
Synthesis, Anticancer Activity and Molecular Docking Studies of Novel N-Mannich Bases of 1,3,4-Oxadiazole Based on 4,6-Dimethylpyridine Scaffold

 , ,
, ,  ,
,  , , and
, , and
Abstract
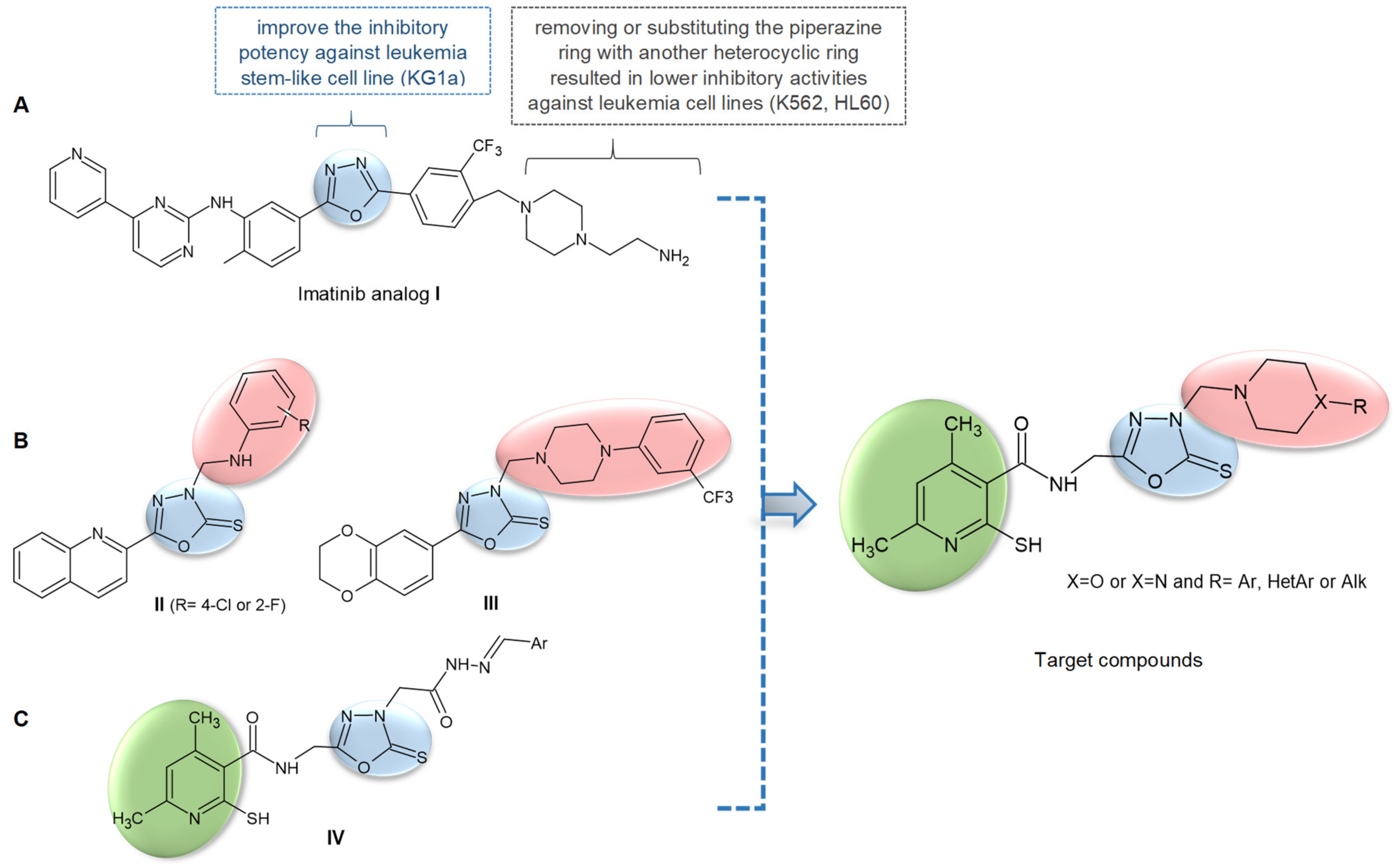
:1. Introduction
2. Results and Discussion
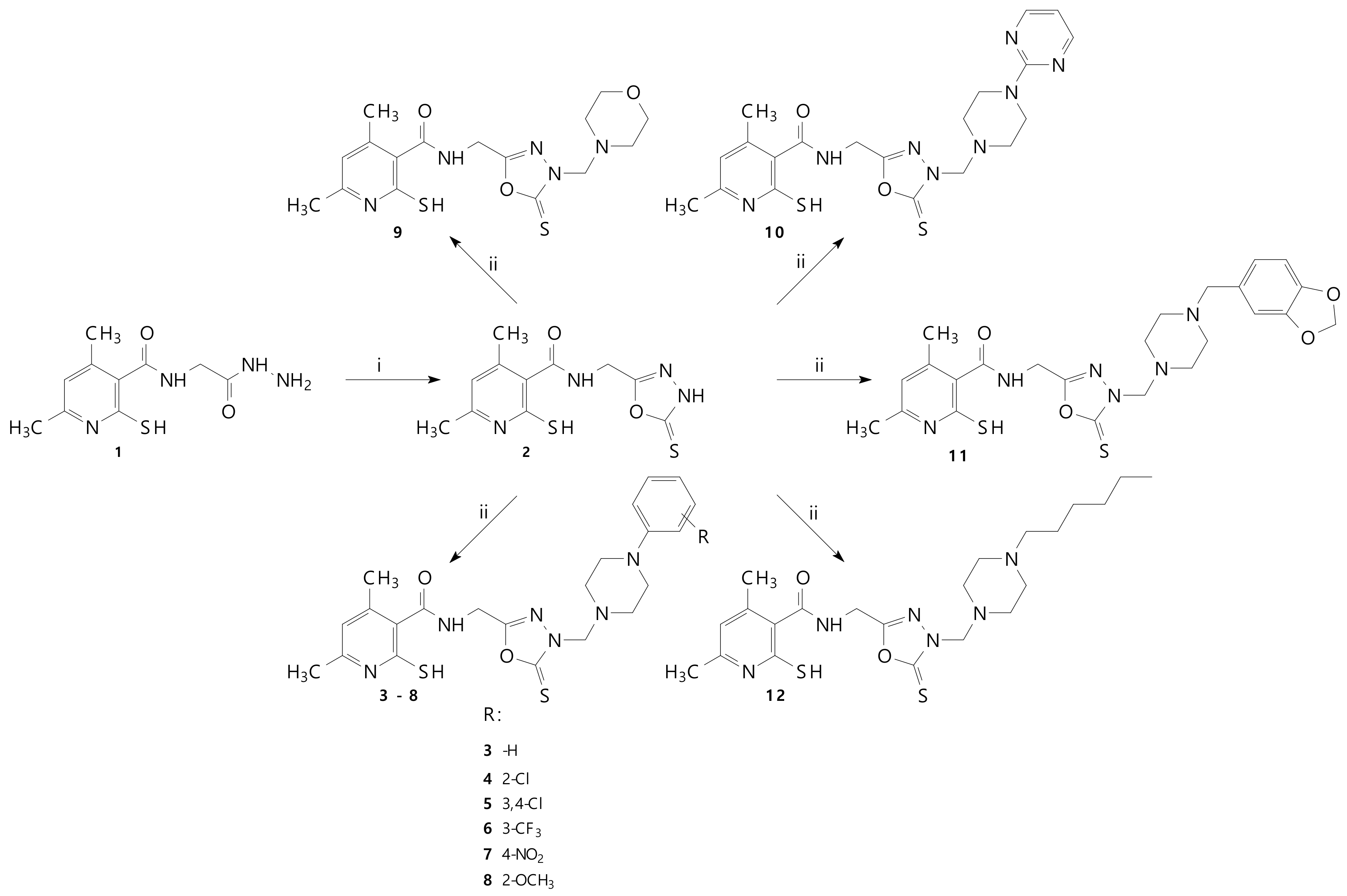
2.1. Chemistry
