
Investigation of Transcriptome Patterns in Endometrial Cancers from Obese and Lean Women
 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
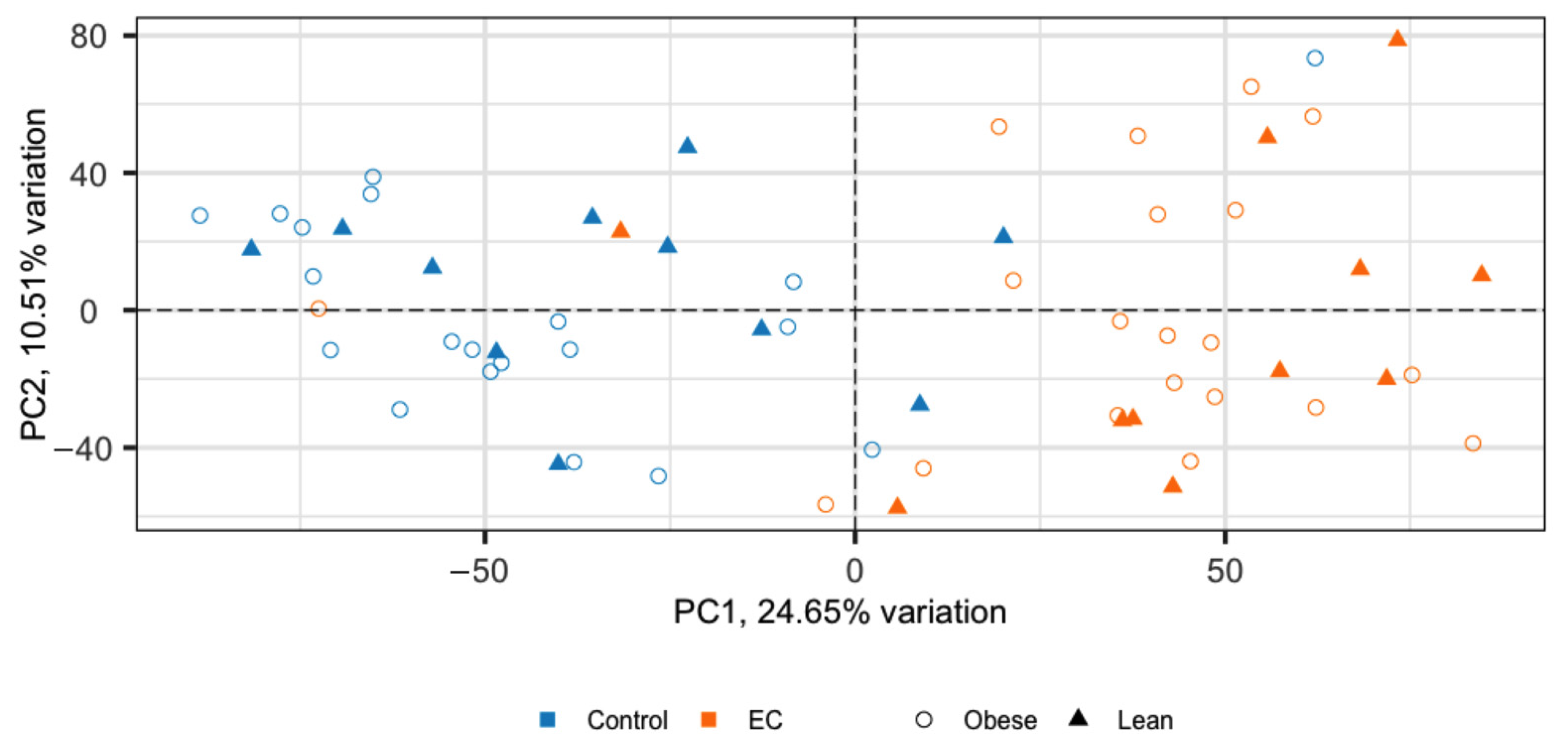
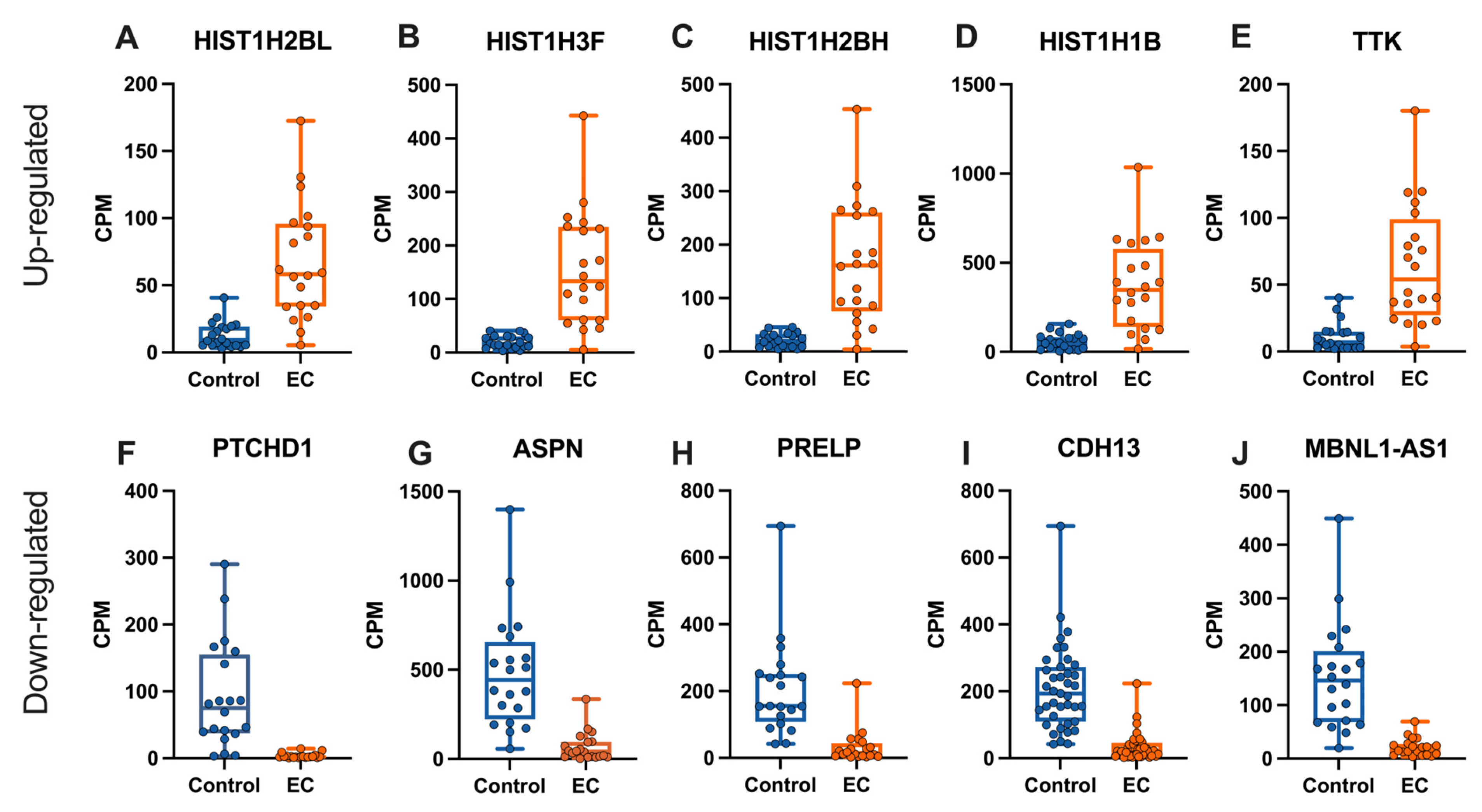
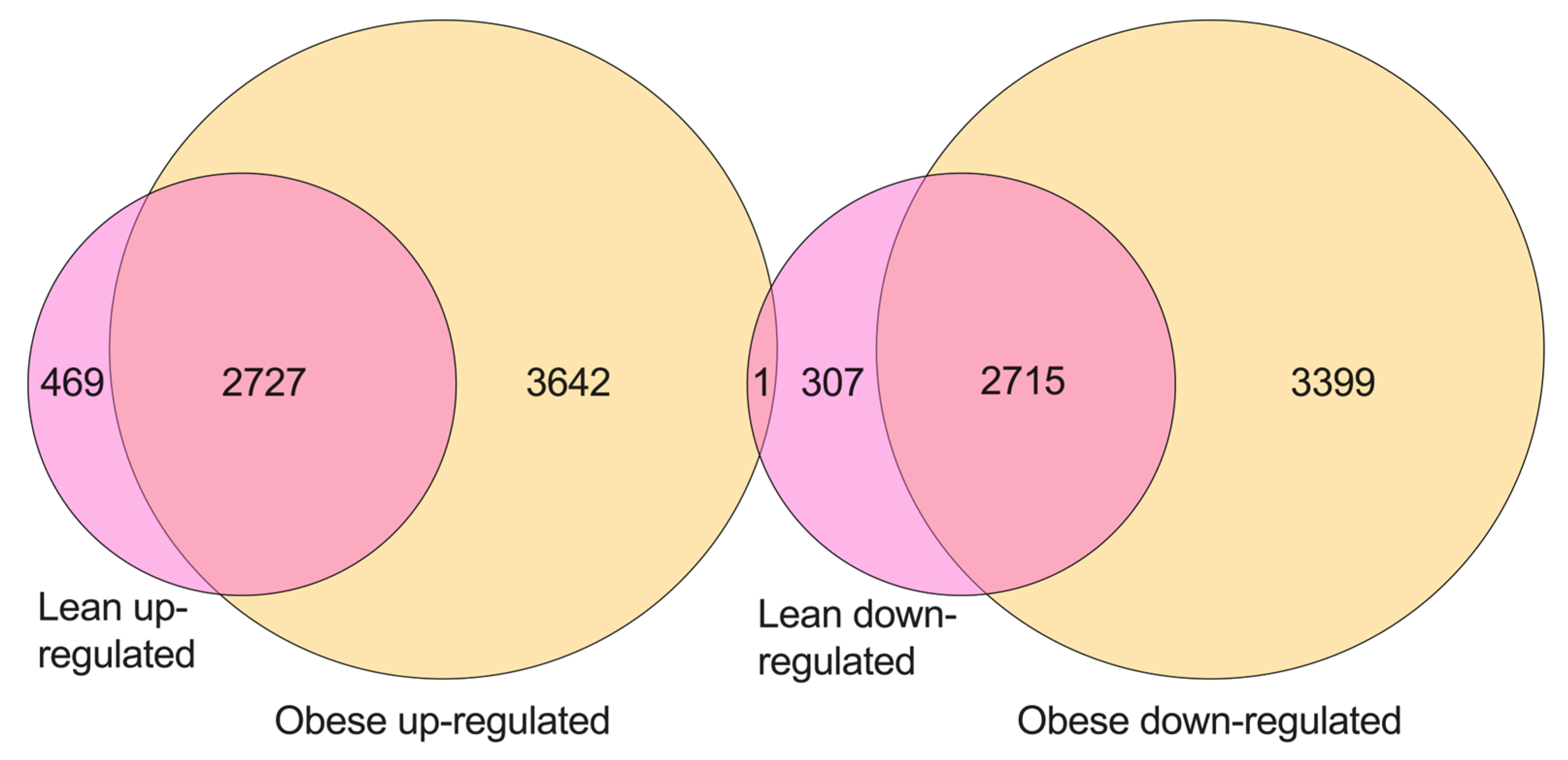
2. Results
3. Discussion
4. Materials and Methods
4.1. Endometrial Cancer and Control Tissue Samples
4.2. RNA Template Preparation and Sequencing
4.3. Sequence Read Alignment and Transcript Assembly
4.4. Principal Component Analysis
4.5. Differential Expression
4.6. Gene Ontology Enrichment Analysis
4.7. Venn Diagram for Common Differentially Expressed Genes between Lean and Obese Cohort
4.8. Hypergeometric Testing
4.9. Validation with the Cancer Genome Atlas (TCGA) Data
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Lortet-Tieulent, J.; Ferlay, J.; Bray, F.; Jemal, A. International Patterns and Trends in Endometrial Cancer Incidence, 1978–2013. JNCI J. Natl. Cancer Inst. 2017, 110, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Morice, P.; Leary, A.; Creutzberg, C.; Abu-Rustum, N.; Darai, E. Endometrial cancer. Lancet 2016, 387, 1094–1108. [Google Scholar] [CrossRef]
- Bokhman, J.V. Two pathogenetic types of endometrial carcinoma. Gynecol. Oncol. 1983, 15, 10–17. [Google Scholar] [CrossRef]
- Levine, D.A.; Getz, G.; Gabriel, S.B.; Cibulskis, K.; Lander, E.; Sivachenko, A.; Sougnez, C.; Lawrence, M.; Kandoth, C.; Dooling, D.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef]
- Chen, B.J.; Byrne, F.L.; Takenaka, K.; Modesitt, S.C.; Olzomer, E.M.; Mills, J.D.; Farrell, R.; Hoehn, K.L.; Janitz, M. Transcriptome landscape of long intergenic non-coding RNAs in endometrial cancer. Gynecol. Oncol. 2017, 147, 654–662. [Google Scholar] [CrossRef]
- Townsend, M.H.; Ence, Z.E.; Felsted, A.M.; Parker, A.C.; Piccolo, S.R.; Robison, R.A.; O’Neill, K.L. Potential new biomarkers for endometrial cancer. Cancer Cell Int. 2019, 19, 19. [Google Scholar] [CrossRef]
