1. Introduction
Testosterone is the main sex hormone produced by the mature testis. It is essential for establishing biological maleness in typical XY males: development of the male urogenital tract during fetal development, acquisition of male secondary sex characteristics at puberty, and fertility in adults. Testosterone is synthesized and secreted by Leydig cells present in the testicular interstitium through the process of steroidogenesis. Steroidogenesis is the multistep conversion of cholesterol into steroid hormones via the sequential action of multiple proteins/enzymes. Steroidogenesis needs to be tightly regulated as too little or too much steroid hormone production can lead to incidences of differences of sex development (DSD) or pathologies such as congenital adrenal hyperplasia (CAH), osteopenia, and hormone-related cancers (reviewed in [
1]). The regulation of testicular steroidogenesis is complex and involves many regulatory molecules and mechanisms such as luteinizing hormone (LH), its signaling pathways, and the factors that interpret these signals [
1]. The latter include most notably transcription factors acting on the expression of genes that encode steroidogenic enzymes and other proteins involved in the biosynthesis of testosterone (reviewed in [
2,
3]).
Members of the GATA family of transcription factors are emerging as important regulators of steroidogenesis (reviewed in [
4,
5,
6]). The GATA family is comprised of six factors (GATA1 to 6) that recognize and bind to the DNA motif (A/T)GATA(A/G) through their two conserved zinc finger domains. GATA factors are found in a broad array of tissues where they participate in cell differentiation, organogenesis, and the control of tissue-specific gene expression (reviewed in [
4,
7]). In both male and female gonads, GATA factors, especially GATA4 and/or GATA6, are crucial for the formation of the urogenital ridge, sex determination (gonad differentiation), fertility, and most likely steroidogenesis (reviewed in [
4,
7,
8]). Insights into the GATA-dependence of steroidogenesis have come mainly from studies done in whole testis or immortalized Leydig cell lines. For example, GATA4 knockdown in either MA-10 or MLTC-1 Leydig cells represses the steroidogenic gene expression program and ultimately the production of sex steroid precursors [
9,
10]. In mice, loss of GATA4 and/or GATA6 function in the testis appears to block steroidogenic cell development and concomitant undermasculization of male embryos [
11]. One of the testicular genes shown to be prominently affected by a modulation of GATA4/6 gene expression or activity is the gene encoding the steroidogenic acute regulatory protein (STAR).
Steroidogenic acute regulatory protein (STAR or STARD1) is a member of the larger START domain family, a group of transport proteins that share a common functional domain, the STAR-related lipid-transfer domain. In humans, 15 members (STARTD1-15) of the START domain family are known to transport cholesterol, ceramide, phosphatidylcholine, phosphatidylethanolamine (PE), and bile acids (reviewed in [
12]). The first STARD protein discovered was the STAR or STARD1 protein which transports cholesterol from the outer to the inner mitochondrial membrane in steroidogenic cells of both the adrenals and gonads (reviewed in [
13,
14]). Cholesterol transport into the mitochondria is the rate-limiting step of steroidogenesis. STAR function in steroidogenesis was confirmed by both human disease (lipoid congenital adrenal hyperplasia/lipoid CAH) where the
STAR gene is mutated [
14,
15], and in
Star null mice which have a similar steroidogenic defect as seen in human lipoid CAH [
16]. Being the rate-limiting step in steroidogenesis, STAR expression and activity is acutely regulated at both the transcriptional and post-transcriptional levels (reviewed in [
13]).
The transcriptional control of the
STAR gene has been intensely studied (reviewed in [
13]). The proximal
STAR promoter contains many tightly clustered regulatory motifs for the binding of different transcription factors that have been shown to modulate
STAR promoter activity in vitro. These include motifs for the binding of NR5A factors (SF1/NR5A1 and LRH1/NR5A2), NR4A factors (NUR77/NGFI-B/NR4A1, NURR1/NR4A2, and NOR1/NR4A3), CCAAT/enhancer binding protein β (C/EBP β), CREB family factors (cAMP response element (CRE)-binding protein (CREB), CRE modulator (CREM), and sterol element binding protein (SREBP)), activator protein 1 (AP1), dosage-sensitive sex reversal, adrenal hypoplasia critical region, on chromosome X, gene 1 (DAX1), and Ying Yang 1 (YY1) (reviewed in [
13]). As previously mentioned, GATA factors, mostly notably GATA4, are known to bind to the proximal
Star promoter and enhance its transcription in vitro both basally and in response to acute hormonal stimulation [
17,
18]. The endogenous
Star gene, however, has not yet been validated as a direct target for GATA binding or other transcription factors proposed to modulate its expression.