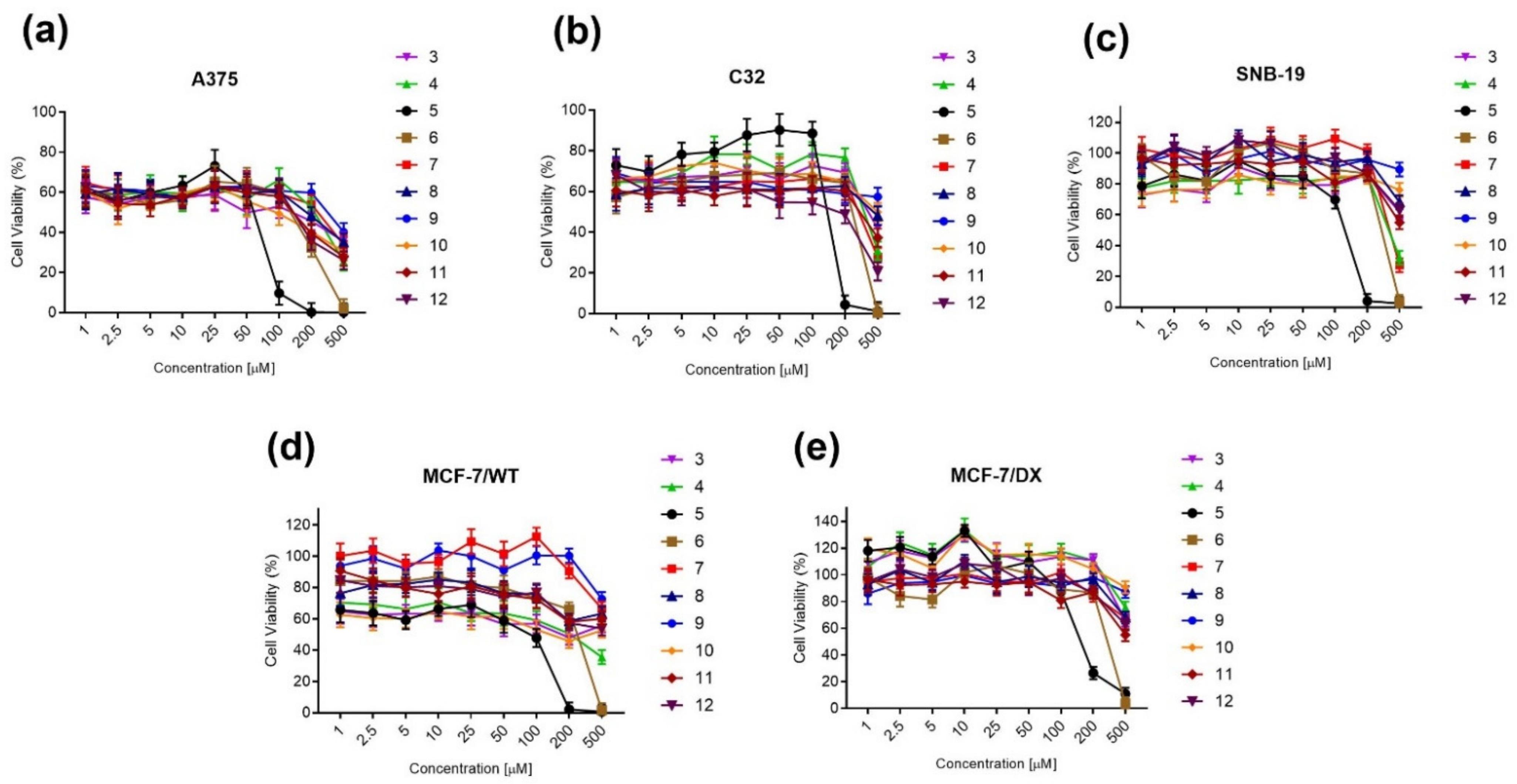
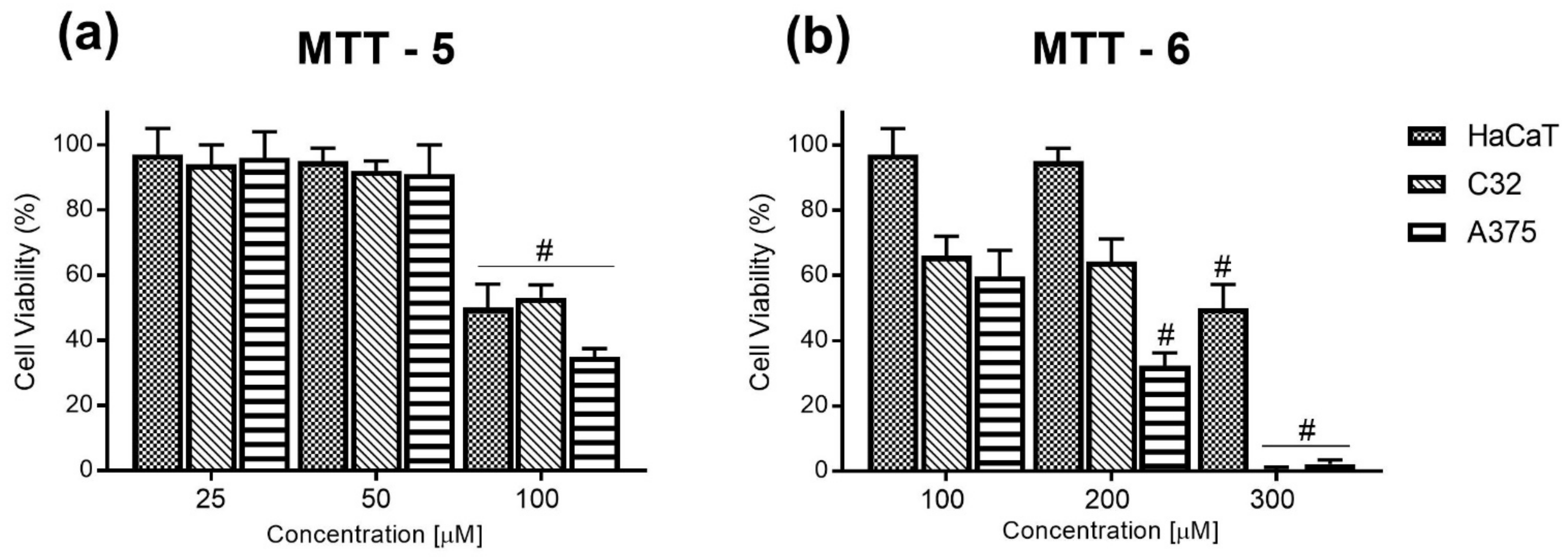
2.2. Biological Tests
2.2.1. MTT Cell Viability Assay
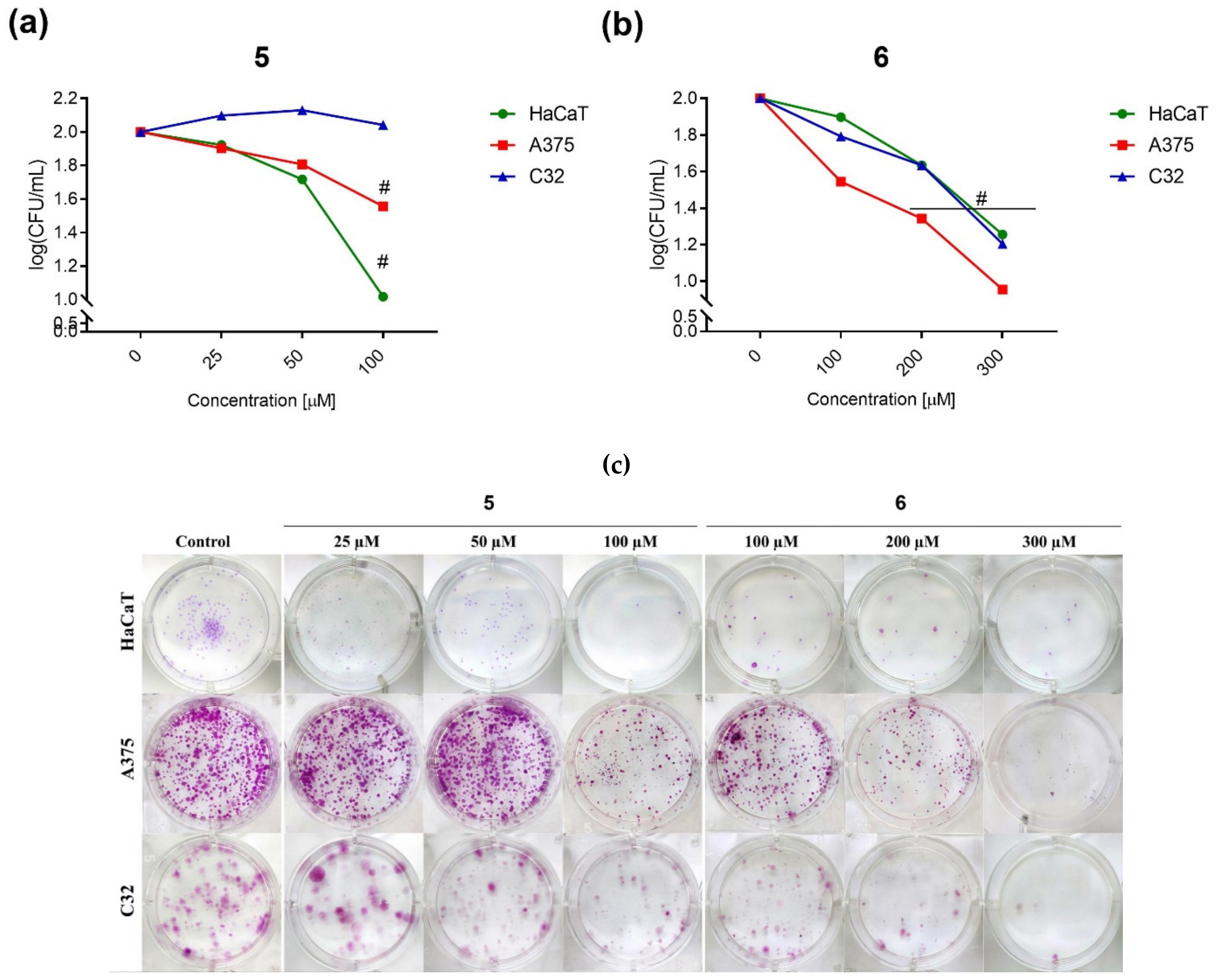
2.2.2. Clonogenic Assay
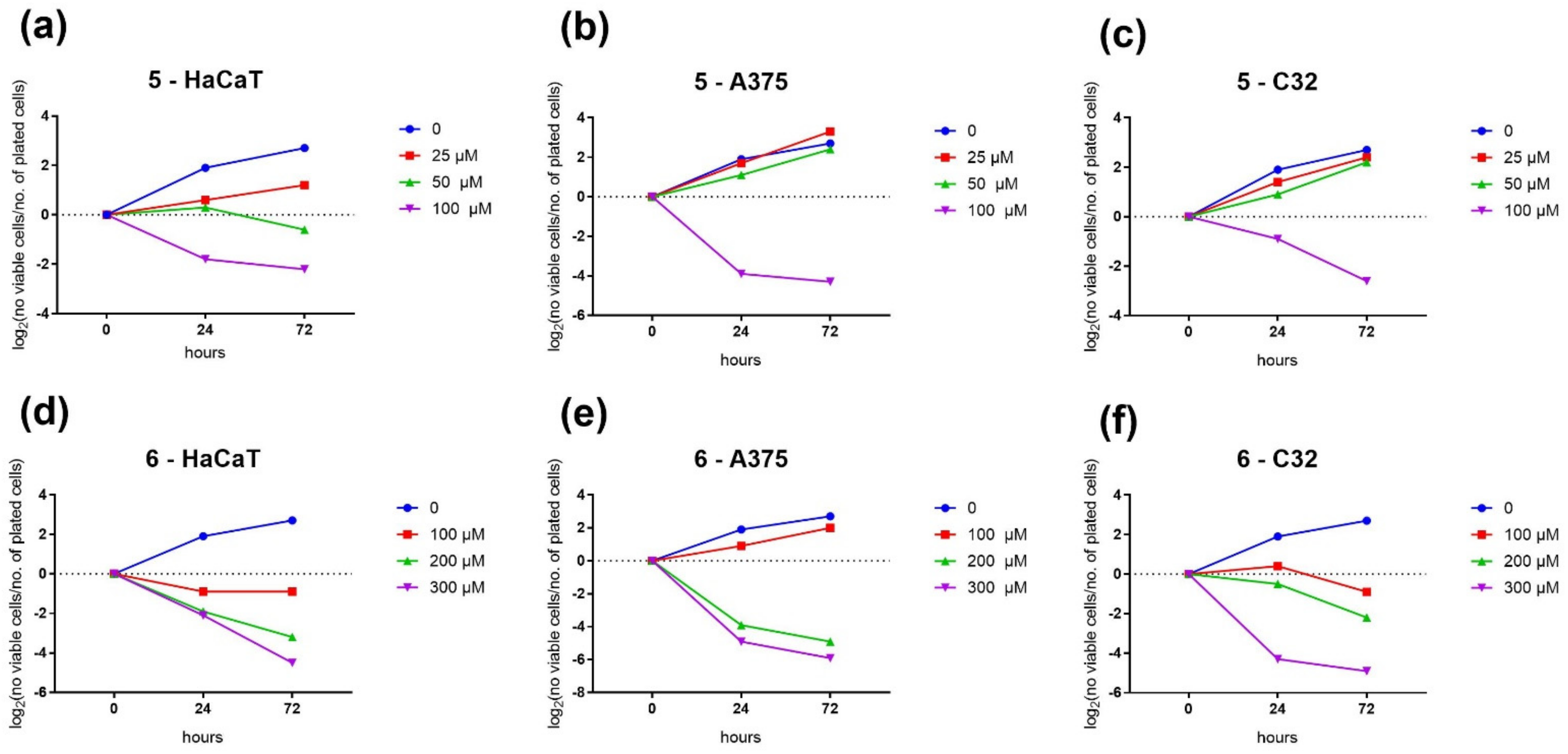
2.2.3. Population Doubling Time
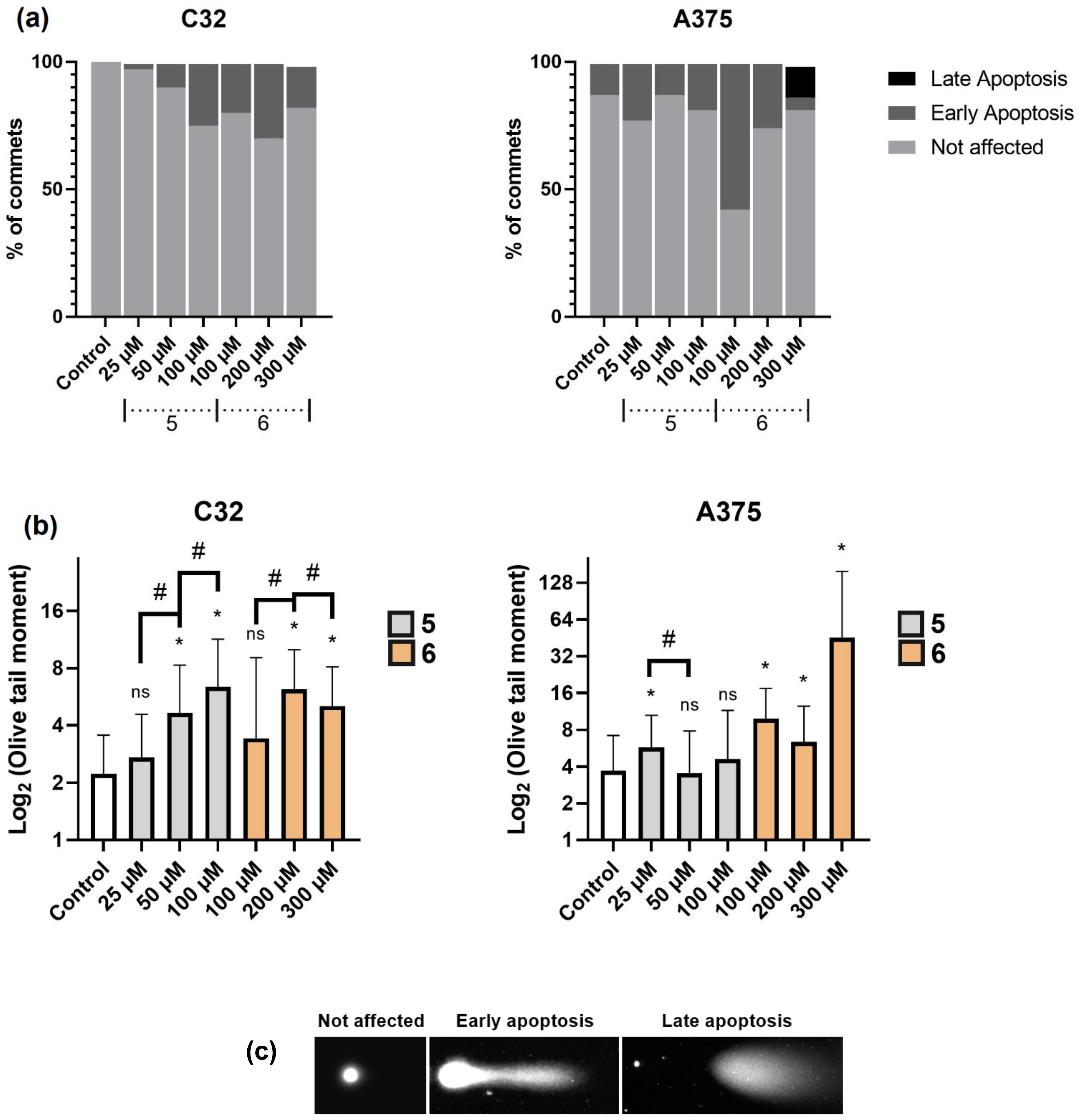