- Treeck, O.; Skrzypczak, M.; Schüler-Toprak, S.; Weber, F.; Ortmann, O. Long non-coding RNA CCAT1 is overexpressed in endometrial cancer and regulates growth and transcriptome of endometrial adenocarcinoma cells. Int. J. Biochem. Cell Biol. 2020, 122, 105740. [Google Scholar] [CrossRef]
- Liu, L.; Chen, X.; Zhang, Y.; Hu, Y.; Shen, X.; Zhu, W. Long non-coding RNA TUG1 promotes endometrial cancer development via inhibiting miR-299 and miR-34a-5p. Oncotarget 2017, 8, 31386–31394. [Google Scholar] [CrossRef]
- You, Z.; Liu, C.; Wang, C.; Ling, Z.; Wang, Y.; Wang, Y.; Zhang, M.; Chen, S.; Xu, B.; Guan, H.A.-O.; et al. LncRNA CCAT1 Promotes Prostate Cancer Cell Proliferation by Interacting with DDX5 and MIR-28-5P. Mol. Cancer Ther. 2019, 18, 2469–2479. [Google Scholar] [CrossRef] [Green Version]
- Nissan, A.; Stojadinovic, A.; Mitrani-Rosenbaum, S.; Halle, D.; Grinbaum, R.; Roistacher, M.; Bochem, A.; Dayanc, B.E.; Ritter, G.; Gomceli, I.; et al. Colon cancer associated transcript-1: A novel RNA expressed in malignant and pre-malignant human tissues. Int. J. Cancer 2012, 130, 1598–1606. [Google Scholar] [CrossRef] [PubMed]
- Cui, B.; Li, B.; Liu, Q.; Cui, Y. lncRNA CCAT1 Promotes Glioma Tumorigenesis by Sponging miR-181b. J. Cell. Biochem. 2017, 118, 4548–4557. [Google Scholar] [CrossRef] [PubMed]
- Cho-Clark, M.J.; Sukumar, G.; Vidal, N.M.; Raiciulescu, S.; Oyola, M.G.; Olsen, C.; Mariño-Ramírez, L.; Dalgard, C.L.; Wu, T.J. Comparative transcriptome analysis between patient and endometrial cancer cell lines to determine common signaling pathways and markers linked to cancer progression. Oncotarget 2021, 12, 2500–2513. [Google Scholar] [CrossRef] [PubMed]
- Peterson, C.L.; Laniel, M.-A. Histones and histone modifications. Curr. Biol. 2004, 14, R546–R551. [Google Scholar] [CrossRef]
- Nayak, S.R.; Harrington, E.; Boone, D.; Hartmaier, R.; Chen, J.; Pathiraja, T.N.; Cooper, K.L.; Fine, J.L.; Sanfilippo, J.; Davidson, N.E.; et al. A Role for Histone H2B Variants in Endocrine-Resistant Breast Cancer. Horm. Cancer 2015, 6, 214–224. [Google Scholar] [CrossRef]