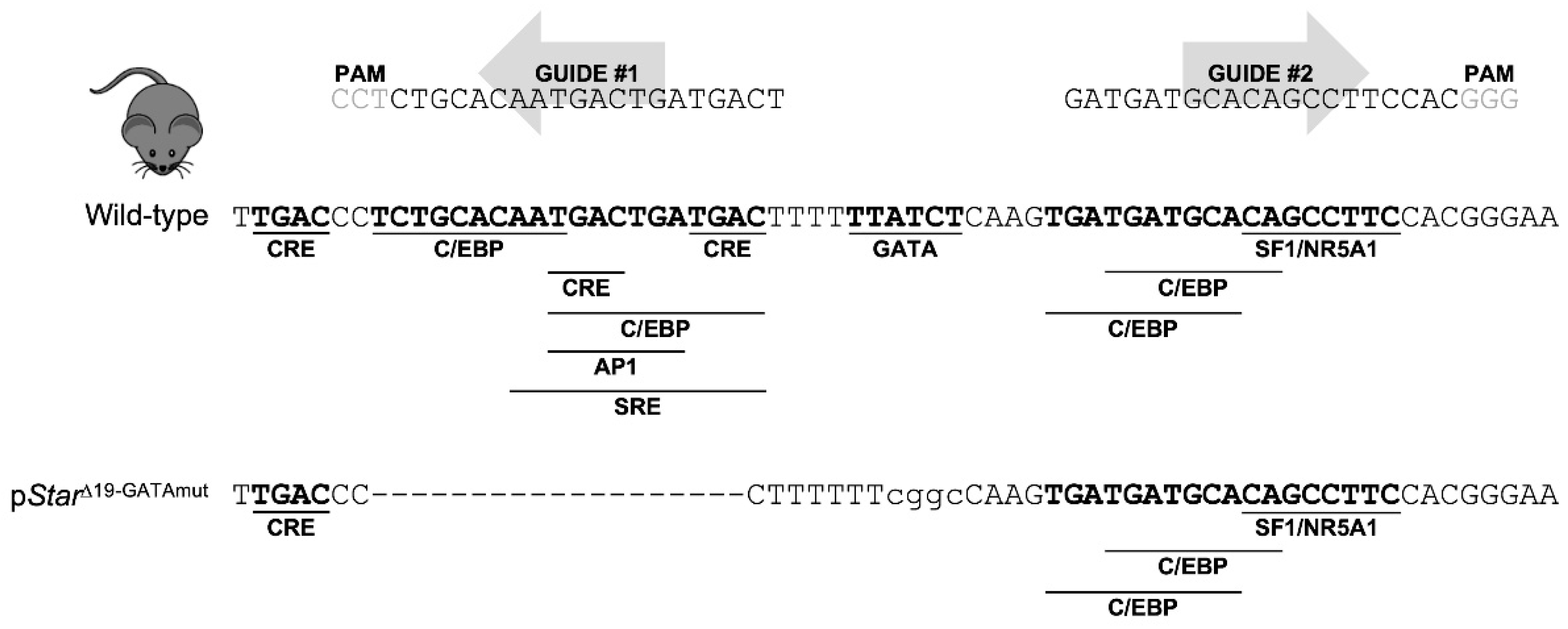
In this study, we report a novel mouse model created by CRISPR/Cas9 genome editing that modifies the mouse proximal Star promoter region to inactivate the critical GATA-binding motif as well as an adjacent 19-bp deletion that removes additional CRE, C/EBP, and AP1 binding motifs. Our results show that this short promoter region containing the GATA motif is required for endogenous Star expression in both fetal and postnatal mouse testes.
