
Mucopolysaccharidosis-Plus Syndrome: Report on a Polish Patient with a Novel VPS33A Variant with Comparison with Other Described Patients

 ,
, 
 , and
, and 
Abstract
:1. Introduction
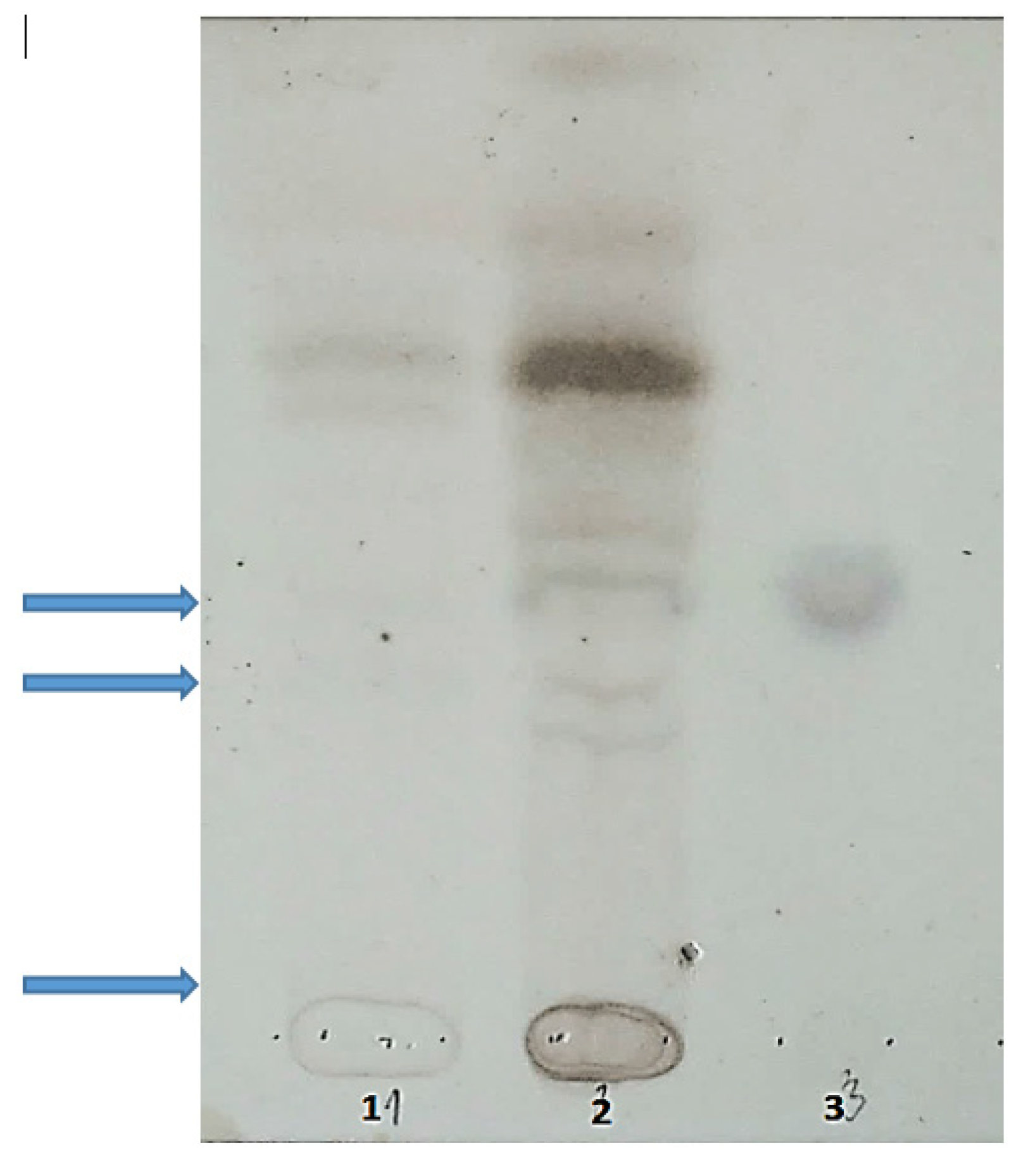
2. Case Report
3. Comparison with Other Reported MPS-PS Cases
4. Discussion
5. Material and Methods
5.1. Clinical Case
5.2. Lysosomal Enzymes and Lysosphingolipids in Dried Blood Spot (DBS)
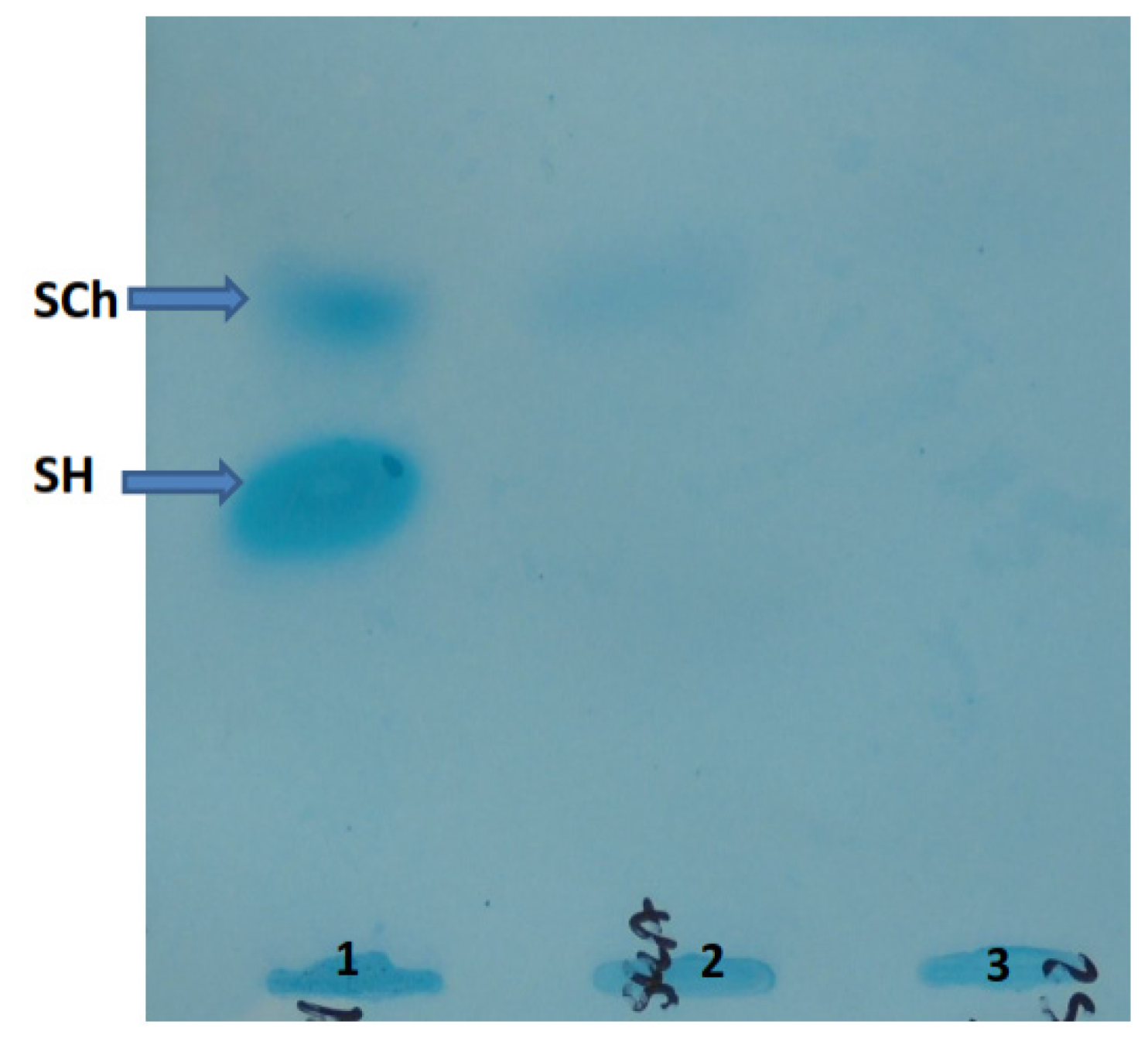
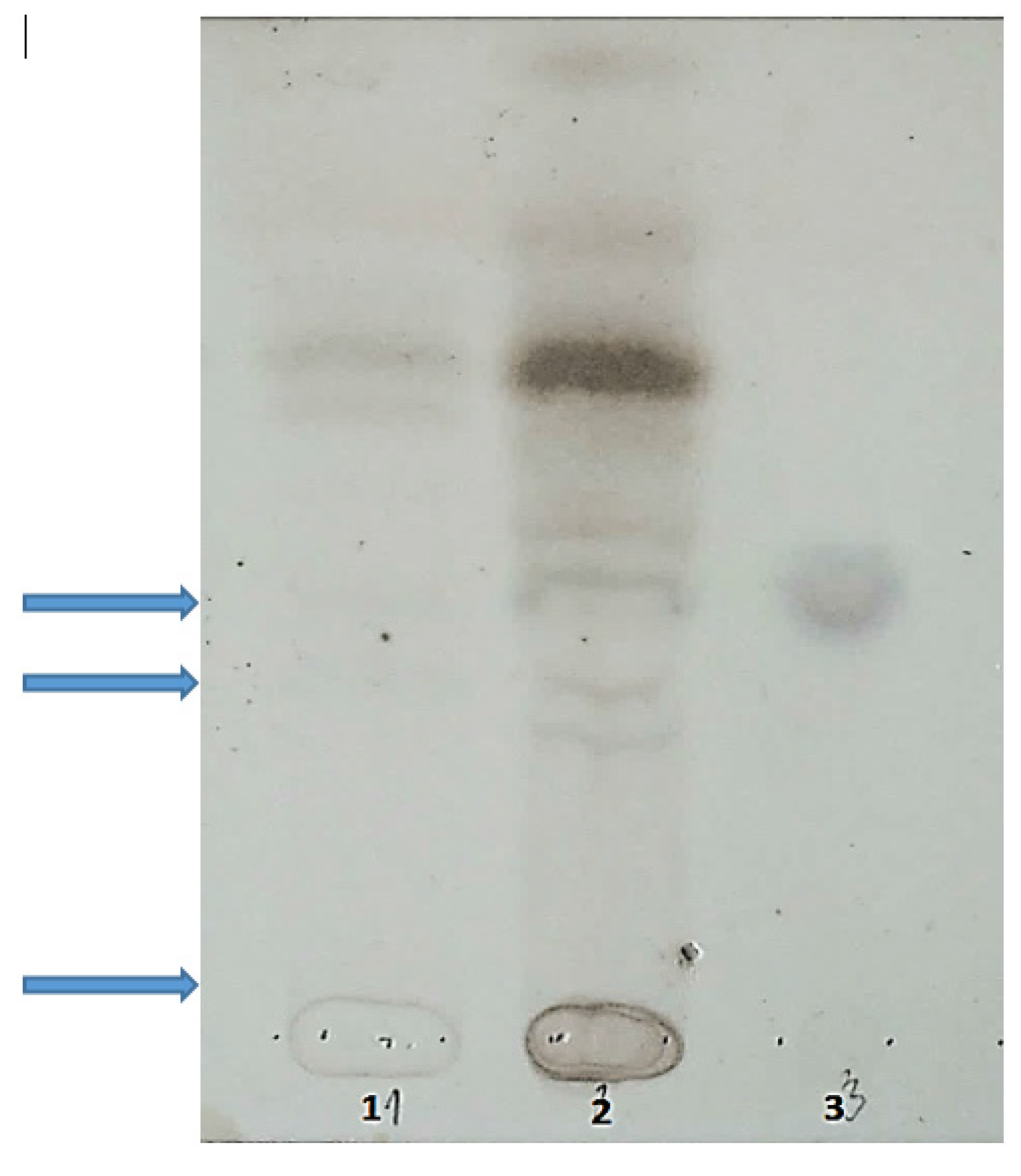
