ± 1 | 12.3 ± 0.3 |
| IPP | 100 | 1.45 | 0.40 | 1.45 | 223 ± 9 | 46 ± 2 | 84 ± 3 | 11.6 ± 0.5 |
| ACE Inhibition (%) | |||
|---|---|---|---|
| Concentrations | Native | CL113 NP | CZ NP |
| IPP (10 µM) | 79 ± 2 | 81 ± 3 | 83 ± 9 |
| LKP (10 µM) | 80 ± 1 | 83 ± 2 | 84 ± 11 |
| Controls | |||
| Captopril (10 µM) | 86 ± 4 | - | - |
| Unloaded NP | - | 5 ± 2 | 6 ± 5 |
| Coefficients | Estimated | t-Value | p-Value |
|---|---|---|---|
| IPP NP | |||
| ks1 | 14.7 ± 0.5 | 27.3 | 0.000 |
| ks2 | −0.7 ± 0.1 | −12.5 | 0.000 |
| ki1 | 1.4 ± 0.2 | 9.1 | 0.000 |
| LKP NP | |||
| ks1 | −2.3 ± 0.6 | −3.8 | 0.000 |
| ks2 | 0.2 ± 0.1 | 3.1 | 0.002 |
| ki1 | 1.4 ± 0.2 | 9.1 | 0.000 |
| CZ NP (IPP and LKP) | |||
| ks2-ks2.(CZ) | 0.2 ± 0.1 | 4.4 | 0.000 |
| ki1-ki1.(CZ) | 3.7 ± 0.2 | 24.6 | 0.000 |
| Time (Hours) | PBS | Captopril | Unloaded NP | IPP | IPP-NP | LKP | LKP-NP |
|---|---|---|---|---|---|---|---|
| 0 | −1.7 ± 1.8 | −1.2 ± 1.9 | −0.1 ± 2.7 | −2.9 ± 2.7 | −0.3 ± 2.4 | −3.4 ± 1.8 | −4.9 ± 3.2 |
| 1 | −4.7 ± 2.2 | −14 ± 2.4 *** | −6.7 ± 1.2 | −12.6 ± 1.8 * | −4.4 ± 1.8 | −12.8 ± 2.2 * | −2.5 ± 2.3 |
| 2 | −6.7 ± 2.2 | −27 ± 1.6 *** | −5.4 ± 1.8 | −22.5 ± 1.9 *** | −21.8 ± 1.6 *** | −21.2 ± 2.9 *** | −23.1 ± 2.8 *** |
| 3 | −6.6 ± 1.7 | −28.2 ± 1.6 *** | −5.8 ± 2.5 | −26.5 ± 2.6 *** | −23.5 ± 1.4 *** | 26.5 ± 1.9 *** | −26 ± 2.4 *** |
| 4 | −4.8 ± 1.8 | −29.2 ± 2.9 *** | −4.7 ± 1.3 | −16.6 ± 3.1 ** | −19.8 ± 1.8 *** | −16.5 ± 2.4 *** | −19.6 ± 3.5 *** |
| 6 | −6.8 ± 1.3 | −21.9 ± 2.5 *** | −4.2 ± 1.2 | −15.1 ± 4.3 | −17.4 ± 1.1 ** | −9.2 ± 1.7 | −19.4 ± 1.3 *** |
| 8 | −6.7 ± 1.1 | −17.5 ± 2.2 * | −5.7 ± 1.6 | −7.8 ± 2.6 | −15.7 ± 1.8 * | −3.6 ± 1.2 | −16.3 ± 1.1 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khalid Danish, M.; Gleeson, J.P.; Brayden, D.J.; Byrne, H.J.; Frías, J.M.; Ryan, S.M. Formulation, Characterisation and Evaluation of the Antihypertensive Peptides, Isoleucine-Proline-Proline and Leucine-Lysine-Proline in Chitosan Nanoparticles Coated with Zein for Oral Drug Delivery. Int. J. Mol. Sci. 2022, 23, 11160. https://doi.org/10.3390/ijms231911160
Khalid Danish M, Gleeson JP, Brayden DJ, Byrne HJ, Frías JM, Ryan SM. Formulation, Characterisation and Evaluation of the Antihypertensive Peptides, Isoleucine-Proline-Proline and Leucine-Lysine-Proline in Chitosan Nanoparticles Coated with Zein for Oral Drug Delivery. International Journal of Molecular Sciences. 2022; 23(19):11160. https://doi.org/10.3390/ijms231911160
Chicago/Turabian StyleKhalid Danish, Minna, John P. Gleeson, David J. Brayden, Hugh J. Byrne, Jesus M. Frías, and Sinéad M. Ryan. 2022. "Formulation, Characterisation and Evaluation of the Antihypertensive Peptides, Isoleucine-Proline-Proline and Leucine-Lysine-Proline in Chitosan Nanoparticles Coated with Zein for Oral Drug Delivery" International Journal of Molecular Sciences 23, no. 19: 11160. https://doi.org/10.3390/ijms231911160