3. Discussion
Steroidogenic acute regulatory protein (STAR), identified nearly three decades ago, is an essential cholesterol transporter in all steroidogenic tissues [
21]. The delivery of cholesterol from the outer to the inner mitochondrial membrane mediated by STAR is the rate-limiting step in steroidogenesis [
13]. As such, many studies have been devoted to understanding how
STAR gene expression and protein activity are regulated in steroid producing tissues such as the adrenals and gonads. At the transcriptional level, the regulation of the
STAR gene is complex, involving the interaction of numerous transcription factors to species-conserved binding motifs that are tightly clustered, and sometimes overlapping, located within the first few hundred base pairs upstream of the transcription initiation site (reviewed in [
13]). Although many transcription factors have been shown to participate in
STAR transcription across many species, to our knowledge, this evidence has been essentially limited to in vitro studies performed in cell lines or isolated steroidogenic tissues. No studies have yet probed the importance of these transcription factor binding sites for
Star transcription in an in vivo whole animal context. In the present study, we have generated the first such mouse model (p
StarΔ19-GATAmut), using CRISPR/Cas9 genome editing, to inactivate a short region of the mouse
Star promoter containing a subset of these transcription factor binding sites. Analysis of the mutant mice confirmed that they are indeed essential for maximal expression of the endogenous
Star gene but not basal or hormone-activated testosterone production.
The initial goal of our genome editing effort was to inactivate the lone GATA regulatory motif in the mouse
Star promoter that has been long proposed to be critical for
Star transcription [
18]. However, even after screening more than 100 founder mice, we were unable to exclusively alter the
Star GATA-binding motif. This was not totally unexpected given the low rate of homology directed repair (HDR) [
22]. Despite this, we were successful in creating a mouse (p
StarΔ19-GATAmut) with a short 19-bp deletion in tandem with a mutated GATA motif (see
Figure 1). The deleted region contains six additional regulatory motifs—many of them overlapping—for various transcription factors, including members of the C/EBP and CREB families. The sequence covered by the Δ19-GATA mutation spans one of two critical regions that largely govern the positive modulation of
STAR expression as documented through in vitro studies (reviewed in [
20,
23]). The analysis of our p
StarΔ19-GATAmut mice confirmed that this region is indeed important for maximal
Star expression in fetal, peripubertal, and adult testes. However, we cannot at present formally attribute the decrease in
Star expression to any one of these specific motifs since they were simultaneously disrupted in p
StarΔ19-GATAmut mice. Contrasting our in vivo findings with existing in vitro data do suggest that the CRE motifs are likely important—MA-10 Leydig cells transfected with CREB increases
Star mRNA levels while mutation of at least the middle CRE motif (CRE2 in ref. [
24]) reduces
Star promoter activity by approximately 50% [
24], a decrease comparable to what we observed for
Star mRNA in p
StarΔ19-GATAmut testes. The proximal
Star promoter also contains two C/EBP binding sites; one of these sites (site C2 identified in ref. [
25]) was deleted in our p
StarΔ19-GATAmut mice. Mutation of this C/EBP motif significantly reduces
Star promoter activity in MA-10 Leydig cells [
25]. The same can be said for the GATA motif—the
Star promoter is potently activated by GATA factors in cells, an effect that is completely lost when motif is mutated [
18]. Considering the short length of the Δ19-GATAmut region as well as the very close proximity of the regulatory motifs present therein, it is reasonable to speculate that these motifs are not simultaneously bound by different transcription factors but rather are occupied by dynamic binding. Moreover, the proximity of the regulatory motifs further implies a physical closeness of the various DNA binding proteins that would permit the proteins to directly interact. Indeed, the existence of regulatory complexes among the different transcription factors has been well-documented. For example, C/EBP and CREB proteins interact with GATA4 to increase
Star promoter activity, at least in vitro [
17,
26]. Therefore, we can now conclude that the integrity of this short region is critical not only for in vitro
Star promoter activity but also for
Star transcription in both fetal and postnatal testis in the mouse.