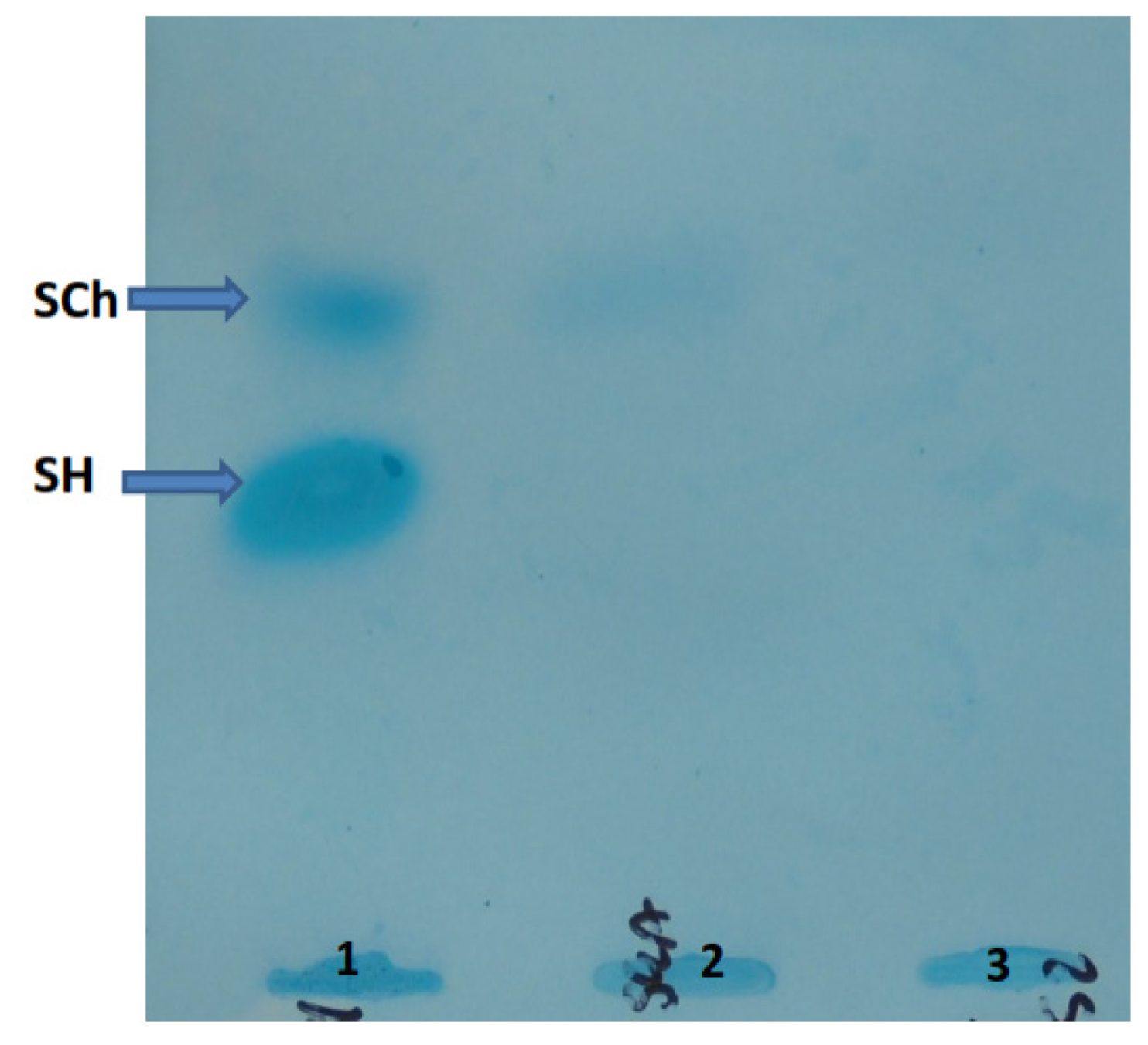
5.3. Oligosaccharides and Sialooligosaccharides in Urine
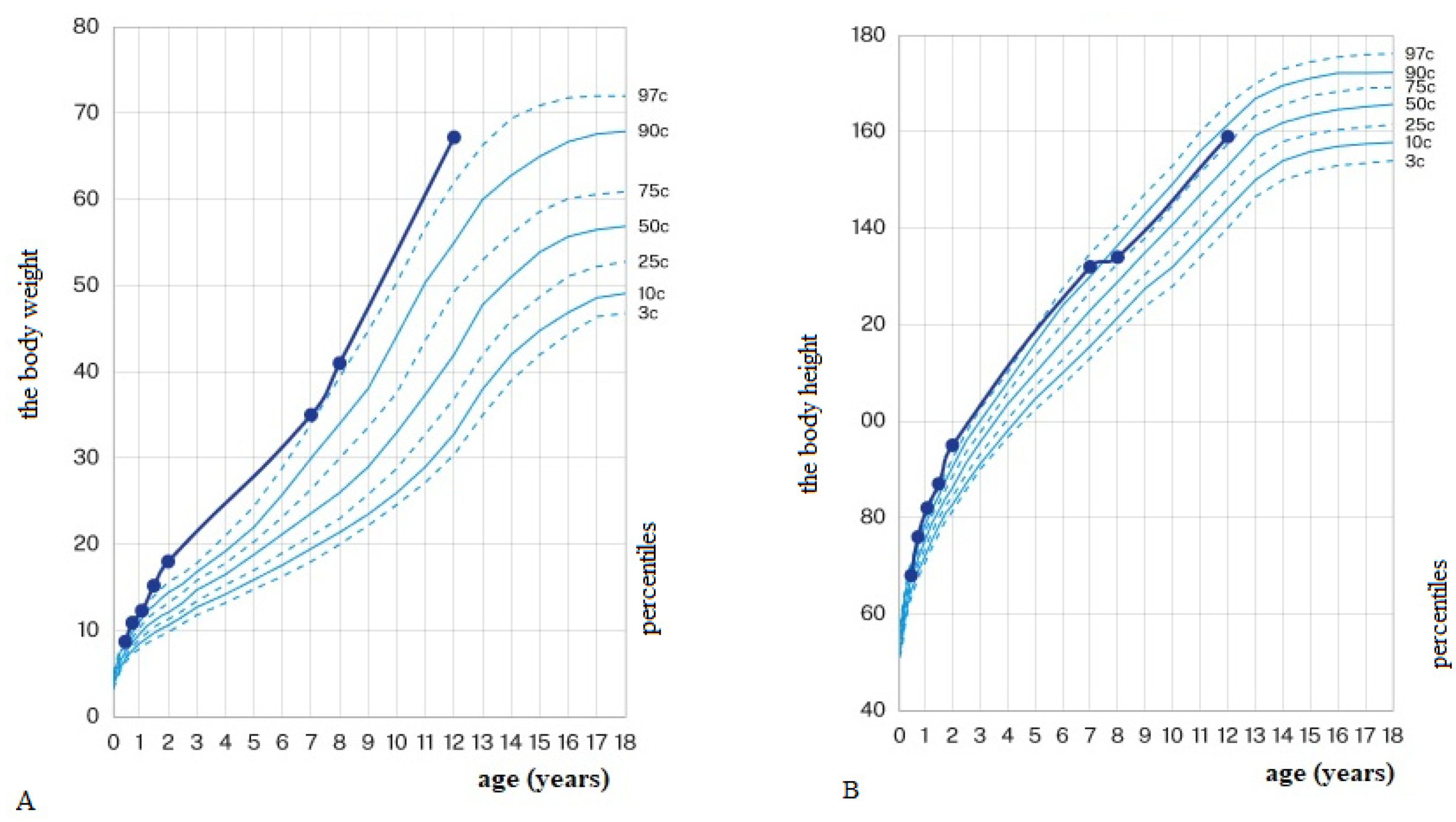
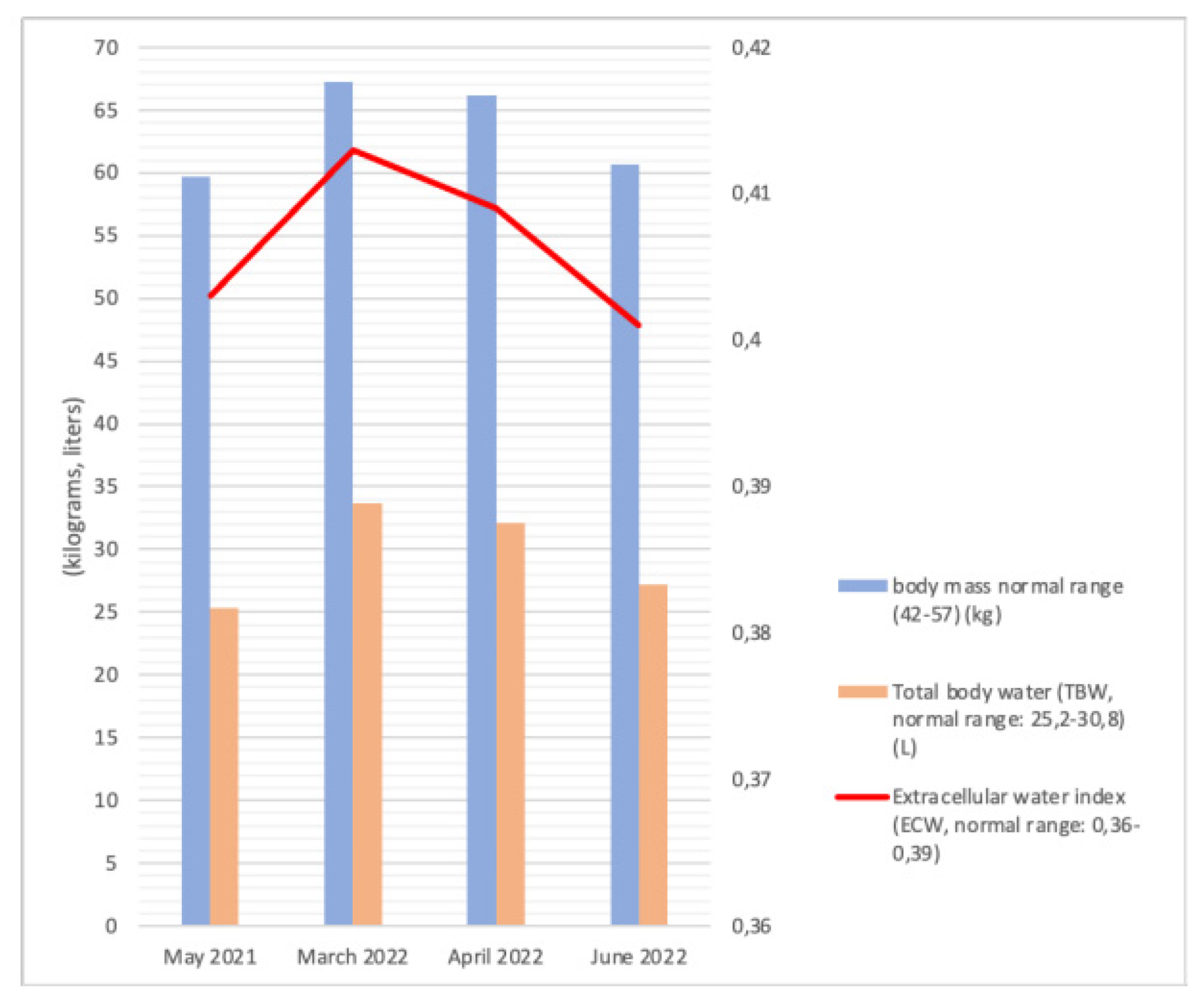
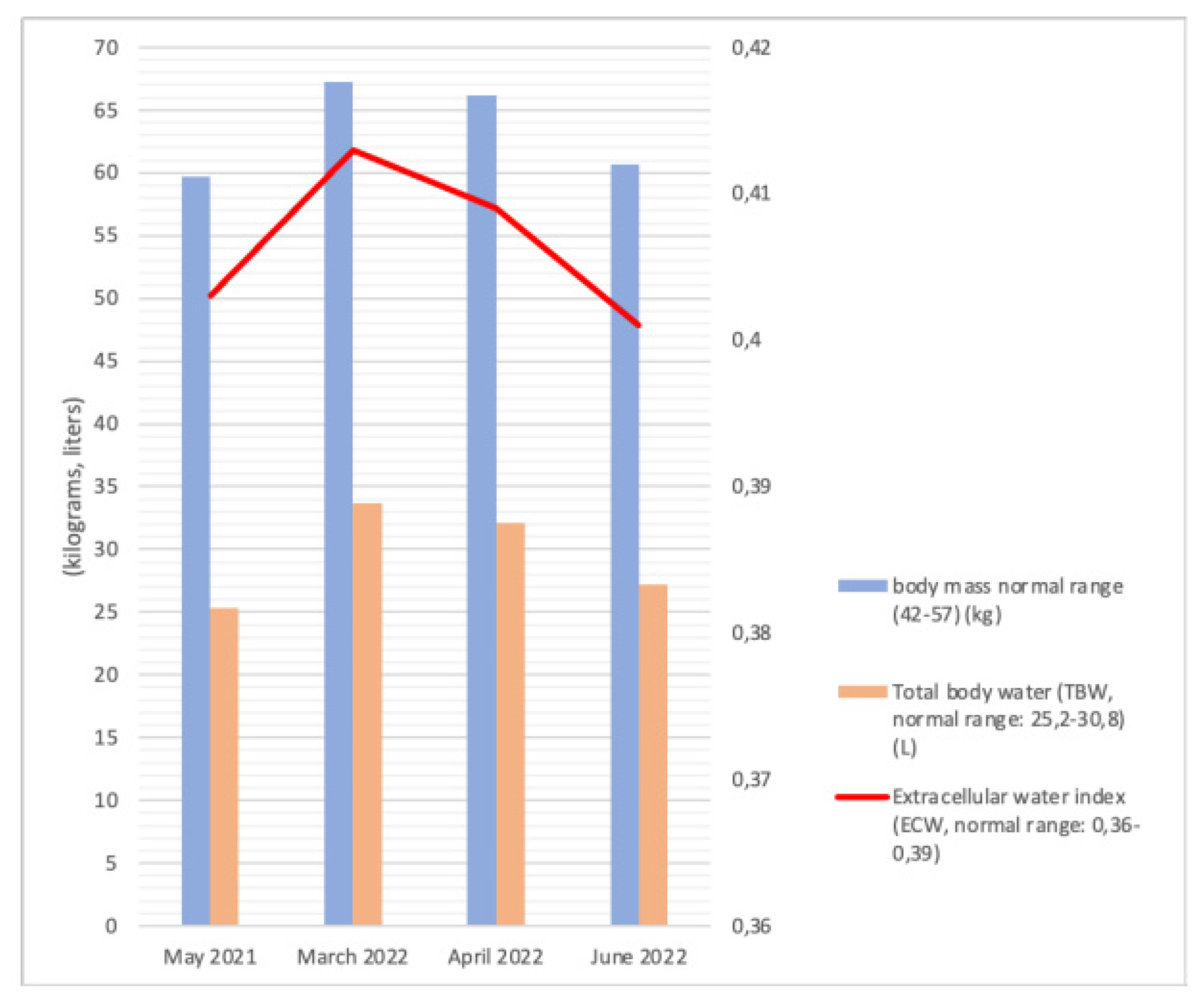
5.4. Bioelectric Impedance Analysis
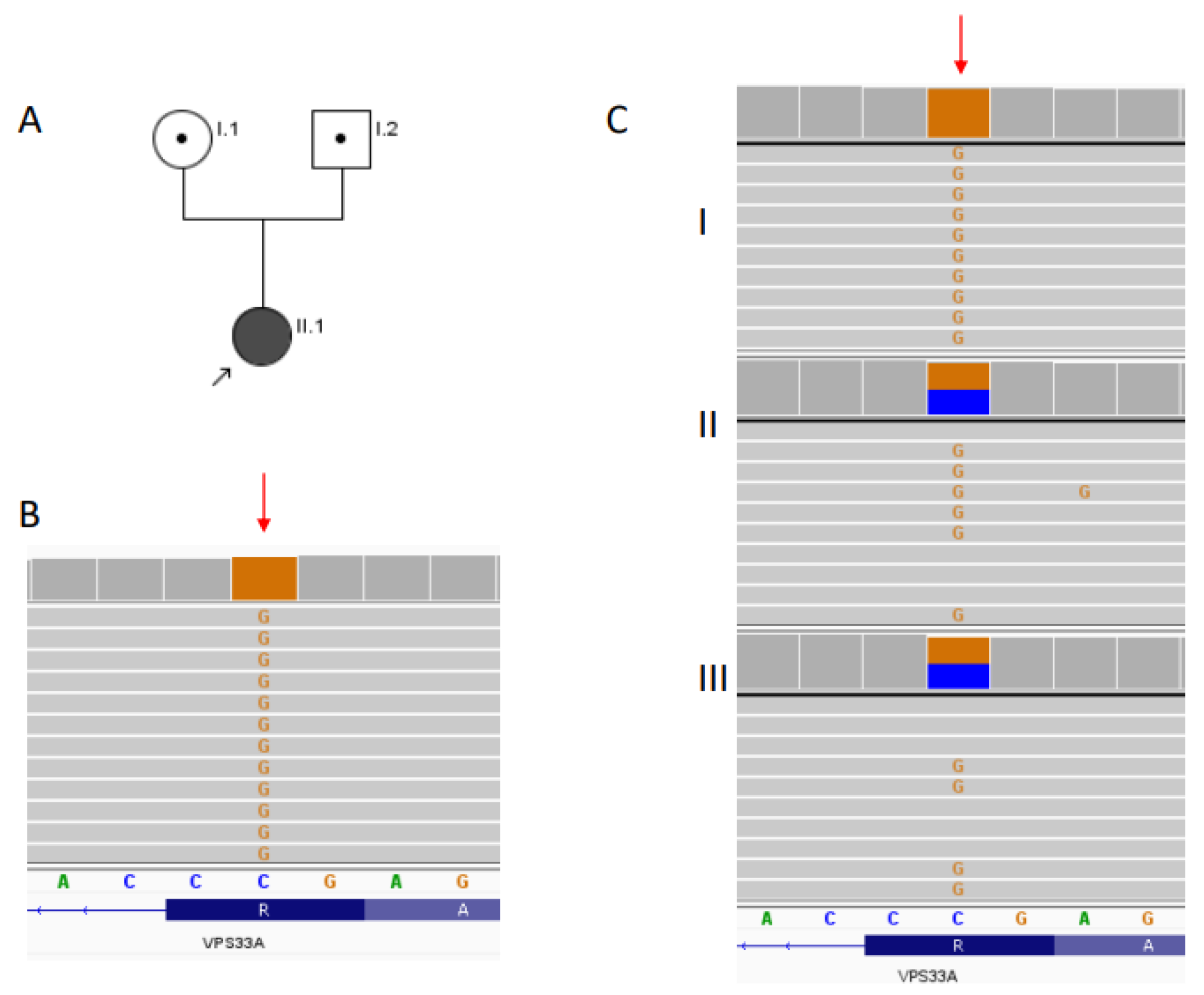
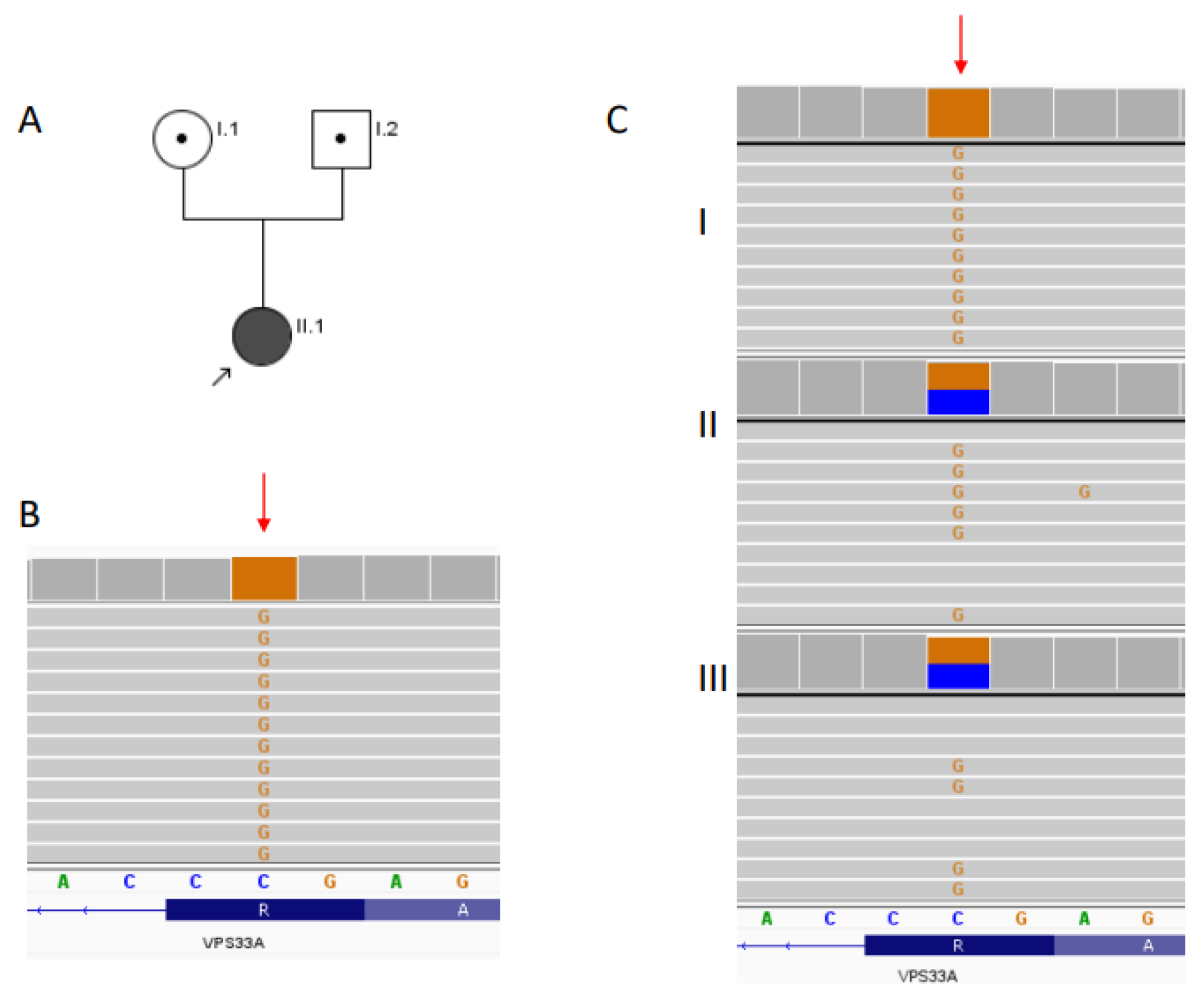
5.5. Genetic Analysis
5.6. Comparison with Other Reported MPS-PS Cases
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| BIA | Bioelectric impedance analysis |
