
Safe and Efficient Sigma1 Ligand: A Potential Drug Candidate for Multiple Sclerosis
 ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Results
2.1. In Vitro Drug Candidate Assessment
2.1.1. Metabolic Stability and Metabolite Identification and Profiling
2.1.2. Preliminary ADME Studies
2.1.3. Safety Pharmacology and Toxicology
2.2. Evaluation of Compound 7i as a Sigma1 Receptor Agonist
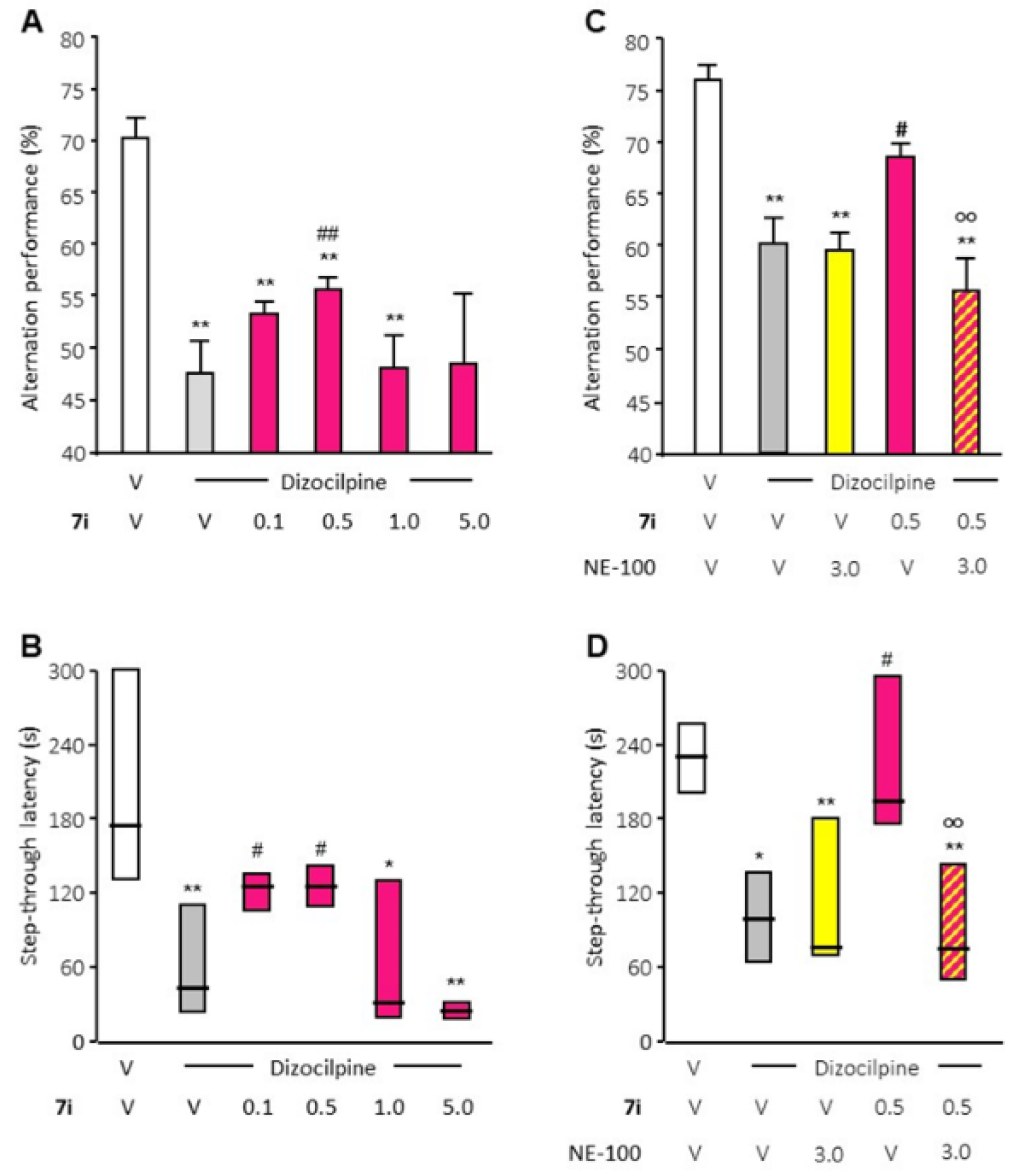
2.3. In Vivo Pharmacology
2.3.1. Preliminary Pharmacokinetic Analysis
2.3.2. Effects of Systemic Administration of the Selective 7i Drug on the Cardiovascular System
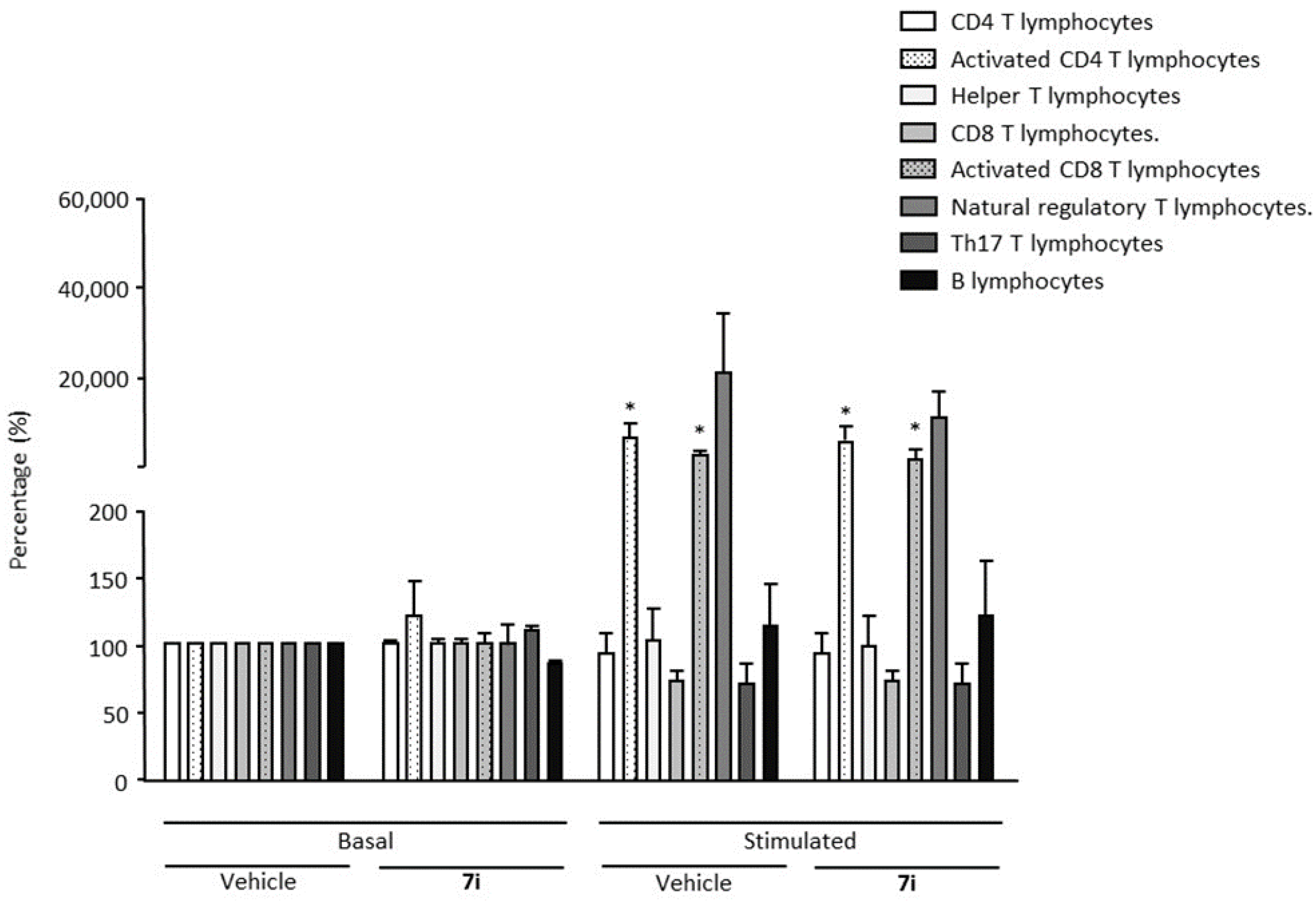
2.4. In Vivo Efficacy of the Selective 7i Agonist in an MS Experimental Model
3. Discussion
4. Materials and Methods
4.1. Chemical
4.2. In Vitro Metabolic Stability and Metabolite Identification
4.3. Assay for Binding to Sigma Receptors
4.4. Physicochemical Properties, Absorption, Distribution and Toxicity
4.5. Toxicology
4.6. In Vitro Evaluation of S1R Functionality
4.7. In Vivo Evaluation of S1R Functionality
4.8. Phamacokinetic Analyses
4.9. Animals
4.10. Pro-Arrhythmic Effects
4.11. EAE Induction and Evaluation
4.12. Statistical Analysis
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gupta, M.; Weaver, D.F. Alzheimer’s: The ABCDE Paradigm. ACS Chem. Neurosci. 2022, 13, 1355–1357. [Google Scholar] [CrossRef]
- Elsbernd, P.M.; Carter, J.L. Using Monoclonal Antibody Therapies for Multiple Sclerosis: A Review. Biol. Targets Ther. 2021, 15, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Hauser, S.L.; Cree, B.A. Treatment of Multiple Sclerosis: A Review. Am. J. Med. 2020, 133, 1380–1390. [Google Scholar] [CrossRef] [PubMed]
- Zadeh, A.R.; Askari, M.; Azadani, N.N.; Ataei, A.; Ghadimi, K.; Tavoosi, N.; Falahatian, M. Mechanism and adverse effects of multiple sclerosis drugs: A review article. Part 1. Int. J. Physiol. Pathophysiol. Pharmacol. 2019, 11, 95–104. [Google Scholar]
- Makurvet, F.D. Biologics vs. small molecules: Drug costs and patient access. Med. Drug Discov. 2021, 9, 100075. [Google Scholar] [CrossRef]
- Campbell, G.; Worrall, J.; Mahad, D.J. The central role of mitochondria in axonal degeneration in multiple sclerosis. Mult. Scler. J. 2014, 20, 1806–1813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, W.R.; Eades, C.G.; Thompson, J.A.; Huppler, R.E.; Gilbert, P.E. The effects of morphine- and nalorphine- like drugs in the nondependent and morphine-dependent chronic spinal dog. J. Pharmacol. Exp. Ther. 1976, 197, 517–532. [Google Scholar] [PubMed]
- Su, T.P. Evidence for sigma opioid receptor: Binding of [3H]SKF-10047 to etorphine-inaccessible sites in guinea-pig brain. J. Pharmacol. Exp. Ther. 1982, 223, 284–290. [Google Scholar]
- Hanner, M.; Moebius, F.F.; Flandorfer, A.; Knaus, H.G.; Striessnig, J.; Kempner, E.; Glossmann, H. Purification, molecular cloning, and expression of the mammalian sigma1-binding site. Proc. Natl. Acad. Sci. USA 1996, 93, 8072–8077. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Tsai, S.-Y.; Mori, T.; Fujimoto, M.; Su, T.-P. Targeting ligand-operated chaperone sigma-1 receptors in the treatment of neuropsychiatric disorders. Expert Opin. Ther. Targets 2011, 15, 557–577. [Google Scholar] [CrossRef] [Green Version]