2.2.4. Cell Death Detection by Comet Assay
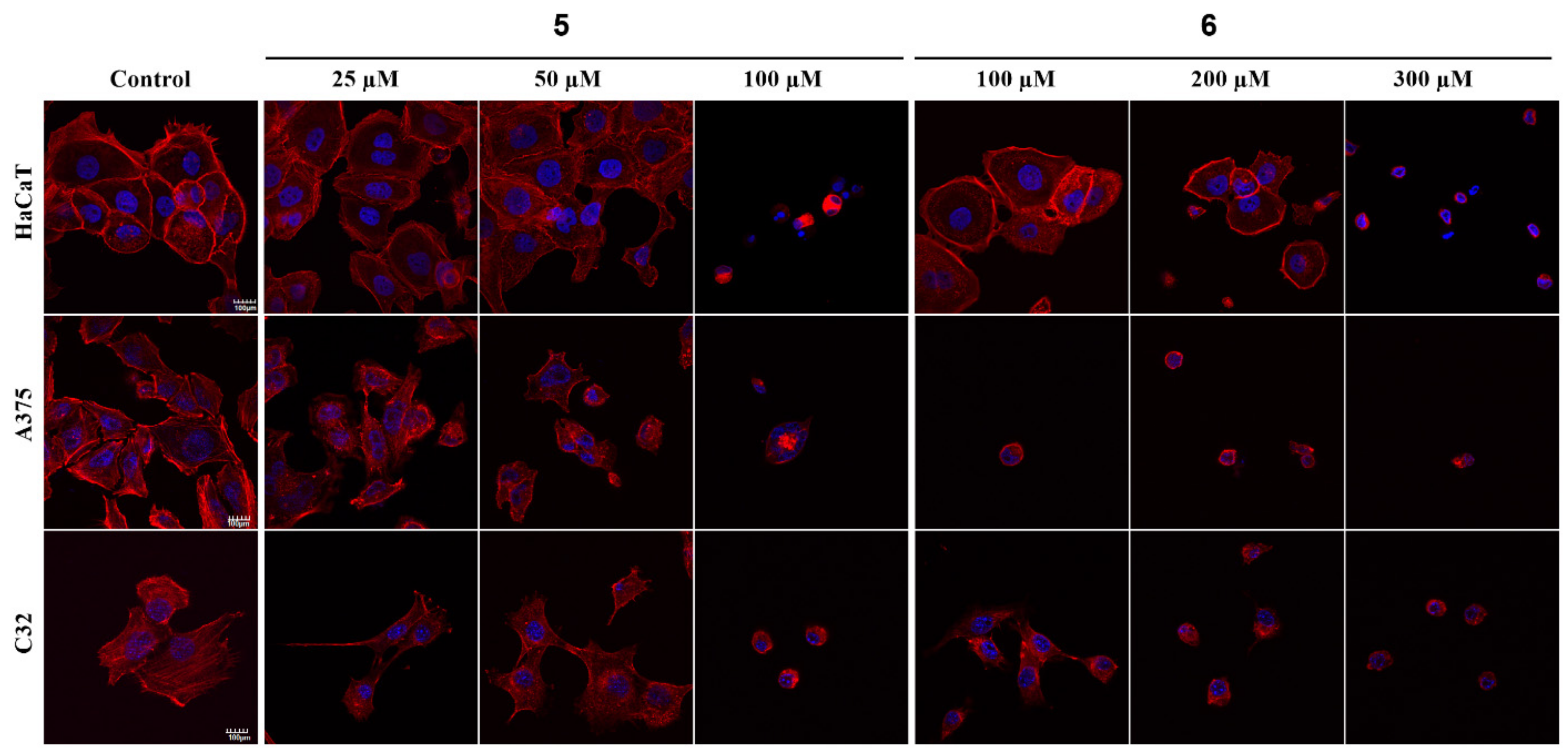
2.2.5. Fluorescent Staining of Actin Filaments
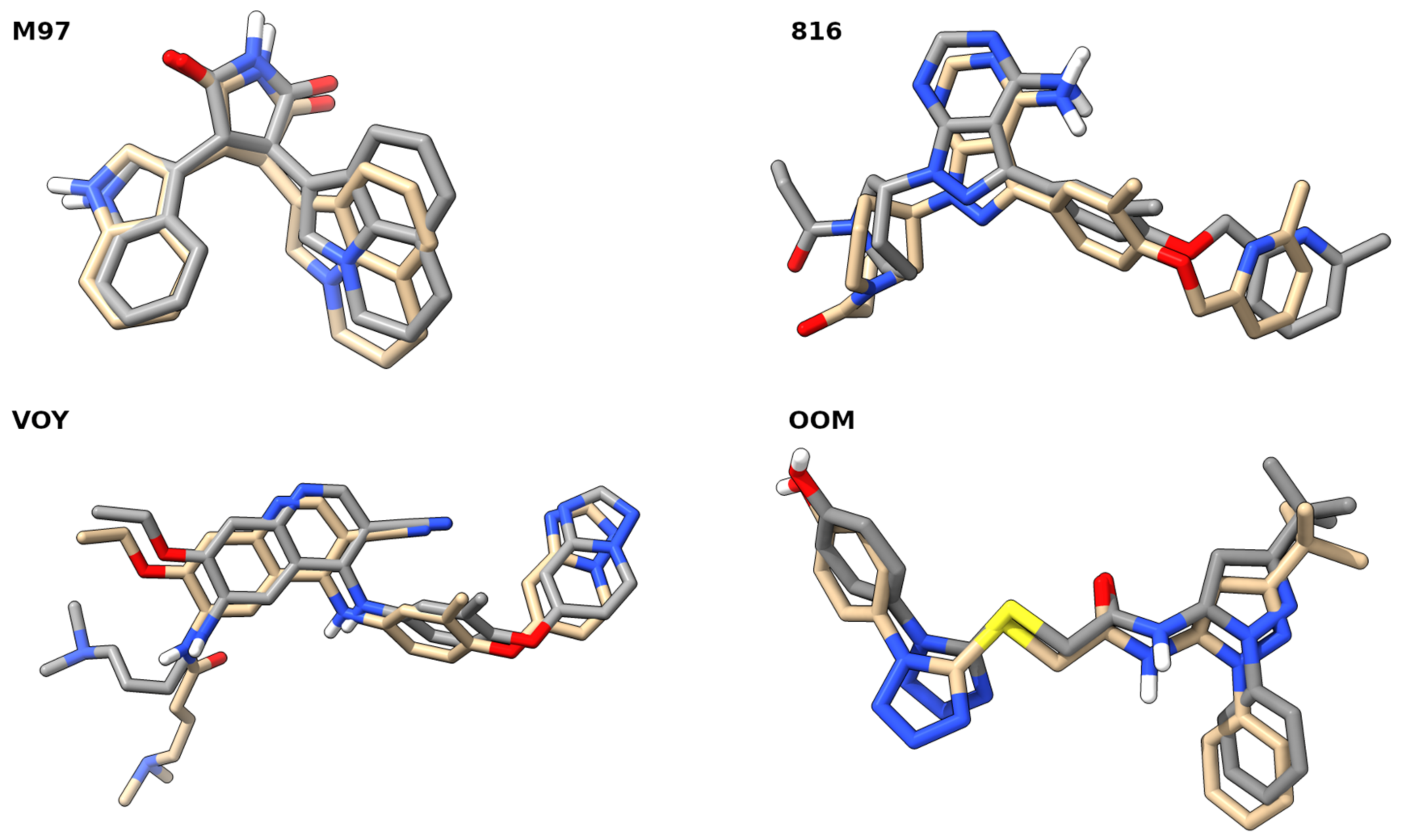
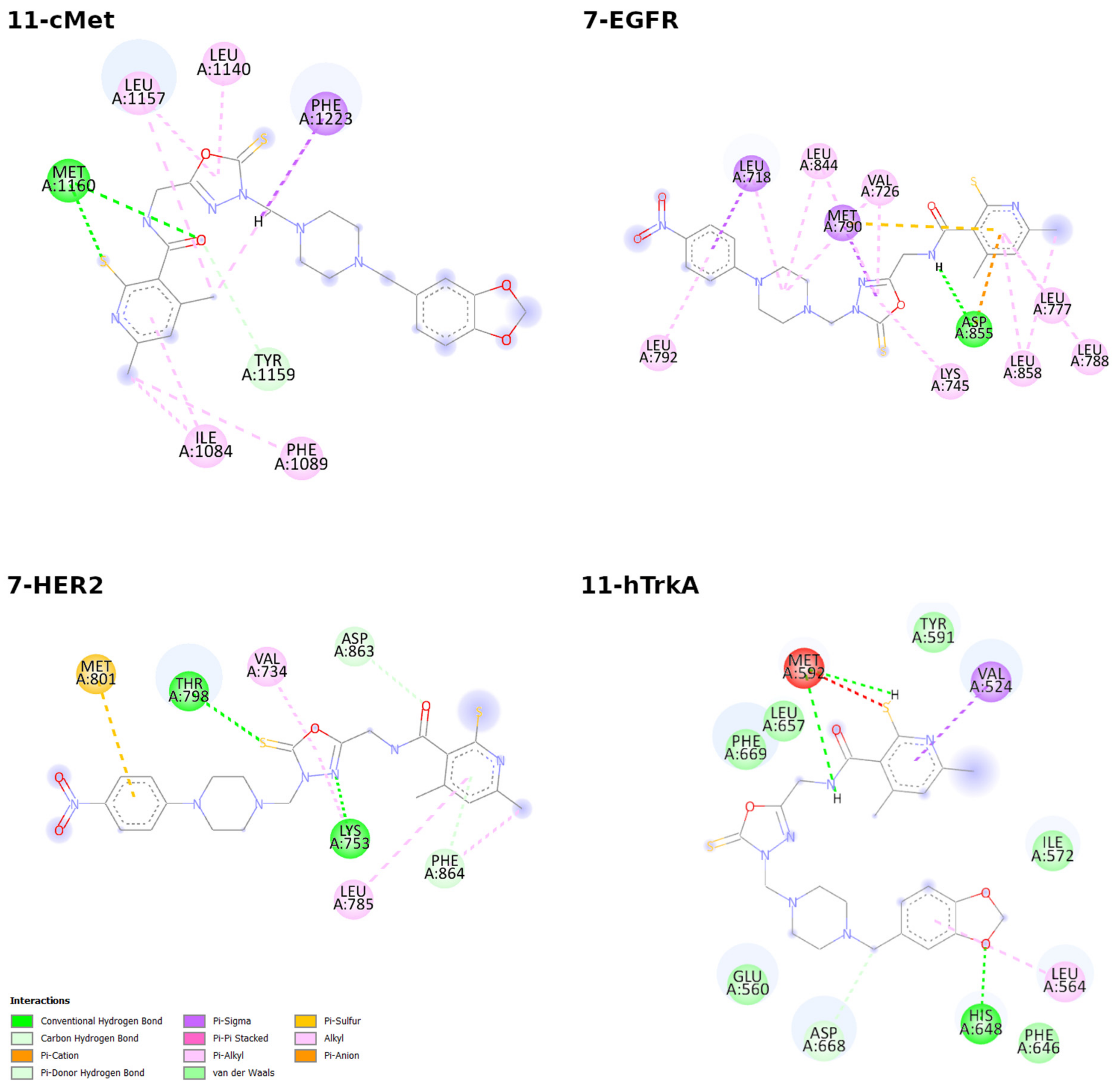
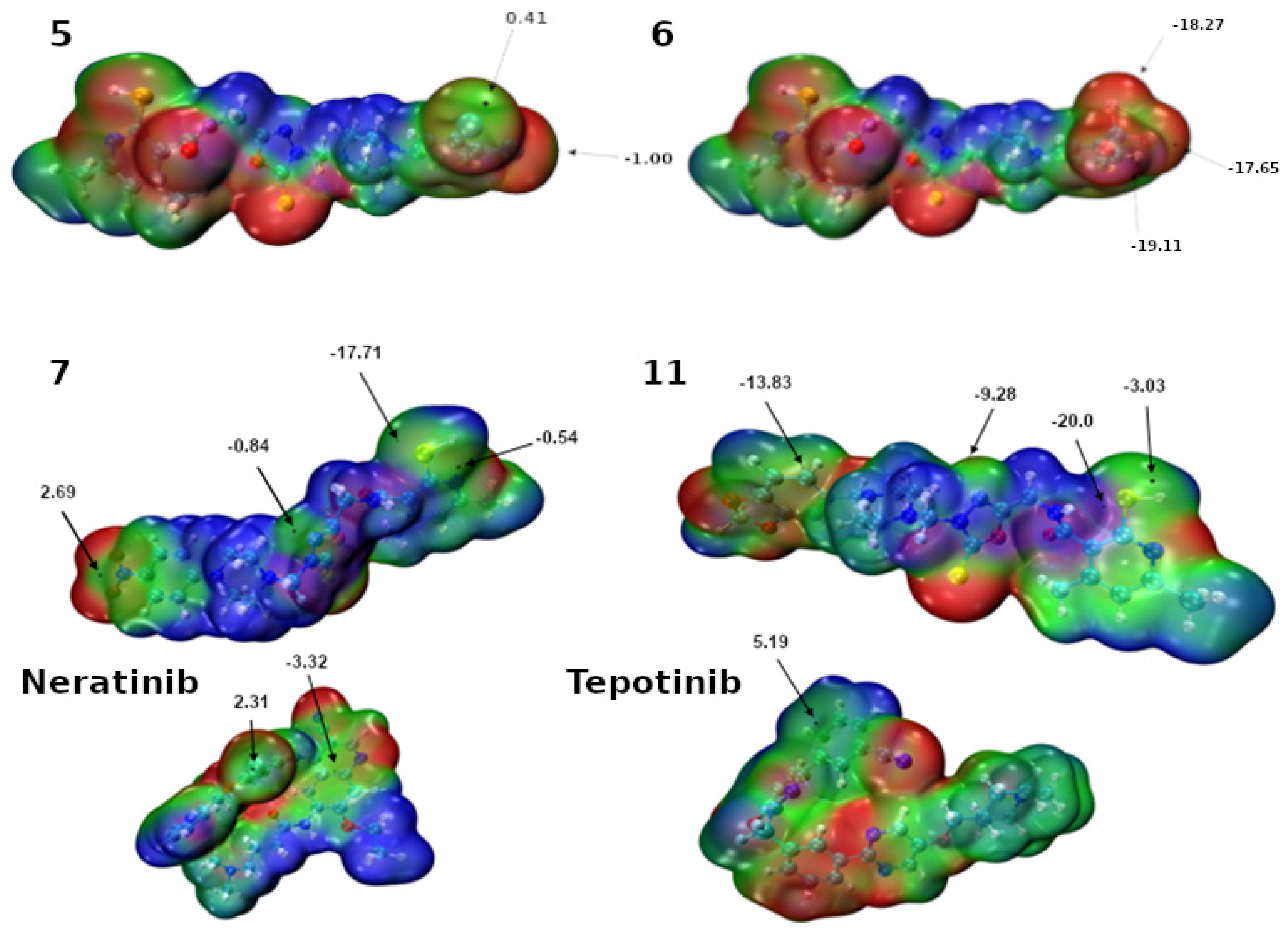
2.3. Molecular Docking Studies
3. Materials and Methods
3.1. Chemistry
3.1.1. Instruments and Chemicals
3.1.2. Preparation and Experimental Properties of Compounds 3–12
3.2. Biological Section
3.2.1. Cell Lines
3.2.2. MTT Cell Viability Assay
3.2.3. Clonogenic Assay
3.2.4. Population Doubling Time
3.2.5. Cell Death Evaluation by Neutral Comet Assay
3.2.6. Fluorescent Staining of Actin Filaments
3.3. Molecular Modeling—Computational Methodology
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 14 July 2022).
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Side Effects of Cancer Treatment|CDC. Available online: https://www.cdc.gov/cancer/survivors/patients/side-effects-of-treatment.htm (accessed on 14 July 2022).