| CS | Chondroitin sulfate |
| DBS | Dry blood spot |
| DS | Dermatan sulfate |
| GAGs | Glycosaminoglycans |
| HOPS | Homotypic fusion and vacuole protein sorting |
| HS | Heparan sulfate |
| IS | Internal standard |
| KS | Keratan sulfate |
| LC-MS/MS | Liquid chromatography-tandem mass spectrometry (LC-MS/MS) |
| LSD | Lysosomal storage diseases |
| MPS | Mucopolysaccharidoses |
| MPS-PS | Mucopolysaccharidosis-plus syndrome |
| pc | Percentile |
| TBW | Total body water |
| TLC | Thin-layer chromatography |
| WES | Whole-exome sequencing |
References
- Clarke, L.A.; Winchester, B.; Giugliani, R.; Tylki-Szymańska, A.; Amartino, H. Biomarkers for the mucopolysaccharidoses: Discovery and clinical utility. Mol. Genet. Metab. 2012, 106, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Tomatsu, S.; Lavery, C.; Giugliani, R.; Harmatz, P.; Scarpa, M.; Węgrzyn, G.; Orii, T. (Eds.) Mucopolysaccharidoses Update. In Metabolic Diseases-Laboratory and Clinical Research; Nova Science Publishers: New York, NY, USA, 2018; Volume 2. [Google Scholar]
- Neufeld, E.F.; Muenzer, J. The Mucopolysaccharidoses. In The Online Metabolic and Molecular Bases of Inherited Disease; Valle, D.L., Antonarakis, S., Ballabio, A., Beaudet, A.L., Mitchell, G.A., Eds.; McGraw Hill: New York, NY, USA, 2019. [Google Scholar]
- Stapleton, M.; Arunkumar, N.; Kubaski, F.; Mason, R.W.; Tadao, O.; Tomatsu, S. Clinical presentation and diagnosis of mucopolysaccharidoses. Mol. Genet. Metab. 2018, 125, 4–17. [Google Scholar] [CrossRef] [PubMed]
- McBride, K.L.; Flanigan, K.M. Update in the Mucopolysaccharidoses. Semin. Pediatr. Neurol. 2021, 37, 100874. [Google Scholar] [CrossRef] [PubMed]
- Parenti, G.; Medina, D.L.; Ballabio, A. The rapidly evolving view of lysosomal storage diseases. EMBO Mol. Med. 2021, 13, e12836. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://omim.org/entry/617303 (accessed on 19 August 2022).