- A Tsai, S.-Y.; Pokrass, M.J.; Klauer, N.R.; E De Credico, N.; Su, T.-P. Sigma-1 receptor chaperones in neurodegenerative and psychiatric disorders. Expert Opin. Ther. Targets 2014, 18, 1461–1476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurice, T.; Goguadze, N. Sigma-1 (σ1) Receptor in Memory and Neurodegenerative Diseases. Handb. Exp. Pharmacol. 2017, 244, 81–108. [Google Scholar]
- Hayashi, T.; Su, T.-P. Sigma-1 Receptor Chaperones at the ER- Mitochondrion Interface Regulate Ca2+ Signaling and Cell Survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef] [PubMed]
- Delprat, B.; Crouzier, L.; Su, T.P.; Maurice, T. At the Crossing of ER Stress and MAMs: A Key Role of Sigma-1 Receptor? Adv. Exp. Med. Biol. 2020, 1131, 699–718. [Google Scholar] [PubMed]
- Maurice, T. Bi-phasic dose response in the preclinical and clinical developments of sigma-1 receptor ligands for the treatment of neurodegenerative disorders. Expert Opin. Drug Discov. 2021, 16, 373–389. [Google Scholar] [CrossRef] [PubMed]
- Su, T.-P.; Hayashi, T.; Maurice, T.; Buch, S.; Ruoho, A.E. The sigma-1 receptor chaperone as an inter-organelle signaling modulator. Trends Pharmacol. Sci. 2010, 31, 557–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, T.P.; Su, T.C.; Nakamura, Y.; Tsai, S.Y. The Sigma-1 Receptor as a Pluripotent Modulator in Living Systems. Trends Pharmacol. Sci. 2016, 37, 262–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, H.R.; Kruse, A.C. The Molecular Function of σ Receptors: Past, Present, and Future. Trends Pharmacol. Sci. 2019, 40, 636–654. [Google Scholar] [CrossRef]
- Vavers, E.; Zvejniece, L.; Maurice, T.; Dambrova, M. Allosteric Modulators of Sigma-1 Receptor: A Review. Front. Pharmacol. 2019, 10, 223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurice, T.; Su, T.P. The pharmacology of sigma-1 receptors. Pharmacol. Ther. 2009, 124, 195–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, S.-Y.; Hayashi, T.; Mori, T.; Su, T.-P. Sigma-1 receptor chaperones and diseases. Cent. Nerv. Syst. Agents Med. Chem. 2009, 9, 184–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.; Lucke-Wold, B.P.; Mookerjee, S.; Kaushal, N.; Matsumoto, R.R. Sigma-1 Receptors and Neurodegenerative Diseases: Towards a Hypothesis of Sigma-1 Receptors as Amplifiers of Neurodegeneration and Neuroprotection. Adv. Exp. Med. Biol. 2017, 964, 133–152. [Google Scholar] [PubMed] [Green Version]
- Wu, N.H.; Ye, Y.; Wan, B.; Yu, Y.; Liu, C.; Chen, Q. Emerging benefits: Pathophysiological functions and target drugs of the Sigma-1 receptor in neurodegenerative diseases. Mol. Neurobiol. 2021, 58, 5649–5666. [Google Scholar] [CrossRef] [PubMed]
- Collina, S.; Rui, M.; Stotani, S. Are sigma receptor modulators a weapon against multiple sclerosis disease? Future Med. Chem. 2017, 9, 2029–2051. [Google Scholar] [CrossRef] [PubMed]
- Ye, N.; Qin, W.; Tian, S.; Xu, Q.; Wold, E.A.; Zhou, J.; Zhen, X.-C. Small Molecules Selectively Targeting Sigma-1 Receptor for the Treatment of Neurological Diseases. J. Med. Chem. 2020, 63, 15187–15217. [Google Scholar] [CrossRef] [PubMed]
- Toussaint, M.; Delair, B.; Foulon, C.; Lempereur, N.; Vaccher, C.; Maurice, T.; Melnyk, P. Tic hydantoin sigma-1 agonist: Pharmacological characterization on cocaine-induced stimulant and appetitive effects. Eur. Neuropsychopharmacol. 2009, 19, 504–515. [Google Scholar] [CrossRef]