- Xie, W.; Zhang, J.; Zhong, P.; Qin, S.; Zhang, H.; Fan, X.; Yin, Y.; Liang, R.; Han, Y.; Liao, Y.; et al. Expression and potential prognostic value of histone family gene signature in breast cancer. Exp. Med. 2019, 18, 4893–4903. [Google Scholar] [CrossRef]
- Mirisola, V.; Mora, R.; Esposito, A.I.; Guastini, L.; Tabacchiera, F.; Paleari, L.; Amaro, A.; Angelini, G.; Dellepiane, M.; Pfeffer, U.; et al. A prognostic multigene classifier for squamous cell carcinomas of the larynx. Cancer Lett. 2011, 307, 37–46. [Google Scholar] [CrossRef]
- Liu, S.; Shi, J.; Liu, Y.; Wang, L.; Zhang, J.; Huang, Y.; Chen, Z.; Yang, J. Analysis of mRNA expression differences in bladder cancer metastasis based on TCGA datasets. J. Int. Med. Res. 2021, 49, 0300060521996929. [Google Scholar] [CrossRef]
- Zhang, Y.-Q.; Yuan, Y.; Zhang, J.; Lin, C.-Y.; Guo, J.-L.; Liu, H.-S.; Guo, Q. Evaluation of the roles and regulatory mechanisms of PD-1 target molecules in NSCLC progression. Ann. Transl. Med. 2021, 9, 1168. [Google Scholar] [CrossRef]
- Liao, R.; Chen, X.; Cao, Q.; Wang, Y.; Miao, Z.; Lei, X.; Jiang, Q.; Chen, J.; Wu, X.; Li, X.; et al. HIST1H1B Promotes Basal-Like Breast Cancer Progression by Modulating CSF2 Expression. Front. Oncol. 2021, 11, 780094. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Pang, L.; Wei, C. Identification of a Four-Gene Signature with Prognostic Significance in Endometrial Cancer Using Weighted-Gene Correlation Network Analysis. Front. Genet. 2021, 12, 678780. [Google Scholar] [CrossRef]
- Tsai, Y.-M.; Wu, K.-L.; Chang, Y.-Y.; Hung, J.-Y.; Chang, W.-A.; Chang, C.-Y.; Jian, S.-F.; Tsai, P.-H.; Huang, Y.-C.; Chong, I.-W.; et al. Upregulation of Thr/Tyr kinase Increases the Cancer Progression by Neurotensin and Dihydropyrimidinase-Like 3 in Lung Cancer. Int. J. Mol. Sci. 2020, 21, 1640. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Yang, Y.; Zhang, W.; Liu, X.; Yang, G. TTK regulates proliferation and apoptosis of gastric cancer cells through the Akt-mTOR pathway. FEBS Open Bio 2020, 10, 1542–1549. [Google Scholar] [CrossRef] [PubMed]