- Gurinova, E.E.; Maksimova, N.R.; Sukhomyasova, A.L. Clinical Description of a Rare Autosomal Recessive Syndrome in the Yakut Children. Yakut. Med. J. 2014, 2, 14–18. [Google Scholar]
- Dursun, A.; Yalnizoglu, D.; Gerdan, O.F.; Yucel-Yilmaz, D.; Sagiroglu, M.S.; Yuksel, B.; Gucer, S.; Sivri, S.; Ozgul, R.K. A probable new syndrome with the storage disease phenotype caused by the VPS33A gene mutation. Clin. Dysmorphol. 2017, 26, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kondo, H.; Maksimova, N.; Otomo, T.; Kato, H.; Imai, A.; Asano, Y.; Kobayashi, K.; Nojima, S.; Nakaya, A.; Hamada, Y.; et al. Mutation in VPS33A affects metabolism of glycosaminoglycans: A new type of mucopolysaccharidosis with severe systemic symptoms. Hum. Mol. Genet. 2017, 26, 173–183. [Google Scholar] [PubMed]
- Pavlova, E.V.; Shatunov, A.; Wartosch, L.; Moskvina, A.I.; Nikolaeva, L.E.; Bright, N.A.; Tylee, K.L.; Church, H.J.; Ballabio, A.; Luzio, J.P.; et al. The lysosomal disease caused by mutant VPS33A. Hum. Mol. Genet. 2019, 28, 2514–2530. [Google Scholar] [CrossRef] [PubMed]
- Vasilev, F.; Sukhomyasova, A.; Otomo, T. Mucopolysaccharidosis-Plus Syndrome. Int. J. Mol. Sci. 2020, 21, 421. [Google Scholar] [CrossRef] [PubMed]
- Faraguna, M.C.; Musto, F.; Crescitelli, V.; Iascone, M.; Spaccini, L.; Tonduti, D.; Fedeli, T.; Kullmann, G.; Canonico, F.; Cattoni, A.; et al. Mucopolysaccharidosis-Plus Syndrome, a Rapidly Progressive Disease: Favorable Impact of a Very Prolonged Steroid Treatment on the Clinical Course in a Child. Genes 2022, 13, 442. [Google Scholar] [CrossRef] [PubMed]
- Sofronova, V.; Iwata, R.; Moriya, T.; Loskutova, K.; Gurinova, E.; Chernova, M.; Timofeeva, A.; Shvedova, A.; Vasilev, F.; Novgorodova, S.; et al. Hematopoietic Disorders, Renal Impairment and Growth in Mucopolysaccharidosis-Plus Syndrome. Int. J. Mol. Sci. 2022, 23, 5851. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.medrxiv.org/content/10.1101/2022.08.27.22279208v1 (accessed on 21 September 2022).
- Tarskaia, L.A.; Zinchenko, R.A.; El’chinova, G.I.; Egorova, A.G.; Korotov, M.N.; Basova, E.V.; Prokop’eva, A.M.; Sivtseva, E.N.; Nikolaeva, E.E.; Banshchinkova, E.S.; et al. The structure and diversiity of hereditary pathology in Sakha Republic (Yakutia). Genetika 2004, 40, 1530–1539. [Google Scholar] [PubMed]
- Yıldız, Y.; Koşukcu, C.; Aygün, D.; Akçaboy, M.; Öztek Çelebi, F.Z.; Taşcı Yıldız, Y.; Şahin, G.; Aytekin, C.; Yüksel, D.; Lay, İ.; et al. Homozygous missense VPS16 variant is associated with a novel disease, resembling mucopolysaccharidosis-plus syndrome in two siblings. Clin. Genet. 2021, 100, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Sofou, K.; Meier, K.; Sanderson, L.E.; Kaminski, D.; Montoliu-Gaya, L.; Samuelsson, E.; Blomqvist, M.; Agholme, L.; Gärtner, J.; Mühlhausen, C.; et al. Bi-allelic VPS16 variants limit HOPS/CORVET levels and cause a mucopolysaccharidosis-like disease. EMBO Mol. Med. 2021, 13, e13376. [Google Scholar] [CrossRef] [PubMed]
- Blom, W.; Luteyn, J.C.; Kelholt-Dijkman, H.H.; Huijmans, J.G.; Loonen, M.C. Thin-layer chromatography of oligosaccharides in urine as a rapid indication for the diagnosis of lysosomal acid maltase deficiency (Pompe’s disease). Clin. Chim. Acta 1983, 134, 221–227. [Google Scholar] [CrossRef]
- Humbel, R.; Collart, M. Oligosaccharides in urine of patients with glycoprotein storage diseases. I. Rapid detection by thin-layer chromatography. Clin. Chim. Acta 1975, 60, 143–145. [Google Scholar] [CrossRef]
- Svenneholm, L. Quantitative estimation of sialic acids. II. A colorimetric resorcinol-hydrochloric acid method. Biochim. Biophys. Acta 1957, 24, 604–611. [Google Scholar]
- Ługowska, A.; Tylki-Szymańska, A.; Sawnor-Korszyńska, D. Thin-layer chromatography of urine oligosaccharides in the diagnosis of some lysosomal storage disorders. Pediatria. Polska. 1995, 70, 847–855. [Google Scholar] [PubMed]
- Lee, S.W.; Song, J.H.; Kim, G.A.; Lee, K.J.; Kim, M. Assessment of total body water from anthropometry-based equations using bioelectrical impedance as reference in Korean adult control and haemodialysis subjects. Nephrol. Dial. Transpl. 2001, 16, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Rydzanicz, M.; Zwoliński, P.; Gasperowicz, P.; Pollak, A.; Kostrzewa, G.; Walczak, A.; Konarzewska, M.; Płoski, R. A recurrent de novo variant supports KCNC2 involvement in the pathogenesis of developmental and epileptic encephalopathy. Am. J. Med. Genet. A 2021, 185, 3384–3389. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lysosomal Enzyme/Lysosphingolipid | Enzyme Activity/Biomarker Concentration in Dried Blood Spot | Reference Values |
|---|---|---|
| Lysosphingolipid concentration (ng/mL) | ||
| globotriaosylsphingosine (lyso-Gb3) | 0.9 | 0.05–3.0 |
| lysosphingomyelin (lyso-SM) | 6.04 | 0.2–15 |
| lysosphingomyelin-509 (lyso-SM-509) | 2.9 | 0.15–3.7 |
| hexosylsphingosines (galactosylsphingosine + glucosylsphingosine) | 7.79 | 0.2–10 |
| lyso-monosialoganglioside GM1 (lyso-GM1) | 0 | 0–10 |
| lyso-monosialoganglioside GM2 (lyso-GM2) | 0 | 0–10 |
| lysosulfatide | 0 | 0–10 |
| Lysosomal enzymes activity (µmol/hr/L) | ||
| galactosylceramidase (GALC) | 2.84 | 0.7–10 |
| acid alpha-glucosidase (GAA) | 5.12 | 1–25 |
| alpha-galactosidase A (GLA) | 5.53 | 0.8–15 |
| beta-glucosidase (ABG) | 4.12 | 1.5–25 |
| acid sphingomyelinase (ASM) | 5.68 | 1.5–25 |
| alpha-L-iduronidase (IDUA) | 4.86 | 1–25 |
| N-Acetyl-Alpha-Glucosaminidase (NAGLU) | 5.52 | 1–20 |
| N-acetylgalactosamine 6-sulfatase (GALNS) | 1.33 | 0.5–5 |
| arylsulfatase B (ARSB) | 4.10 | 1–15 |
| beta-galactosidase (GLB1) | 6.61 | 2–30 |