- Oxombre, B.; Lee-Chang, C.; Duhamel, A.; Toussaint, M. High-Affinity σ1 protein agonist reduces clinical and pathological signs of experimental autoimmune encephalomyelitis. Br. J. Pharmacol. 2015, 172, 1769–1782. [Google Scholar] [CrossRef] [Green Version]
- Donnier-Maréchal, M.; Carato, P.; Larchanché, P.-E.; Ravez, S.; Boulahjar, R.; Barczyk, A.; Oxombre, B.; Vermersch, P.; Melnyk, P. Synthesis and pharmacological evaluation of benzamide derivatives as potent and selective sigma-1 protein ligands. Eur. J. Med. Chem. 2017, 138, 964–978. [Google Scholar] [CrossRef]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef]
- Keogh, J. Membrane Transporters in Drug Development. Adv. Pharmacol. 2012, 63, 1–42. [Google Scholar]
- Montanari, F.; Ecker, G.F. Prediction of drug-ABC-transporter interaction-Recent advances and future challenges. Adv. Drug Deliv. Rev. 2015, 86, 17–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowes, J.; Brown, A.J.; Hamon, J.; Jarolimek, W.; Sridhar, A.; Waldron, G.; Whitebread, S. Reducing safety-related drug attrition: The use of in vitro pharmacological profiling. Nat. Rev. Drug Discov. 2012, 11, 909–922. [Google Scholar] [CrossRef] [PubMed]
- Baldrick, P. Genotoxicity test battery–An assessment of its utility in early drug development. Mutat. Res. Toxicol. Environ. Mutagen. 2021, 868–869, 503388. [Google Scholar] [CrossRef] [PubMed]
- Wager, T.T.; Chandrasekaran, R.Y.; Hou, X.; Troutman, M.D.; Verhoest, P.R.; Villalobos, A.; Will, Y. Defining Desirable Central Nervous System Drug Space through the Alignment of Molecular Properties, in Vitro ADME, and Safety Attributes. ACS Chem. Neurosci. 2010, 1, 420–434. [Google Scholar] [CrossRef] [PubMed]
- Sanguinetti, M.C.; Tristani-Firouzi, M. hERG potassium channels and cardiac arrhythmia. Nature 2006, 440, 463–469. [Google Scholar] [CrossRef]
- Bebo, B.; Vandenbark, A.; Offner, H. Male SJL mice do not relapse after induction of EAE with PLP 139-151. J. Neurosci. Res. 1996, 45, 680–689. [Google Scholar] [CrossRef]
- Burrows, D.J.; McGown, A.; A Jain, S.; De Felice, M.; Ramesh, T.M.; Sharrack, B.; Majid, A. Animal models of multiple sclerosis: From rodents to zebrafish. Mult. Scler. J. 2018, 25, 306–324. [Google Scholar] [CrossRef] [Green Version]
- Tuohy, V.K.; Lu, Z.; A Sobel, R.; A Laursen, R.; Lees, M.B. Identification of an encephalitogenic determinant of myelin proteolipid protein for SJL mice. J. Immunol. 1989, 142, 1523–1527. [Google Scholar]
- Lee-Chang, C.; Lefranc, D.; Salleron, J.; Faveeuw, C.; Allet, C.; Vermersch, P.; Oxombre, B.; Prin, L. Susceptibility to experimental autoimmune encephalomyelitis is associated with altered B-cell subsets distribution and decreased serum BAFF levels. Immunol. Lett. 2011, 135, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Meunier, J.; Ieni, J.; Maurice, T. The anti-amnesic and neuroprotective effects of donepezil against amyloid Β 25-35 peptide-induced toxicity in mice involve an interaction with the σ 1 receptor. Br. J. Pharmacol. 2006, 149, 998–1012. [Google Scholar] [CrossRef] [Green Version]
- Recanatini, M.; Poluzzi, E.; Masetti, M.; Cavalli, A.; De Ponti, F. QT prolongation through hERG K+ channel blockade: Current knowledge and strategies for the early prediction during drug development. Med. Res. Rev. 2004, 25, 133–166. [Google Scholar] [CrossRef]