- Kaistha, B.P.; Honstein, T.; Müller, V.; Bielak, S.; Sauer, M.; Kreider, R.; Fassan, M.; Scarpa, A.; Schmees, C.; Volkmer, H.; et al. Key role of dual specificity kinase TTK in proliferation and survival of pancreatic cancer cells. Br. J. Cancer 2014, 111, 1780–1787. [Google Scholar] [CrossRef]
- Zhang, J.; Jiang, Y.; Zhao, Y.; Wang, W.; Xie, Y.; Wang, H.; Yang, Y. Downregulation of tyrosine threonine kinase inhibits tumor growth via G2/M arrest in human endometrioid endometrial adenocarcinoma. Tumor Biol. 2017, 39, 1010428317712444. [Google Scholar] [CrossRef]
- Noor, A.; Whibley, A.; Marshall, C.R.; Gianakopoulos, P.J.; Piton, A.; Carson, A.R.; Orlic-Milacic, M.; Lionel, A.C.; Sato, D.; Pinto, D. Disruption at the PTCHD1 Locus on Xp22. 11 in Autism spectrum disorder and intellectual disability. Sci. Transl. Med. 2010, 2, 49ra68. [Google Scholar] [CrossRef]
- Katayama, M.L.H.; Vieira, R.A.d.C.; Andrade, V.P.; Roela, R.A.; Lima, L.G.C.A.; Kerr, L.M.; Campos, A.P.d.; Pereira, C.A.d.B.; Serio, P.A.d.M.P.; Encinas, G. Stromal cell signature associated with response to neoadjuvant chemotherapy in locally advanced breast cancer. Cells 2019, 8, 1566. [Google Scholar] [CrossRef]
- Kram, V.; Kilts, T.M.; Bhattacharyya, N.; Li, L.; Young, M.F. Small leucine rich proteoglycans, a novel link to osteoclastogenesis. Sci. Rep. 2017, 7, 12627. [Google Scholar] [CrossRef]
- Iozzo, R.V.; Schaefer, L. Proteoglycan form and function: A comprehensive nomenclature of proteoglycans. Matrix Biol. 2015, 42, 11–55. [Google Scholar] [CrossRef]
- Henry, S.P.; Takanosu, M.; Boyd, T.C.; Mayne, P.M.; Eberspaecher, H.; Zhou, W.; de Crombrugghe, B.; Hook, M.; Mayne, R. Expression pattern and gene characterization of asporin. a newly discovered member of the leucine-rich repeat protein family. J. Biol. Chem. 2001, 276, 12212–12221. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, P.; Aspberg, A.; Önnerfjord, P.; Bayliss, M.T.; Neame, P.J.; Heinegård, D. Identification and characterization of asporin: A novel member of the leucine-rich repeat protein family closely related to decorin and biglycan. J. Biol. Chem. 2001, 276, 12201–12211. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yang, J.; Zhou, W.; Chen, Z.; Wu, W.; Zhang, S.; Wang, L. Identification of LncRNA CASC7/miR-26/ASPN/TGF-β/Smad Axis in Endometrial Cancer. Pak. J. Zool. 2022, 1–9. [Google Scholar] [CrossRef]
- Bengtsson, E.; Mörgelin, M.; Sasaki, T.; Timpl, R.; Heinegård, D.; Aspberg, A. The Leucine-rich Repeat Protein PRELP Binds Perlecan and Collagens and May Function as a Basement Membrane Anchor*. J. Biol. Chem. 2002, 277, 15061–15068. [Google Scholar] [CrossRef]
- Andreeva, A.V.; Kutuzov, M.A. Cadherin 13 in cancer. Genes Chromosomes Cancer 2010, 49, 775–790. [Google Scholar] [CrossRef]
- Lee, S.W.; Reimer, C.L.; Campbell, D.B.; Cheresh, P.; Duda, R.B.; Kocher, O. H-cadherin expression inhibits in vitro invasiveness and tumor formation in vivo. Carcinogenesis 1998, 19, 1157–1159. [Google Scholar] [CrossRef]
- Ding, X.; Xu, X.; He, X.-F.; Yuan, Y.; Chen, C.; Shen, X.-Y.; Su, S.; Chen, Z.; Xu, S.-T.; Huang, Y.-H. Muscleblind-like 1 antisense RNA 1 inhibits cell proliferation, invasion, and migration of prostate cancer by sponging miR-181a-5p and regulating PTEN/PI3K/AKT/mTOR signaling. Bioengineered 2021, 12, 803–814. [Google Scholar] [CrossRef]
- Cao, G.; Tan, B.; Wei, S.; Shen, W.; Wang, X.; Chu, Y.; Rong, T.; Gao, C. Down-regulation of MBNL1-AS1 contributes to tumorigenesis of NSCLC via sponging miR-135a-5p. Biomed. Pharmacother. 2020, 125, 109856. [Google Scholar] [CrossRef]
- Kartha, R.V.; Subramanian, S. Competing endogenous RNAs (ceRNAs): New entrants to the intricacies of gene regulation. Front. Genet. 2014, 5, 8. [Google Scholar] [CrossRef]