Although STAR protein is plentiful in Leydig cells, it was technically difficult for us to quantify it in early fetal testes. However, in adult testes, STAR was clearly less abundant in p
StarΔ19-GATAmut than in WT mice. Immunohistochemistry also showed that this was not accompanied by a change in cellular localization, suggesting that although less abundant, it should remain functional. Based on these observations, we expected that this would translate into reduced testosterone production either in plasma or locally within the testis itself. However, for both intratesticular and circulating (plasma) testosterone, levels in p
StarΔ19-GATAmut adult mice were not significantly different from WT. This was somewhat surprising knowing that genetic male
Star null fetal mice have feminized external genitalia which is suggestive of a deficit in the production of androgen precursors from fetal Leydig cells [
16]. Moreover, testosterone levels are 10 times lower at 8 weeks in serum from glucocorticoid-rescued
Star null males when compared to age-matched controls [
27]. This indicates that despite a 50–75% decrease in
Star mRNA or STAR protein observed in p
StarΔ19-GATAmut testes, the amount of STAR remaining must be sufficient to allow for normal steroidogenesis. In steroidogenic tissues,
STAR transcription is rapidly induced by the gonadotropin LH acting via a cAMP signaling pathway [
28]. The short promoter region targeted in our p
StarΔ19-GATAmut mice also harbors regulatory elements known to confer LH responsiveness, including the GATA-binding motif (reviewed in [
20]). Based on these facts, we surmised that a steroidogenic deficit might be more easily captured under conditions of hormonal stimulation. Yet again, we observed that ex vivo testis cultures from adult p
StarΔ19-GATAmut mice still responded to hCG stimulation when compared to WT controls, therefore reinforcing the notion that STAR protein, while diminished significantly in amount, was still adequate to elicit an induction of steroidogenesis when stimulated.
Taken together, our results provide new insights into the transcriptional regulation of the Star gene and highlight the power of CRISPR/Cas9 genome editing for validating the importance of proposed promoter regulatory sequences for both basal and acute hormone-stimulated gene regulation.
4. Materials and Methods
4.1. Animals
All mouse experiments were carried out in accordance with the Canadian Council of Animal Care guidelines for the care and manipulation of animals used in research. Protocols were approved by the Comité de Protection des Animaux de l’Université Laval, Québec, QC, Canada (protocol nos. 2019-149 and CHU-19-046).
4.2. CRISPR/Cas9 Generation of Mouse Star Promoter Mutants
A 100-bp genomic region of the proximal murine
Star promoter spanning a single conserved GATA binding motif was searched using the CRISPOR Web tool (
www.crispor.tefor.net, accessed on 15 June 2019) for potential single-guide RNA (sgRNA) sequences [
29]. Guides were selected based on their low predicted off-target potential. A pX330-U6-Chimeric_BBCBh-hSpCas9 plasmid (no. 42230) purchased from Addgene (Cambridge, MA, USA) was used to generate SpCas9/chimeric sgRNA expression plasmids [
30], as previously described [
19]. A single-strand oligonucleotide (ssODN) was synthesized as a template for HDR of the double-strand breaks created by the sgRNAs. The ssODN contains a mutated GATA motif of the murine
Star promoter flanked on each side by ~90-nucleotide-long homology arms. The oligonucleotides used as primers for creating the sgRNAs as well as the ssODN used as a donor for HDR are shown in
Table 1 (the GATA motif is underlined and the mutated nucleotides are in lowercase). The SpCas9/chimeric sgRNA constructs were first validated in vitro and then microinjected along with the ssODN into fertilized C57BL/6J mouse eggs using the microinjection and transgenesis platform of the Institut de Recherches Cliniques de Montréal. A total of 105 founder mice were born and analyzed. Genomic DNA was isolated from tail tips collected from the founder mice using the HotSHOT method [
31]. DNA screened for genomic rearrangements using genotyping primers (listed in
Table 1) and Taq FroggaMix 2X master mix (FroggaBio, Concord, ON, Canada). PCR conditions were: initial denaturation for 3 min at 95 °C followed by 35 cycles of denaturation (30 s at 95 °C), annealing (30 s at 68 °C), and extension (30 s at 72 °C), and a final extension for 3 min at 72 °C. Amplicons were sequenced and analyzed using the Web tool TIDE (for tracking indels by decomposition) to evaluate the extent of Cas9-mediated rearrangements that occurred [
32] and to identify the desired mutated or deleted alleles. Founder mice that targeted the GATA motif were crossed with C57BL/6J mice (stock no. 000664; The Jackson Laboratory, Bar Harbor, ME) to assess the transmission of the modified alleles. One founder mouse that possessed a combined GATA mutation and 19-bp deletion (named p
StarΔ19-GATAmut) successfully transferred the modified
Star promoter sequence to its offspring. Mice were backcrossed for a minimum of 5 generations with C57BL/6J mice to eliminate potential off-target effects. Heterozygous descendants were crossed to generate homozygous as well as wild-type (WT) control mice for experimentation as well as additional heterozygous mice that were used for colony maintenance.