- Balasuriya, D.; D’Sa, L.; Talker, R.; Dupuis, E.; Maurin, F.; Martin, P.; Borgese, F.; Soriani, O.; Edwardson, J.M. A Direct Interaction between the Sigma-1 Receptor and the hERG Voltage-gated K+ Channel Revealed by Atomic Force Microscopy and Homogeneous Time-resolved Fluorescence (HTRF®). J. Biol. Chem. 2014, 289, 32353–32363. [Google Scholar] [CrossRef] [Green Version]
- Brod, S.A. In MS: Immunosuppression is passé. Mult. Scler. Relat. Disord. 2020, 40, 101967. [Google Scholar] [CrossRef] [PubMed]
- Martins, F.; Sofiya, L.; Sykiotis, G.P.; Lamine, F.; Maillard, M.; Fraga, M.; Shabafrouz, K.; Ribi, C.; Cairoli, A.; Guex-Crosier, Y.; et al. Adverse effects of immune-checkpoint inhibitors: Epidemiology, management and surveillance. Nat. Rev. Clin. Oncol. 2019, 16, 563–580. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, E.J. Hormesis and medicine. Br. J. Clin. Pharmacol. 2008, 66, 594–617. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.K.; Mavlyutov, T.; Singh, D.; Biener, G.; Yang, J.; Oliver, J.A.; Ruoho, A.; Raicu, V. The sigma-1 receptors are present in monomeric and oligomeric forms in living cells in the presence and absence of ligands. Biochem. J. 2015, 466, 263–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gromek, K.A.; Suchy, F.P.; Meddaugh, H.R.; Wrobel, R.L.; LaPointe, L.M.; Chu, U.B.; Primm, J.G.; Ruoho, A.E.; Senes, A.; Fox, B.G. The Oligomeric States of the Purified Sigma-1 Receptor Are Stabilized by Ligands. J. Biol. Chem. 2014, 289, 20333–20344. [Google Scholar] [CrossRef] [Green Version]
- Chu, U.B.; Ruoho, A.E. Biochemical Pharmacology of the Sigma-1 Receptor. Mol. Pharmacol. 2015, 89, 142–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motawe, Z.Y.; Abdelmaboud, S.S.; Cuevas, J.; Breslin, J.W. PRE-084 as a tool to uncover potential therapeutic applications for selective sigma-1 receptor activation. Int. J. Biochem. Cell Biol. 2020, 126, 105803. [Google Scholar] [CrossRef]
- Marra, A.; Rossi, D.; Pignataro, L.; Bigogno, C.; Canta, A.; Oggioni, N.; Malacrida, A.; Corbo, M.; Cavaletti, G.; Peviani, M.; et al. Toward the identification of neuroprotective agents: G-scale synthesis, pharmacokinetic evaluation and CNS distribution of (R)-RC-33, a promising Sigma1 receptor agonist. Futur. Med. Chem. 2016, 8, 287–2955. [Google Scholar] [CrossRef]
- Glatigny, S.; Bettelli, E. Experimental Autoimmune Encephalomyelitis (EAE) as Animal Models of Multiple Sclerosis (MS). Cold Spring Harb. Perspect. Med. 2018, 8, a028977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demerens, C.; Stankoff, B.; Zalc, B.; Lubetzki, C. Eliprodil stimulates CNS myelination: New prospects for multiple sclerosis? Neurology 1999, 52, 346. [Google Scholar] [CrossRef] [PubMed]
- Chechneva, O.V.; Mayrhofer, F.; Daugherty, D.J.; Pleasure, D.E.; Hong, J.S.; Deng, W. Low dose dextromethorphan attenuates moderate experimental autoimmune encephalomyelitis by inhibiting NOX2 and reducing peripheral immune cells infiltration in the spinal cord. Neurobiol. Dis. 2011, 44, 63–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisak, R.P.; Nedelkoska, L.; Benjamins, J.A. Sigma-1 receptor agonists as potential protective therapies in multiple sclerosis. J. Neuroimmunol. 2020, 342, 577188. [Google Scholar] [CrossRef]