- Umene, K.; Yanokura, M.; Banno, K.; Irie, H.; Adachi, M.; Iida, M.; Nakamura, K.; Nogami, Y.; Masuda, K.; Kobayashi, Y.; et al. Aurora kinase A has a significant role as a therapeutic target and clinical biomarker in endometrial cancer. Int. J. Oncol. 2015, 46, 1498–1506. [Google Scholar] [CrossRef] [Green Version]
- Weber, D.G.; Johnen, G.; Casjens, S.; Bryk, O.; Pesch, B.; Jöckel, K.-H.; Kollmeier, J.; Brüning, T. Evaluation of long noncoding RNA MALAT1 as a candidate blood-based biomarker for the diagnosis of non-small cell lung cancer. BMC Res. Notes 2013, 6, 518. [Google Scholar] [CrossRef] [PubMed]
- Adiga, D.; Eswaran, S.; Pandey, D.; Sharan, K.; Kabekkodu, S.P. Molecular landscape of recurrent cervical cancer. Crit. Rev. Oncol./Hematol. 2021, 157, 103178. [Google Scholar] [CrossRef] [PubMed]
- Weng, S.-L.; Wu, W.-J.; Hsiao, Y.-H.; Yang, S.-F.; Hsu, C.-F.; Wang, P.-H. Significant association of long non-coding RNAs HOTAIR genetic polymorphisms with cancer recurrence and patient survival in patients with uterine cervical cancer. Int. J. Med. Sci. 2018, 15, 1312. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, J.; Zhong, Y.; Guo, R.; Chu, D.; Qiu, H.; Yuan, Z. HOTAIR: A key regulator in gynecologic cancers. Cancer Cell Int. 2017, 17, 65. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.-n.; Li, W.; Wang, X.-l.; Hu, Z.; Zhu, D.; Ding, W.-c.; Liu, D.; Li, K.-z.; Ma, D.; Wang, H. CLDN1 expression in cervical cancer cells is related to tumor invasion and metastasis. Oncotarget 2016, 7, 87449. [Google Scholar] [CrossRef] [PubMed]
- Cheung, T.-H.; Lo, K.W.-K.; Yim, S.-F.; Chan, L.K.-Y.; Heung, M.-S.; Chan, C.-S.; Cheung, A.Y.-K.; Chung, T.K.-H.; Wong, Y.-F. Epigenetic and genetic alternation of PTEN in cervical neoplasm. Gynecol. Oncol. 2004, 93, 621–627. [Google Scholar] [CrossRef]
- Lucidi, A.; Buca, D.; Ronsini, C.; Tinari, S.; Bologna, G.; Buca, D.; Leombroni, M.; Liberati, M.; D’antonio, F.; Scambia, G. Role of extracellular vesicles in epithelial ovarian cancer: A systematic review. Int. J. Mol. Sci. 2020, 21, 8762. [Google Scholar] [CrossRef]