4.3. Quantitative Real-Time RT-PCR
Testes were dissected from male mice at various developmental ages: embryonic day 15.5 (E15.5), E18.5, postnatal day 35 (P35, juvenile adult), and P90 (adult). Testes from E15.5, E18.5, and P35 testes were processed for total RNA extraction using TRI Reagent solution (Sigma-Aldrich Canada, Oakville, ON, Canada) in accordance with the manufacturer’s instructions. Testes from adult male mice were halved while frozen. One half was used for protein extraction (described below) and the other half was used for RNA extraction and intratesticular testosterone quantification (described below under
Hormone assay). First-strand cDNA was synthesized from total RNA isolated from tissues using the iScript Advanced cDNA synthesis kit for quantitative real-time RT-PCR (qPCR; Bio-Rad Laboratories, Mississauga, ON, Canada). Assessment of gene expression by qPCR was done using a CFX96 plate thermal cycler and SsoAdvanced Universal SYBR Green Supermix from Bio-Rad Laboratories using their standard protocol. A panel of reference genes known for their stability in the mouse gonad [
33], as well as typically used reference genes, were used for normalization. Primers for qPCR are listed in
Table 2. Primer pairs were optimized beforehand for specificity and efficiency using a temperature gradient to identify the best annealing temperature and by performing a standard curve using a serial dilution of a pool of samples. PCR amplifications were run in duplicate under the following conditions: initial denaturation for 3 min at 95 °C followed by 40 cycles of denaturation (10 s at 95 °C), annealing (20 s at 62.6 °C), and extension (20 s at 72 °C) with a single acquisition of fluorescence level at the end of each extension step. Differences in mRNA levels between the mouse genotypes was determined using the ΔΔCq method [
34].
4.4. Immunohistochemistry
Whole male embryos were collected at E13.5. For E18.5 fetuses as well as juvenile and adult mice, gonads were harvested and prepared for histological analysis. Immunohistochemical (IHC) staining was performed using the Rabbit Specific HRP/AEC IHC Detection Kit-Micro-polymer (ab236468; Abcam, Toronto, ON, Canada) following the manufacturer’s protocol. Tissue sections (4 µM) were deparaffinized and rehydrated in graded ethanols. Tissues were processed for antigen retrieval by treating them with citrate buffer (10 mM Sodium citrate, 0.05% Tween 20, pH 6.0) in a Decloaking Chamber™ NxGen (Biocare Medical, Pacheco, CA, USA) for 10 min at 110 °C. Sections were incubated overnight at 4 °C with primary antibodies for either a rabbit polyclonal anti-STAR IgG (Proteintech Cat# 12225-1-AP, RRID:AB_2115832) diluted 1:200 (1.7 μg/mL) in phosphate buffered saline (PBS) containing 1% bovine serum albumin (BSA) or a rabbit polyclonal anti-GATA4 IgG (Abcam Cat# ab84593, RRID:AB_10670538) diluted 1:500 (1.8 μg/mL) in PBS containing 1% BSA. Sections incubated with rabbit IgG isotype control (Invitrogen Cat # 02-6102, RRID:AB_2532938) diluted 1:2500 (2 μg/mL) in PBS containing 1% BSA were used as negative controls. All sections were counterstained with Harris Modified hematoxylin solution (Thermo Fisher Scientific, Nepean, ON, Canada) and mounted in MOWIOL (EMD Millipore, Gibbstown, NJ, USA). Slides were visualized with a Zeiss Axioscop II microscope (Carl Zeiss Canada, Toronto, ON, Canada) connected to a Spot RT Slider digital camera (Diagnostic Instruments, Sterling Heights, MI, USA). At least three animals per genotype were assessed.