- Obach, R.S.; Baxter, J.; Liston, T.; Silber, B. The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. J. Pharmacol. Exp. Ther. 1997, 283, 46–58. [Google Scholar]
- Ganapathy, M.E.; Prasad, P.D.; Huang, W.; Seth, P.; Leibach, F.H.; Ganapathy, V. Molecular and ligand-binding characterization of the sigma-receptor in the Jurkat human T lymphocyte cell line. J. Pharmacol. Exp. Ther. 1999, 289, 251–260. [Google Scholar]
- Cheng, Y.C.; Prusoff, W.H. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Hidalgo, I.; Raub, T.; Borchardt, R. Characterization of the human colon carcinoma cell line (Caco-2) as a model system for intestinal epithelial permeability. Gastroenterology 1989, 96, 736–749. [Google Scholar] [CrossRef]
- Banker, M.J.; Clark, T.H.; Williams, J.A. Development and validation of a 96-well equilibrium dialysis apparatus for measuring plasma protein binding. J. Pharm. Sci. 2003, 92, 967–974. [Google Scholar] [CrossRef]
- Dierks, E.A.; Stams, K.R.; Lim, H.K.; Cornelius, G.; Zhang, H.; Ball, S.E. A method for the simultaneous evaluation of the activities of seven major human drug-metabolizing cytochrome P450s using an in vitro cocktail of probe substrates and fast gradient liquid chromatography tandem mass spectrometry. Drug Metab. Dispos. 2001, 29, 23–29. [Google Scholar]
- Kido, Y.; Matsson, P.; Giacomini, K.M. Profiling of a Prescription Drug Library for Potential Renal Drug–Drug Interactions Mediated by the Organic Cation Transporter 2. J. Med. Chem. 2011, 54, 4548–4558. [Google Scholar] [CrossRef] [Green Version]
- Craddock, A.L.; Love, M.W.; Daniel, R.W.; Kirby, L.C.; Walters, H.C.; Wong, M.H.; Dawson, P.A. Expression and transport properties of the human ileal and renal sodium-dependent bile acid transporter. Am. J. Physiol. Liver Physiol. 1998, 274, G157–G169. [Google Scholar] [CrossRef]
- Paturi, D.K.; Kwatra, D.; Ananthula, H.K.; Pal, D.; Mitra, A.K. Identification and functional characterization of breast cancer resistance protein in human bronchial epithelial cells (Calu-3). Int. J. Pharm. 2010, 384, 32–38. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, O.K.; Topp, E.; Makagiansar, I.; Siahaan, T.; Yazdanian, M.; Audus, K.L. Multidrug resistance-associated protein-1 functional activity in Calu-3 cells. J. Pharmacol. Exp. Ther. 2001, 298, 1199–1205. [Google Scholar]
- Matsson, P.; Pedersen, J.M.; Norinder, U.; Bergström, C.A.S.; Artursson, P. Identification of novel specific and general inhibitors of the three major human ATP-binding cassette transporters P-gp, BCRP and MRP2 among registered drugs. Pharm. Research. 2009, 26, 1816–1831. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.; Theile, D.; Ketabi-Kiyanvash, N.; Lindenmaier, H.; Haefeli, W.E. Inhibition of MRP1/ABCC1, MRP2/ABCC2, and MRP3/ABCC3 by nucleoside, nucleotide, and non-nucleoside reverse transcriptase inhibitors. Drug Metab. Dispos. 2007, 35, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.B.; Leake, B.; Cvetkovic, M.; Roden, M.M.; Nadeau, J.; Walubo, A.; Wilkinson, G.R. Modulation by drugs of human hepatic sodium-dependent bile acid transporter (sodium taurocholate cotransporting polypeptide) activity. J. Pharmacol. Exp. Ther. 1999, 291, 1204–1209. [Google Scholar] [PubMed]