- Assaraf, Y.G.; Brozovic, A.; Gonçalves, A.C.; Jurkovicova, D.; Linē, A.; Machuqueiro, M.; Saponara, S.; Sarmento-Ribeiro, A.B.; Xavier, C.P.R.; Vasconcelos, M.H. The Multi-Factorial Nature of Clinical Multidrug Resistance in Cancer. Drug Resist. Updates 2019, 46, 100645. [Google Scholar] [CrossRef] [PubMed]
- Bukowski, K.; Kciuk, M.; Kontek, R. Mechanisms of Multidrug Resistance in Cancer Chemotherapy. Int. J. Mol. Sci. 2020, 21, 3233. [Google Scholar] [CrossRef] [PubMed]
- Sochacka-Ćwikła, A.; Mączyński, M.; Regiec, A. FDA-Approved Small Molecule Compounds as Drugs for Solid Cancers from Early 2011 to the End of 2021. Molecules 2022, 27, 2259. [Google Scholar] [CrossRef]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging Functions of the EGFR in Cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef]
- Massicano, A.V.F.; Marquez-Nostra, B.V.; Lapi, S.E. Targeting HER2 in Nuclear Medicine for Imaging and Therapy. Mol. Imaging 2018, 17, 153601211774538. [Google Scholar] [CrossRef]
- Zhang, Y.; Xia, M.; Jin, K.; Wang, S.; Wei, H.; Fan, C.; Wu, Y.; Li, X.; Li, X.; Li, G.; et al. Function of the C-Met Receptor Tyrosine Kinase in Carcinogenesis and Associated Therapeutic Opportunities. Mol. Cancer 2018, 17, 45. [Google Scholar] [CrossRef]
- Miao, Q.; Ma, K.; Chen, D.; Wu, X.; Jiang, S. Targeting Tropomyosin Receptor Kinase for Cancer Therapy. Eur. J. Med. Chem. 2019, 175, 129–148. [Google Scholar] [CrossRef]
- Debela, D.T.; Muzazu, S.G.; Heraro, K.D.; Ndalama, M.T.; Mesele, B.W.; Haile, D.C.; Kitui, S.K.; Manyazewal, T. New Approaches and Procedures for Cancer Treatment: Current Perspectives. SAGE Open Med. 2021, 9, 205031212110343. [Google Scholar] [CrossRef]
- Kottschade, L.A. The Future of Immunotherapy in the Treatment of Cancer. Semin. Oncol. Nurs. 2019, 35, 150934. [Google Scholar] [CrossRef]
- Roman, G. Mannich Bases in Medicinal Chemistry and Drug Design. Eur. J. Med. Chem. 2015, 89, 743–816. [Google Scholar] [CrossRef]
- Tugrak, M.; Gul, H.I.; Bandow, K.; Sakagami, H.; Gulcin, I.; Ozkay, Y.; Supuran, C.T. Synthesis and Biological Evaluation of Some New Mono Mannich Bases with Piperazines as Possible Anticancer Agents and Carbonic Anhydrase Inhibitors. Bioorg. Chem. 2019, 90, 103095. [Google Scholar] [CrossRef]
- Ma, L.; Xiao, Y.; Li, C.; Xie, Z.L.; Li, D.D.; Wang, Y.T.; Ma, H.T.; Zhu, H.L.; Wang, M.H.; Ye, Y.H. Synthesis and Antioxidant Activity of Novel Mannich Base of 1,3,4-Oxadiazole Derivatives Possessing 1,4-Benzodioxan. Bioorg. Med. Chem. 2013, 21, 6763–6770. [Google Scholar] [CrossRef]
- Palkar, M.B.; Singhai, A.S.; Ronad, P.M.; Vishwanathswamy, A.H.M.; Boreddy, T.S.; Veerapur, V.P.; Shaikh, M.S.; Rane, R.A.; Karpoormath, R. Synthesis, Pharmacological Screening and in Silico Studies of New Class of Diclofenac Analogues as a Promising Anti-Inflammatory Agents. Bioorg. Med. Chem. 2014, 22, 2855–2866. [Google Scholar] [CrossRef]
- Köksal, M.; Gökhan, N.; Küpeli, E.; Yesilada, E.; Erdogan, H. Analgesic and Antiinflammatory Activities of Some New Mannich Bases of 5-Nitro-2-Benzoxazolinones. Arch. Pharm. Res. 2007, 30, 419–424. [Google Scholar] [CrossRef]
- Szczukowski, Ł.; Krzyżak, E.; Wiatrak, B.; Jawień, P.; Marciniak, A.; Kotynia, A.; Świątek, P. New N-Substituted-1,2,4-Triazole Derivatives of Pyrrolo [3,4-d]Pyridazinone with Significant Anti-Inflammatory Activity—Design, Synthesis and Complementary In Vitro, Computational and Spectroscopic Studies. Int. J. Mol. Sci. 2021, 22, 11235. [Google Scholar] [CrossRef]
- Patel, A.B.; Rohit, J.V. Development of 1,3,4-Thiadiazole and Piperazine Fused Hybrid Quinazoline Derivatives as Dynamic Antimycobacterial Agents. Polycycl. Aromat. Compd. 2021, 1–12. [Google Scholar] [CrossRef]
- Pandeya, S.N.; Sriram, D.; Nath, G.; De Clercq, E. Synthesis, Antibacterial, Antifungal and Anti-HIV Activities of Norfloxacin Mannich Bases. Eur. J. Med. Chem. 2000, 35, 249–255. [Google Scholar] [CrossRef]
- Abrão, P.H.O.; Pizi, R.B.; de Souza, T.B.; Silva, N.C.; Fregnan, A.M.; Silva, F.N.; Coelho, L.F.L.; Malaquias, L.C.C.; Dias, A.L.T.; Dias, D.F.; et al. Synthesis and Biological Evaluation of New Eugenol Mannich Bases as Promising Antifungal Agents. Chem. Biol. Drug Des. 2015, 86, 459–465. [Google Scholar] [CrossRef]
- Rybka, S.; Obniska, J.; Rapacz, A.; Filipek, B.; Żmudzki, P. Synthesis and Evaluation of Anticonvulsant Properties of New N-Mannich Bases Derived from Pyrrolidine-2,5-Dione and Its 3-Methyl-, 3-Isopropyl, and 3-Benzhydryl Analogs. Bioorg. Med. Chem. Lett. 2017, 27, 1412–1415. [Google Scholar] [CrossRef]
- Kamiński, K.; Obniska, J.; Chlebek, I.; Wiklik, B.; Rzepka, S. Design, Synthesis and Anticonvulsant Properties of New N-Mannich Bases Derived from 3-Phenylpyrrolidine-2,5-Diones. Bioorg. Med. Chem. 2013, 21, 6821–6830. [Google Scholar] [CrossRef]
- Kaur, P.; Wakode, S. Synthesis and In Vitro Evaluation of Anticancer Activity of Mannich Bases of Benzimidazole Derivatives. Int. J. Sci. Res. ISSN 2016, 5, 1096–1099. [Google Scholar]
- Demirci, S.; Demirbaş, N. Anticancer Activities of Novel Mannich Bases against Prostate Cancer Cells. Med. Chem. Res. 2019, 28, 1945–1958. [Google Scholar] [CrossRef]
- Sun, J.; Zhu, H.; Yang, Z.M.; Zhu, H.L. Synthesis, Molecular Modeling and Biological Evaluation of 2-Aminomethyl-5-(Quinolin-2-Yl)-1,3,4-Oxadiazole-2(3H)-Thione Quinolone Derivatives as Novel Anticancer Agent. Eur. J. Med. Chem. 2013, 60, 23–28. [Google Scholar] [CrossRef]
- Savariz, F.C.; Foglio, M.A.; Goes Ruiz, A.L.T.; Da Costa, W.F.; De Magalhães Silva, M.; Santos, J.C.C.; Figueiredo, I.M.; Meyer, E.; De Carvalho, J.E.; Sarragiotto, M.H. Synthesis and Antitumor Activity of Novel 1-Substituted Phenyl 3-(2-Oxo-1,3,4-Oxadiazol-5-Yl) β-Carbolines and Their Mannich Bases. Bioorg. Med. Chem. 2014, 22, 6867–6875. [Google Scholar] [CrossRef]
- Sun, J.; Ren, S.Z.; Lu, X.Y.; Li, J.J.; Shen, F.Q.; Xu, C.; Zhu, H.L. Discovery of a Series of 1,3,4-Oxadiazole-2(3H)-Thione Derivatives Containing Piperazine Skeleton as Potential FAK Inhibitors. Bioorg. Med. Chem. 2017, 25, 2593–2600. [Google Scholar] [CrossRef]
- Luczynski, M.; Kudelko, A. Synthesis and Biological Activity of 1,3,4-Oxadiazoles Used in Medicine and Agriculture. Appl. Sci. 2022, 12, 3756. [Google Scholar] [CrossRef]
- Bąchor, U.; Drozd-Szczygieł, E.; Bąchor, R.; Jerzykiewicz, L.; Wieczorek, R.; Mączyński, M. New Water-Soluble Isoxazole-Linked 1,3,4-Oxadiazole Derivative with Delocalized Positive Charge. RSC Adv. 2021, 11, 29668–29674. [Google Scholar] [CrossRef]
- Verma, S.K.; Verma, R.; Verma, S.; Vaishnav, Y.; Tiwari, S.P.; Rakesh, K.P. Anti-Tuberculosis Activity and Its Structure-Activity Relationship (SAR) Studies of Oxadiazole Derivatives: A Key Review. Eur. J. Med. Chem. 2021, 209, 112886. [Google Scholar] [CrossRef] [PubMed]
- Boström, J.; Hogner, A.; Llinàs, A.; Wellner, E.; Plowright, A.T. Oxadiazoles in Medicinal Chemistry. J. Med. Chem. 2012, 55, 1817–1830. [Google Scholar] [CrossRef]