- Atallah, G.A.; Aziz, N.H.A.; Teik, C.K.; Shafiee, M.N.; Kampan, N.C. New predictive biomarkers for ovarian cancer. Diagnostics 2021, 11, 465. [Google Scholar] [CrossRef]
- Kaakoush, N.O.; Olzomer, E.M.; Kosasih, M.; Martin, A.R.; Fargah, F.; Lambie, N.; Susic, D.; Hoehn, K.A.-O.; Farrell, R.A.-O.; Byrne, F.A.-O. Differences in the Active Endometrial Microbiota across Body Weight and Cancer in Humans and Mice. Cancers 2022, 14, 2141. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Bioinformatics; Babraham Institute: Cambridge, UK, 2010. [Google Scholar]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- GENCODE. Statistics about the GENCODE Release 31. Available online: https://www.gencodegenes.org/human/stats_31.html (accessed on 2 July 2022).
- matplotlib-venn. matplotlib-venn. Available online: https://pypi.org/project/matplotlib-venn/ (accessed on 25 June 2022).
- Blighe, K.; Lun, A.; Blighe, K.; Lun, A. PCAtools: Everything Principal Components Analysis. R Package Version 2.6.0. 2021. Available online: https://github.com/kevinblighe/PCAtools (accessed on 10 August 2022).
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, D.J.; Chen, Y.; Smyth, G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takenaka, K.; Curry-Hyde, A.; Olzomer, E.M.; Farrell, R.; Byrne, F.L.; Janitz, M. Investigation of Transcriptome Patterns in Endometrial Cancers from Obese and Lean Women. Int. J. Mol. Sci. 2022, 23, 11471. https://doi.org/10.3390/ijms231911471
Takenaka K, Curry-Hyde A, Olzomer EM, Farrell R, Byrne FL, Janitz M. Investigation of Transcriptome Patterns in Endometrial Cancers from Obese and Lean Women. International Journal of Molecular Sciences. 2022; 23(19):11471. https://doi.org/10.3390/ijms231911471
Chicago/Turabian StyleTakenaka, Konii, Ashton Curry-Hyde, Ellen M. Olzomer, Rhonda Farrell, Frances L. Byrne, and Michael Janitz. 2022. "Investigation of Transcriptome Patterns in Endometrial Cancers from Obese and Lean Women" International Journal of Molecular Sciences 23, no. 19: 11471. https://doi.org/10.3390/ijms231911471