4.5. Protein Extraction and Western Blot Analysis
For western blot analysis, half of a testis was homogenized while still frozen in ice-cold extraction buffer (50 mM Tris pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.5% Igepal, 1 mM DTT, 0.5 mM phenylmethylsulfonyl fluoride (Sigma-Aldrich), and proteinase inhibitors (Sigma-Aldrich): 5 μg/mL aprotinin, 5 μg/mL leupeptin, and 5 μg/mL pepstatin. Homogenates were incubated on ice for 15 min after which the samples were briefly sonicated to break genomic DNA. Protein concentration was evaluated by Bradford assay [
35], using Bio-Rad protein assay dye reagent concentrate (Bio-Rad Laboratories, Mississauga, ON, Canada) and BSA as protein standard. Aliquots (40 μg) of testicular homogenates were separated by SDS-PAGE and then electrotransferred to nitrocellulose membrane (Bio-Rad Laboratories, Mississauga, ON, Canada). Non-specific antibody binding was prevented by blocking for 1 h at RT using 5% non-fat dry milk in Tris-buffered saline (TBS: 20 mM Tris, 150 mM NaCl, pH 7.6) with 0.1% Tween 20 (TBS-T). Proteins were detected using commercially available primary antibodies for either a rabbit monoclonal anti-STAR IgG (Cell Signaling Technology Cat# 8449, RRID:AB_10889737) at a dilution of 1:5000 in 5% non-fat dry milk in TBS-T, a mouse monoclonal anti-α-tubulin IgG (used as a loading control, Sigma-Aldrich Cat# T5168, RRID:AB_477579) at a dilution of 1:10,000 in 5% non-fat dry milk in TBS-T. After washing in TBS-T, membranes were incubated with horseradish peroxidase-labeled secondary antibodies: goat anti-rabbit IgG (Vector Laboratories Cat# PI-1000, RRID:AB_2336198) diluted 1:5000 5% non-fat dry milk in TBS-T for STAR detection or horse anti-mouse IgG (Vector Laboratories Cat# PI-2000, RRID:AB_2336177) diluted 1:5000 in 5% non-fat dry milk in TBS-T for α-tubulin. After washing in TBS-T, membranes were finally incubated with Clarity Western ECL Substrate (Bio-Rad Laboratories) for 5 min. The chemiluminescent signal was detected on a ChemiDoc Imaging System (Bio-Rad Laboratories).
4.6. Hormone Assay
At the time of sacrifice, blood was drawn from adult mice by cardiac puncture. Blood was collected in EDTA-coated microtubes to prevent clotting, and plasma was isolated by centrifugation for 3 min at 2400 g and stored at −80 °C until further needed. For intratesticular testosterone quantification and RNA extraction, half a testis from each animal was homogenized while still frozen in cold PBS on ice and then centrifuged at 3000 rpm for 5 min at 4°C. Supernatant was collected and assayed for testosterone. The pellet was resuspended in TRI reagent to isolate total RNA for qPCR analysis (described above). For ex vivo stimulation of testosterone, testes were harvested and placed in 500 μL of DMEM containing 100 IU/mL penicillin and 100 μg/mL streptomycin. Testes were detunicated and treated with either 1 IU/mL of human chorionic gonadotropin (hCG, Sigma-Aldrich) or vehicle (H2O) and incubated for 4 h at 32 °C. After incubation, tissues were centrifuged for 5 min at 2400 g and the culture medium was collected. Testosterone quantification in plasma, testicular homogenates or culture medium was performed using an ELISA kit purchased from Cayman Chemical (Cayman Chemical, Ann Arbor, MI, USA, Cat# 582701, RRID:AB_2895148) following the instructions recommended by the manufacturer. To obtain readings within the range of the standard curve, samples were diluted in EIA buffer (provided by the kit manufacturer) as follows: plasma (1:20 and 1:50), testicular homogenates (1:100 and 1:200) and culture medium (1:200 and 1:500). Microplates were read using a Tecan Spark® 10M multimode plate reader (Tecan, Morrisville, NC, USA). The assay has a cross reactivity of 100% for testosterone. Cross-reactivity is negligible for other sex steroids with the exception of 5α-dihydrotestosterone (DHT) at 27.4% (Cayman Chemical). However, since mouse intratesticular and plasma DHT levels are very low, this would not impact the testosterone measurements.
4.7. Statistical Analysis
Statistical analyses were done using JASP version 0.16.3 (
https://jasp-stats.org, accessed on 10 October 2021; JASP Team (2022), Amsterdam, The Netherlands). Quantitative comparisons between wild-type and mutant mice (
Star mRNA, plasma and intratesticular testosterone) were analyzed using a parametric Student’s
t-test. Measurement of ex vivo testosterone production was analyzed by one-way ANOVA followed by Tukey multiple comparisons tests. For all statistical analyses,
p < 0.05 was considered significant.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}