- Rana, K.; Salahuddin; Sahu, J.K. Significance of 1,3,4-Oxadiazole Containing Compounds in New Drug Development. Curr. Drug Res. Rev. 2021, 13, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Hughes, T.V.; Xu, G.; Wetter, S.K.; Connolly, P.J.; Emanuel, S.L.; Karnachi, P.; Pollack, S.R.; Pandey, N.; Adams, M.; Moreno-Mazza, S.; et al. A Novel 5-[1,3,4-Oxadiazol-2-Yl]-N-Aryl-4,6-Pyrimidine Diamine Having Dual EGFR/HER2 Kinase Activity: Design, Synthesis, and Biological Activity. Bioorg. Med. Chem. Lett. 2008, 18, 4896–4899. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Lu, X.; Zhang, H.J.; Sun, J.; Zhu, H.L. Synthesis, Molecular Modeling and Biological Evaluation of 2-(Benzylthio)-5-Aryloxadiazole Derivatives as Anti-Tumor Agents. Eur. J. Med. Chem. 2012, 47, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, M.J.; Siddiqui, A.A.; Khan, A.A.; Ali, Z.; Dewangan, R.P.; Pasha, S.; Yar, M.S. Design, Synthesis, Docking and QSAR Study of Substituted Benzimidazole Linked Oxadiazole as Cytotoxic Agents, EGFR and ErbB2 Receptor Inhibitors. Eur. J. Med. Chem. 2017, 126, 853–869. [Google Scholar] [CrossRef]
- Osmaniye, D.; Görgülü, Ş.; Sağlık, B.N.; Levent, S.; Özkay, Y.; Kaplancıklı, Z.A. Synthesis and Biological Evaluation of Novel 1,3,4-oxadiazole Derivatives as Anticancer Agents and Potential EGFR Inhibitors. J. Heterocycl. Chem. 2022, 59, 518–532. [Google Scholar] [CrossRef]
- Ibrahim, H.A.; Awadallah, F.M.; Refaat, H.M.; Amin, K.M. Molecular Docking Simulation, Synthesis and 3D Pharmacophore Studies of Novel 2-Substituted-5-Nitro-Benzimidazole Derivatives as Anticancer Agents Targeting VEGFR-2 and c-Met. Bioorg. Chem. 2018, 77, 457–470. [Google Scholar] [CrossRef]
- Tian, C.; Huang, S.; Xu, Z.; Liu, W.; Li, D.; Liu, M.; Zhu, C.; Wu, L.; Jiang, X.; Ding, H.; et al. Design, Synthesis, and Biological Evaluation of β-Carboline 1,3,4-Oxadiazole Based Hybrids as HDAC Inhibitors with Potential Antitumor Effects. Bioorg. Med. Chem. Lett. 2022, 64, 128663. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, X.L.; Shi, J.; Wang, S.F.; Yin, Y.; Yang, Y.S.; Zhang, W.M.; Zhu, H.L. Synthesis, Molecular Modeling and Biological Evaluation of N-Benzylidene-2-((5-(Pyridin-4-Yl)-1,3,4-Oxadiazol-2-Yl)Thio)Acetohydrazide Derivatives as Potential Anticancer Agents. Bioorg. Med. Chem. 2014, 22, 468–477. [Google Scholar] [CrossRef]
- Alzhrani, Z.M.M.; Alam, M.M.; Neamatallah, T.; Nazreen, S. Design, Synthesis and in Vitro Antiproliferative Activity of New Thiazolidinedione-1,3,4-Oxadiazole Hybrids as Thymidylate Synthase Inhibitors. J. Enzyme Inhib. Med. Chem. 2020, 35, 1116–1123. [Google Scholar] [CrossRef]
- Tantak, M.P.; Malik, M.; Klingler, L.; Olson, Z.; Kumar, A.; Sadana, R.; Kumar, D. Indolyl-α-Keto-1,3,4-Oxadiazoles: Synthesis, Anti-Cell Proliferation Activity, and Inhibition of Tubulin Polymerization. Bioorg. Med. Chem. Lett. 2021, 37, 127842. [Google Scholar] [CrossRef]
- Tagad, H.D.; Hamada, Y.; Nguyen, J.T.; Hamada, T.; Abdel-Rahman, H.; Yamani, A.; Nagamine, A.; Ikari, H.; Igawa, N.; Hidaka, K.; et al. Design of Pentapeptidic BACE1 Inhibitors with Carboxylic Acid Bioisosteres at P1′ and P4 Positions. Bioorg. Med. Chem. 2010, 18, 3175–3186. [Google Scholar] [CrossRef]
- Goldberg, K.; Groombridge, S.; Hudson, J.; Leach, A.G.; MacFaul, P.A.; Pickup, A.; Poultney, R.; Scott, J.S.; Svensson, P.H.; Sweeney, J. Oxadiazole Isomers: All Bioisosteres Are Not Created Equal. MedChemComm 2012, 3, 600–604. [Google Scholar] [CrossRef]
- Li, Y.T.; Wang, J.H.; Pan, C.W.; Meng, F.F.; Chu, X.Q.; Ding, Y.H.; Qu, W.Z.; Li, H.Y.; Yang, C.; Zhang, Q.; et al. Syntheses and Biological Evaluation of 1,2,3-Triazole and 1,3,4-Oxadiazole Derivatives of Imatinib. Bioorg. Med. Chem. Lett. 2016, 26, 1419–1427. [Google Scholar] [CrossRef]
- Świątek, P.; Glomb, T.; Dobosz, A.; Gębarowski, T.; Wojtkowiak, K.; Jezierska, A.; Panek, J.J.; Świątek, M.; Strzelecka, M. Biological Evaluation and Molecular Docking Studies of Novel 1,3,4-Oxadiazole Derivatives of 4,6-Dimethyl-2-Sulfanylpyridine-3-Carboxamide. Int. J. Mol. Sci. 2022, 23, 549. [Google Scholar] [CrossRef]
- Świątek, P.; Saczko, J.; Rembiałkowska, N.; Kulbacka, J. Synthesis of New Hydrazone Derivatives and Evaluation of Their Efficacy as Proliferation Inhibitors in Human Cancer Cells. Med. Chem. 2019, 15, 903–910. [Google Scholar] [CrossRef]
- Ahmed, E.M.; Bandopadhyay, G.; Coyle, B.; Grabowska, A. A HIF-Independent, CD133-Mediated Mechanism of Cisplatin Resistance in Glioblastoma Cells. Cell. Oncol. 2018, 41, 319–328. [Google Scholar] [CrossRef]
- Suberu, J.O.; Romero-Canelón, I.; Sullivan, N.; Lapkin, A.A.; Barker, G.C. Comparative Cytotoxicity of Artemisinin and Cisplatin and Their Interactions with Chlorogenic Acids in MCF7 Breast Cancer Cells. ChemMedChem 2014, 9, 2791–2797. [Google Scholar] [CrossRef]
- Elias, S.T.; Borges, G.A.; Rêgo, D.F.; E Silva, L.F.O.; Avelino, S.; Neto, J.N.D.M.; Simeoni, L.A.; Guerra, E.N.S. Combined Paclitaxel, Cisplatin and Fluorouracil Therapy Enhances Ionizing Radiation Effects, Inhibits Migration and Induces G0/G1 Cell Cycle Arrest and Apoptosis in Oral Carcinoma Cell Lines. Oncol. Lett. 2015, 10, 1721–1727. [Google Scholar] [CrossRef]
- Cortés-Gutiérrez, E.I.; Hernández-Garza, F.; García-Pérez, J.O.; Dávila-Rodríguez, M.I.; Aguado-Barrera, M.E.; Cerda-Flores, R.M. Evaluation of DNA Single and Double Strand Breaks in Women with Cervical Neoplasia Based on Alkaline and Neutral Comet Assay Techniques. J. Biomed. Biotechnol. 2012, 2012, 385245. [Google Scholar] [CrossRef]
- Eathiraj, S.; Palma, R.; Volckova, E.; Hirschi, M.; France, D.S.; Ashwell, M.A.; Chan, T.C.K. Discovery of a Novel Mode of Protein Kinase Inhibition Characterized by the Mechanism of Inhibition of Human Mesenchymal-Epithelial Transition Factor (c-Met) Protein Autophosphorylation by ARQ 197. J. Biol. Chem. 2011, 286, 20666–20676. [Google Scholar] [CrossRef]
- Hu, C.; Wang, A.; Wu, H.; Qi, Z.; Li, X.; Yan, X.E.; Chen, C.; Yu, K.; Zou, F.; Wang, W.; et al. Discovery and Characterization of a Novel Irreversible EGFR Mutants Selective and Potent Kinase Inhibitor CHMFL-EGFR-26 with a Distinct Binding Mode. Oncotarget 2017, 8, 18359–18372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, J.; Jang, J.; Beyett, T.S.; Eum, Y.; Haikala, H.M.; Verano, A.; Lin, M.; Hatcher, J.M.; Kwiatkowski, N.P.; Eser, P.; et al. A Novel HER2-Selective Kinase Inhibitor Is Effective in HER2 Mutant and Amplified Non-Small Cell Lung Cancer. Cancer Res. 2022, 82, 1633–1645. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, G.; Bowen, S.J.; Zhu, Y.; Roush, N.; Zachary, T.; Javens, C.; Williams, T.; Janssen, A.; Gonzales, A. Type 2 Inhibitor Leads of Human Tropomyosin Receptor Kinase (HTrkA). Bioorg. Med. Chem. Lett. 2019, 29, 126624. [Google Scholar] [CrossRef] [PubMed]
- Pecina, A.; Haldar, S.; Fanfrlík, J.; Meier, R.; Řezáč, J.; Lepšík, M.; Hobza, P. SQM/COSMO Scoring Function at the DFTB3-D3H4 Level: Unique Identification of Native Protein-Ligand Poses. J. Chem. Inf. Model. 2017, 57, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.R. The Comet Assay. Principles, Applications, and Limitations. In Methods in Molecular Biology; Humana Press: Clifton, NJ, USA, 2002; pp. 163–177. [Google Scholar] [CrossRef]
- Nadin, S.B.; Vargas-Roig, L.M.; Ciocca, D.R. A Silver Staining Method for Single-Cell Gel Assay. J. Histochem. Cytochem. 2001, 49, 1183–1186. [Google Scholar] [CrossRef]
- RCSB PDB: Homepage. Available online: https://www.rcsb.org/ (accessed on 27 June 2022).