- Cihlar, T.; Ho, E.S. Fluorescence-based assay for the interaction of small molecules with the human renal organic anion transporter 1. Anal. Biochem. 2000, 283, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Gui, C.; Obaidat, A.; Chaguturu, R.; Hagenbuch, B. Development of a Cell-Based High-Throughput Assay to Screen for Inhibitors of Organic Anion Transporting Polypeptides 1B1 and 1B3. Curr. Chem. Genom. 2010, 4, 1–8. [Google Scholar] [CrossRef]
- Polli, J.; A Wring, S.; E Humphreys, J.; Huang, L.; Morgan, J.B.; O Webster, L.; Serabjit-Singh, C.S. Rational use of in vitro P-glycoprotein assays in drug discovery. J. Pharmacol. Exp. Ther. 2001, 299, 620–628. [Google Scholar]
- Fahmi, O.A.; Kish, M.; Boldt, S.; Obach, R.S. Cytochrome P450 3A4 mRNA Is a More Reliable Marker than CYP3A4 Activity for Detecting Pregnane X Receptor-Activated Induction of Drug-Metabolizing Enzymes. Drug Metab. Dispos. 2010, 38, 1605–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathes, C. QPatch: The past, present and future of automated patch clamp. Expert Opin. Ther. Targets 2006, 10, 319–327. [Google Scholar] [CrossRef]
- Abraham, V. High content screening applied to large-scale cell biology. Trends Biotechnol. 2004, 22, 15–22. [Google Scholar] [CrossRef]
- Ames, B.N.; McCann, J.; Yamasaki, E. Methods for detecting carcinogens and mutagens with the salmonella/mammalian-microsome mutagenicity test. Mutat. Res. Environ. Mutagenesis Subj. 1975, 31, 347–363. [Google Scholar] [CrossRef]
- Fenech, M.; Morley, A.A. Measurement of micronuclei in lymphocytes. Mutat. Res. 1985, 147, 29–36. [Google Scholar] [CrossRef]
- Nesslany, F. A micromethod for the in vitro micronucleus assay. Mutagenesis 1999, 14, 403–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mrizak, D.; Martin, N.; Barjon, C.; Jimens, A. Effect of Nasopharyngeal Carcinoma-Derived Exosomes on Human Regulatory T Cells. JNCI 2015, 107, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, P.; de Witte, P.A.; Maurice, T.; Gammaitoni, A.; Farfel, G.; Galer, B. Fenfluramine acts as a positive modulator of sigma-1 receptors. Epilepsy Behav. 2020, 105, 106989. [Google Scholar] [CrossRef] [PubMed]
- Maurice, T.; Hiramatsu, M.; Itoh, J.; Kameyama, T.; Hasegawa, T.; Nabeshima, T. Behavioral Evidence for a Modulating Role of Sigma Ligands in Memory Processes. I. Attenuation of Dizocilpine (MK-801)-Induced Amnesia. Brain Res. 1994, 647, 44–56. [Google Scholar] [CrossRef]
- Maurice, T. Protection by sigma-1 receptor agonists is synergic with donepezil, but not with memantine, in a mouse model of amyloid-induced memory impairments. Behav. Brain Res. 2016, 296, 270–278. [Google Scholar] [CrossRef] [PubMed]
- du Sert, N.P.; Hurst, V.; Ahluwalia, A.; Wurbel, H. The arrive guidelines 2.0: Updated guidelines for reporting animal research. PLoS Biol. 2020, 18, e3000410. [Google Scholar]
- Fleming, K.K.; Bovaird, J.A.; Mosier, M.C.; Emerson, M.R.; LeVine, S.M.; Marquis, J.G. Statistical analysis of data from studies on experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2005, 170, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.; Amor, S. Publication guidelines for refereeing and reporting on animal use in experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2012, 242, 78–83. [Google Scholar] [CrossRef] [PubMed]
- el Behi, M.; Zéphir, H.; Lefranc, D.; Dutoit, V. Changes in self-reactive IgG antibody repertoire after treatment of experimental autoimmune encephalomyelitis with anti-allergic drugs. J. Neuroimmunol. 2007, 182, 80–88. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| mLM | hLM | ||||||