| beta-glucuronidase (GUSB) | 14.28 | 10–65 |
| iduronate-2-sulfatase (ID2S) | 18.5 | 10–50 |
| tripeptidyl peptidase 1 (TPP1) | 40.09 | 15–85 |
| References | No. of Patients | Ethnicity | Consanguinity | Renal Phenotype | Hematological Phenotype | Cardiological Phenotype | Immunological Phenotype | Other | Follow-Up |
|---|---|---|---|---|---|---|---|---|---|
| This report | 1 | Caucasian | Probably yes | Slight proteinuria with normal kidney function | Normal results of hematological studies | Stable heart disease—mild mitral stenosis | Decreased serum IgG concentration | Recurrent peripheral edemas, autism spectrum disorder, visceral obesity, fetal ascites | Alive; last follow-up: 12 years |
| Gurinova et al., 2014 [8] | 11 | Yakut | Yes | Nephromegaly in 3/11 | Not reported | Congenital heart defects in 7/11; heart failure and pulmonary hypertension | Not reported | Not reported | 9/11 died till 2 y due to cardiorespiratory failure |
| Dursun et al., 2016 [9] | 2 | Turkish/ Yakut | Yes | Proteinuria in 2/2, renal biopsy—segmental/global sclerosis, periglomelular fibrosis | Anemia in 2/2 | Not reported | Not reported | Not reported | 2/2 died at the age of 6 years and 3 months (respiratory and renal failure) and 6 months (cardiopulmonary failure), respectively |
| Kondo et al., 2017 [10] | 13 | Yakut | Yes | Proteinuria in 13/13 while nephritic syndrome in 4/13; autopsy findings in 1 of them—significant grade of glomerular hyalinization, accumulation of lymphocytes in the renal interstitium | Anemia in 13/13, thrombocytopenia in 12/13, leukocytopenia in 8/13; bone marrow hypoplasia in 2/3 | Congenital heart defects: patent ductus arteriosus in 7/13, atrial septal defect in 7/13; hypertrophic cardiomyopathy in 9/13 | Not reported | Not reported | 11/13 died of cardiorespiratory failure at approximately 1 to 2 y |
| Pavlova et al., 2019 [11] | 5 | Yakut | Yes | Nephrotic syndrome (full-blown) in 4/5 | Anemia in 5/5, thrombocytopenia in 4/13, leukopenia in 4/5; coagulation defects with episodic intestinal bleeding in 2/5 | Congenital heart defects: patent ductus arteriosus in 2/5 | Low serum IgG concentration in 4/5 | Not reported | |
| Vasilev et al., 2020 [12] | 1 | Yakut | No | Not reported | Anemia | Insufficiency of aortic valve, mitral and tricuspid valve regurgitation, pulmonary hypertension (1 y 9 months) | Not reported | Not reported | Death at 1 y 10 months due to respiratory insufficiency followed by multiple organ failure |
| Faraguna et al., 2022 [13] | 1 | Moroccan | Yes | Tubulopathy with low molecular weight proteinuria | Iron-refractory microcytic anemia, transient mild thrombo-cytopenia | Severe mitral insufficiency with atrial dilatation | Secondary hemophagocytic lymphohistiocytosis | Nonautoimmune subclinical hypothyroidism | Pneumonia complicated by respiratory insufficiency requiring orotracheal intubation at 2 y, development of a secondary hemophagocytic lymphohistiocytosis during septic shock due to pneumonia |
| Sofronova et al., 2022 [14] | 5 | Yakut | Yes | Proteinuria in 5/5, mild kidney damage (defined as eGFR < 90) in 4/5, elevated serum uric acid level in 4/5; autopsy findings—foamy podocytes, chronic interstitial inflammation, periglomelular fibrosis | Progressive anemia in 5/5, low within reference range platelets count in 5/5, bone marrow histology—hypocellular fatty marrow, absence of erythroblastic islands and megakaryocytes | Not reported | Not reported | Growth retardation in several patients; 5/5 patients had below-average weight | 5/5 died—precise cause of death not known |
| Pavlova et al., preprint published on 30 August 2022 [15] | 1 | Southern European/Mediterranean | Not reported | Proteinuria in childhood | Normal results of hematological studies | Not reported | Normal serum IgG concentration | Autism spectrum disorder, intellectual disability | Alive early 20s |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lipiński, P.; Szczałuba, K.; Buda, P.; Zakharova, E.Y.; Baydakova, G.; Ługowska, A.; Różdzyńska-Świątkowska, A.; Cyske, Z.; Węgrzyn, G.; Pollak, A.; et al. Mucopolysaccharidosis-Plus Syndrome: Report on a Polish Patient with a Novel VPS33A Variant with Comparison with Other Described Patients. Int. J. Mol. Sci. 2022, 23, 11424. https://doi.org/10.3390/ijms231911424
Lipiński P, Szczałuba K, Buda P, Zakharova EY, Baydakova G, Ługowska A, Różdzyńska-Świątkowska A, Cyske Z, Węgrzyn G, Pollak A, et al. Mucopolysaccharidosis-Plus Syndrome: Report on a Polish Patient with a Novel VPS33A Variant with Comparison with Other Described Patients. International Journal of Molecular Sciences. 2022; 23(19):11424. https://doi.org/10.3390/ijms231911424
Chicago/Turabian StyleLipiński, Patryk, Krzysztof Szczałuba, Piotr Buda, Ekaterina Y. Zakharova, Galina Baydakova, Agnieszka Ługowska, Agnieszka Różdzyńska-Świątkowska, Zuzanna Cyske, Grzegorz Węgrzyn, Agnieszka Pollak, and et al. 2022. "Mucopolysaccharidosis-Plus Syndrome: Report on a Polish Patient with a Novel VPS33A Variant with Comparison with Other Described Patients" International Journal of Molecular Sciences 23, no. 19: 11424. https://doi.org/10.3390/ijms231911424
APA StyleLipiński, P., Szczałuba, K., Buda, P., Zakharova, E. Y., Baydakova, G., Ługowska, A., Różdzyńska-Świątkowska, A., Cyske, Z., Węgrzyn, G., Pollak, A., Płoski, R., & Tylki-Szymańska, A. (2022). Mucopolysaccharidosis-Plus Syndrome: Report on a Polish Patient with a Novel VPS33A Variant with Comparison with Other Described Patients. International Journal of Molecular Sciences, 23(19), 11424. https://doi.org/10.3390/ijms231911424