- Word, J.M.; Lovell, S.C.; Richardson, J.S.; Richardson, D.C. Asparagine and Glutamine: Using Hydrogen Atom Contacts in the Choice of Side-Chain Amide Orientation. J. Mol. Biol. 1999, 285, 1735–1747. [Google Scholar] [CrossRef] [PubMed]
- Downloads—ADFR. Available online: https://ccsb.scripps.edu/adfr/downloads/ (accessed on 27 June 2022).
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational Protein-Ligand Docking and Virtual Drug Screening with the AutoDock Suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Liu, Y.; Lemmon, M.A.; Radhakrishnan, R. Erlotinib Binds Both Inactive and Active Conformations of the EGFR Tyrosine Kinase Domain. Biochem. J. 2012, 448, 417–423. [Google Scholar] [CrossRef]
- Sogabe, S.; Kawakita, Y.; Igaki, S.; Iwata, H.; Miki, H.; Cary, D.R.; Takagi, T.; Takagi, S.; Ohta, Y.; Ishikawa, T. Structure-Based Approach for the Discovery of Pyrrolo [3,2-d]Pyrimidine-Based EGFR T790M/L858R Mutant Inhibitors. ACS Med. Chem. Lett. 2013, 4, 201–205. [Google Scholar] [CrossRef]
- Dorsch, D.; Schadt, O.; Stieber, F.; Meyring, M.; Grädler, U.; Bladt, F.; Friese-Hamim, M.; Knühl, C.; Pehl, U.; Blaukat, A. Identification and Optimization of Pyridazinones as Potent and Selective C-Met Kinase Inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 1597–1602. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Yu, H.S.; He, X.; Li, S.L.; Truhlar, D.G. MN15: A Kohn-Sham Global-Hybrid Exchange-Correlation Density Functional with Broad Accuracy for Multi-Reference and Single-Reference Systems and Noncovalent Interactions. Chem. Sci. 2016, 7, 5032–5051. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Cossi, M.; Barone, V.; Cammi, R.; Tomasi, J. Ab Initio Study of Solvated Molecules: A New Implementation of the Polarizable Continuum Model. Chem. Phys. Lett. 1996, 255, 327–335. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A. Gaussian 16 Revision C.0.1; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Ravindranath, P.A.; Forli, S.; Goodsell, D.S.; Olson, A.J.; Sanner, M.F. AutoDockFR: Advances in Protein-Ligand Docking with Explicitly Specified Binding Site Flexibility. PLoS Comput. Biol. 2015, 11, e1004586. [Google Scholar] [CrossRef]
- DSV: Discover Studio Visualiser, V21.1.0.20298; BIOVIA, Dassault Systèmes: San Diego, CA, USA, 2020.
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure Visualization for Researchers, Educators, and Developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | IC50 [µM] of Compound 5 | IC50 [µM] of Compound 6 | IC50 [µM] of Cisplatin |
|---|---|---|---|
| A375 | 80.79 ± 4.85 | 202.47 ± 10.12 | 15.98 ± 3.31 * |
| C32 | 170.28 ± 10.22 | 304.39 ± 15.21 | 9.79 ± 1.51 * |
| SNB-19 | 126.02 ± 7.56 | 295.81 ± 14.71 | 43.47 [48] |
| MCF-7/WT | 119.29 ± 7.16 | 261.40 ± 13.07 | 5.75 ± 0.02 [49] |
| MCF-7/DOX | 137.31 ± 8.24 | 295.81 ± 14.92 | 47.82 ± 2.45 * |
| HaCaT | 115.12 ± 6.91 | 270.32 ± 13.25 | 56.00 ± 7.27 [50] |
| Compound | cMET | EGRF | HER2 | hTrkA |
|---|---|---|---|---|
| 3 | −11.8 | −12.0 | −12.2 | −13.2 |
| 4 | −12.1 | −11.8 | −12.9 | −13.4 |
| 5 | −12.1 | −12.2 | −12.5 | −13.6 |
| 6 | −11.4 | −11.4 | −12.1 | −13.2 |
| 7 | −11.9 | −12.9 | −13.6 | −14.0 |
| 8 | −11.6 | −10.8 | −11.8 | −13.7 |
| 9 | −9.7 | −11.0 | −10.3 | −11.7 |
| 10 | −11.1 | −11.4 | −12.2 | −13.6 |
| 11 | −12.6 | −12.0 | −12.3 | −14.5 |
| 12 | −10.6 | −11.1 | −11.2 | −13.5 |
| Erlotinib | −9.2 | −10.9 | −10.5 | −10.4 |
| Neratinib | −12.3 | −13.1 | −13.2 | −14.2 |
| Tepotinib | −13.7 | −13.9 | −13.5 | −14.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strzelecka, M.; Glomb, T.; Drąg-Zalesińska, M.; Kulbacka, J.; Szewczyk, A.; Saczko, J.; Kasperkiewicz-Wasilewska, P.; Rembiałkowska, N.; Wojtkowiak, K.; Jezierska, A.; et al. Synthesis, Anticancer Activity and Molecular Docking Studies of Novel N-Mannich Bases of 1,3,4-Oxadiazole Based on 4,6-Dimethylpyridine Scaffold. Int. J. Mol. Sci. 2022, 23, 11173. https://doi.org/10.3390/ijms231911173
Strzelecka M, Glomb T, Drąg-Zalesińska M, Kulbacka J, Szewczyk A, Saczko J, Kasperkiewicz-Wasilewska P, Rembiałkowska N, Wojtkowiak K, Jezierska A, et al. Synthesis, Anticancer Activity and Molecular Docking Studies of Novel N-Mannich Bases of 1,3,4-Oxadiazole Based on 4,6-Dimethylpyridine Scaffold. International Journal of Molecular Sciences. 2022; 23(19):11173. https://doi.org/10.3390/ijms231911173
Chicago/Turabian StyleStrzelecka, Małgorzata, Teresa Glomb, Małgorzata Drąg-Zalesińska, Julita Kulbacka, Anna Szewczyk, Jolanta Saczko, Paulina Kasperkiewicz-Wasilewska, Nina Rembiałkowska, Kamil Wojtkowiak, Aneta Jezierska, and et al. 2022. "Synthesis, Anticancer Activity and Molecular Docking Studies of Novel N-Mannich Bases of 1,3,4-Oxadiazole Based on 4,6-Dimethylpyridine Scaffold" International Journal of Molecular Sciences 23, no. 19: 11173. https://doi.org/10.3390/ijms231911173
APA StyleStrzelecka, M., Glomb, T., Drąg-Zalesińska, M., Kulbacka, J., Szewczyk, A., Saczko, J., Kasperkiewicz-Wasilewska, P., Rembiałkowska, N., Wojtkowiak, K., Jezierska, A., & Świątek, P. (2022). Synthesis, Anticancer Activity and Molecular Docking Studies of Novel N-Mannich Bases of 1,3,4-Oxadiazole Based on 4,6-Dimethylpyridine Scaffold. International Journal of Molecular Sciences, 23(19), 11173. https://doi.org/10.3390/ijms231911173