|---|---|---|---|---|---|---|---|
| t1/2 (h) | CLint (µL/min/pmol) | Compound Remaining (1 h, %) | t1/2 (h) | CLint (µL/min/pmol) | Compound Remaining (1 h, %) | ||
| 7i | 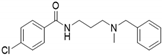 | 1.2 | <115 | 54 | 1.9 | <115 | 66 |
| 7i-deMe | 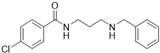 | 3.9 | <115 | 80 | 10.0 | <115 | 83 |
| Ki | Inhibition | |||
|---|---|---|---|---|
| S1R (nM) | S2R (nM) | Ratio S2R/S1R | SH-SY5Y (% at 100 µM) | |
| 7i [28] | 3.2 | 190 | 60 | 28 |
| 7i-deMe | 23 | >1000 | >500 | 77 |
| 7i | 7i-deMe | |
|---|---|---|
| Solubility (µM) | ||
| PBSpH7.4 | 185 | 187 |
| SGF | 193 | 197 |
| SIF | 188 | 196 |
| Permeability (×10−6 cm/s) | ||
| A/B pH6.5/7.4 | 29.1 | 31.1 |
| B/A pH6.5/7.4 | 66.1 | 51.5 |
| e-ratio | 2.3 | 1.7 |
| Plasma t½ (h) | 25.9 | 23 |
| PPB (%) | 94 | 81 |
| CYPs inhibition (%) | ||
| CYP1A | 2.1 | 9.4 |
| CYP2B6 | −4.3 | 15.9 |
| CYP2C8 | −9.2 | −7.2 |
| CYP2C9 | −7.2 | 5.9 |
| CYP2C19 | 36.5 | 18.4 |
| CYP2D6 | 95.1 | 91.6 |
| CYP3A | 36.9 | 3.2 |
| Drug transporters inhibition (%) | ||
| ABC family | ||
| P-gp | 10.6 | 1.0 |
| BCRP | 8.3 | −3.0 |
| MRP1 | 1.9 | −3.4 |
| MRP2 | −18.8 | −11.5 |
| MRP3 | −0.3 | −0.6 |
| SLC family | ||
| OATP1B1 | 12.0 | 8.6 |
| OATP1B3 | 23.1 | 19.1 |
| OAT1 | −9.6 | −6.2 |
| OAT3 | 34.3 | 18.7 |
| OCT1 | 41.2 | 64.6 |
| OCT2 | 49.4 | 85.8 |
| ABST | 5.3 | 5.0 |
| NTCP | −1.7 | 8.7 |
| P-gp substrate (×10−6 cm/s) | ||
| PBS (×10−6 cm/s) | ||
| A/B pH7.4/7.4 | 45.8 | |
| B/A pH7.4/7.4 | 11.7 | |
| e-ratio | 0.3 | |
| Verapamil (×10−6 cm/s) | ||
| A/B pH7.4/7.4 | 28.0 | |
| B/A pH7.4/7.4 | 16.9 | |
| e-ratio | 0.6 | |
| CYPs induction (%) | ||
| CYP1A2 | ||
| #1 (2) | 2.7 | |
| #2 (5) | 1.5 | |
| #3 (2) | 1.9 | |
| CYP2B6 | ||
| #1 (3) | 4.1 | |
| #2 (4) | 4.1 | |
| #3 (2) | 4.2 | |
| CYP3A4 | ||
| #1 (4) | 4.2 | |
| #2 (6) | 1.6 | |
| #3 (3) | 0.8 |
| 7i | |
|---|---|
| Cardiotoxicity | |
| hERG inhibition (mM) | |
| IC50 | 1 |
| Cytotoxicity | |
| HepG2 (% at 10 µM) | |
| Cell number | −3 |
| Intracell free calcium | 0 |
| Nuclear size | 9 |
| Membrane permeability | 0 |
| Mitochondrial membrane potential | 5 |
| TA1535 | TA1537 | TA98 | TA100 | TA102 | ||
|---|---|---|---|---|---|---|
| Dose (µg/plate) | −S9 | −S9 | −S9 | −S9 | −S9 | |
| Positive control | (a) | 85.9 | 8.4 | 23.1 | 13.5 | 6.0 |
| Vehicle control | 0 | - | - | - | - | - |
| 7i | 40 | 1.3 | 0.9 | 1.1 | 0.9 | 0.7 |
| +S9 | +S9 | +S9 | +S9 | +S9 | ||
| Positive control | (b) | 26.9 | 27.6 | 36.0 | 6.0 | 5.7 |
| Vehicle control | 0 | - | - | - | - | - |
| 7i | 40 | 0.9 | 1.1 | 1.1 | 0.9 | 1.0 |
| 3 h Short Treatment With 24 h Recovery Period | 27 h Continuous Treatment | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| −S9 | +S9 | −S9 | ||||||||
| µg/mL | RS (%) | MN/2 × 103 Cells | µg/mL | RS (%) | MN/2 × 103 Cells | µg/mL | RS (%) | MN/2 × 103 Cells | ||
| Positive control | Mytomice C | 0.5 | 62.8 | 153.0 | 0.2 | 67.9 | 62.0 | |||
| Griseofulvin | 5 | 70.5 | 63.0 | |||||||
| Cyclophosphamide | 5 | 88.3 | 35.5 | |||||||
| Vehicle control | 0 | - | 3.0 | 0 | - | 11.0 | 0 | - | 9.5 | |
| 7i | 275 | 80.0 | 8.0 | 275 | 103.7 | 9.0 | 275 | 70.1 | 6.0 | |
| 137.5 | 92.7 | 8.0 | 137.5 | 97.2 | 6.0 | 137.5 | 84.2 | 7.0 | ||
| 68.78 | 94.1 | 6.0 | 68.75 | 106.1 | 10.5 | 68.75 | 86.6 | 1.0 | ||
| i.v | p.o | |
|---|---|---|
| Plasma | ||
| tmax (min) | 0 | ≤30 |
| Cmax (ng/mL) * | 180 | NA |
| C30 (ng/mL) | 79.8 | 6.4 |
| AUC t∞/AUC ∞ | 0.9 | 6.1 |
| Vd (mL/kg) | 6.1 | 168.0 |
| CLT (mL/min) | 3.8 | 74.9 |
| t1/2 el (min) | 44.5 | 25.5 |
| MAT (min) | 0 | 16.5 |
| F (%) | 100 | 5 |
| Brain | ||
| tmax (min) | ≤30 | ≤30 |
| C30 (ng/mL) | 234.3 | 20.7 |
| AUC t∞/AUC ∞ | 5.9 | 7.6 |
| CLbrain (mL/min) | 1.8 | 12.8 |
| t1/2 el (min) | 96.4 | 78.9 |
| D (%) | 61.7 | NA |
| Brain/Plasma | 2.9 | 3.2 |
| QT Interval | |||||||
|---|---|---|---|---|---|---|---|
| Time after Injection | |||||||
| Dose (mg/kg) | Basal | 3 min | 6 min | 10 min | 20 min | 30 min | |
| Vehicle | 51 ± 2 | 46 ± 3 | 45 ± 4 | 56 ± 9 | 62 ± 7 | 63 ± 11 | |
| 7i | 0.5 | 49 ± 3 | 48 ± 4 | 46 ± 4 | 53 ± 3 | 59 ± 4 | 58 ± 3 |
| 1.0 | 48 ± 1 | 45 ± 2 | 46 ± 2 | >49 ± 1 | > | ||
| >5.0 | >46 ± 3 | >43 ± 1 | >42 ± 2 | >43 ± 2 | > | ||
| 10.0 | 46 ± 1 | 43 ± 1 | 45 ± 3 | 49 ± 2 | 52 ± 1 | 54 ± 1 | |
| Quinidine | 10.0 | 46 ± 2 | 50 ± 2 | 64 ± 6 | 65 ± 5 * | ||
| 100.0 | 47 ± 4 | 58 ± 3 | 84 ± 7 * | 76 ± 4 * | |||
| Product | Manufacturer | Reference Number |
|---|---|---|
| CD3 Antibody, anti-human APC-VIO® 770 | Miltenyi Biotec | 130-113-136 |
| CD4 Antibody, anti-human PerCP-Vio® 700 | Miltenyi Biotec | 130-113-228 |
| CD8 Antibody, anti-human VioGreen™ | Miltenyi Biotec | 130-110-684 |
| CD25 Antibody, anti-human, Vio® Bright B515 | Miltenyi Biotec | 130-115-536 |
| CD127 Antibody, anti-human, PE-Vio® 770 | Miltenyi Biotec | 130-113-415 130-113-412 |
| CD19 Antibody, anti-human, PE-Vio® 615 | Miltenyi Biotec | 130-114-522 |
| CD196 (CCR6) Antibody, anti-human, PE | Miltenyi Biotec | 130-120-458 |
| FoxP3 Antibody, anti-human, APC | Miltenyi Biotec | 130-125-580 |
| Viobility 405/452 Fixable Dye | Miltenyi Biotec | 130-110-205 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oxombre, B.; Madouri, F.; Journé, A.-S.; Ravez, S.; Woitrain, E.; Odou, P.; Duhal, N.; Ninni, S.; Montaigne, D.; Delhem, N.; et al. Safe and Efficient Sigma1 Ligand: A Potential Drug Candidate for Multiple Sclerosis. Int. J. Mol. Sci. 2022, 23, 11893. https://doi.org/10.3390/ijms231911893
Oxombre B, Madouri F, Journé A-S, Ravez S, Woitrain E, Odou P, Duhal N, Ninni S, Montaigne D, Delhem N, et al. Safe and Efficient Sigma1 Ligand: A Potential Drug Candidate for Multiple Sclerosis. International Journal of Molecular Sciences. 2022; 23(19):11893. https://doi.org/10.3390/ijms231911893
Chicago/Turabian StyleOxombre, Bénédicte, Fahima Madouri, Anne-Sophie Journé, Séverine Ravez, Eloise Woitrain, Pascal Odou, Nathalie Duhal, Sandro Ninni, David Montaigne, Nadira Delhem, and et al. 2022. "Safe and Efficient Sigma1 Ligand: A Potential Drug Candidate for Multiple Sclerosis" International Journal of Molecular Sciences 23, no. 19: 11893. https://doi.org/10.3390/ijms231911893
APA StyleOxombre, B., Madouri, F., Journé, A. -S., Ravez, S., Woitrain, E., Odou, P., Duhal, N., Ninni, S., Montaigne, D., Delhem, N., Vermersch, P., & Melnyk, P. (2022). Safe and Efficient Sigma1 Ligand: A Potential Drug Candidate for Multiple Sclerosis. International Journal of Molecular Sciences, 23(19), 11893. https://doi.org/10.3390/ijms231911893