
Ectopic Expression of FVIII in HPCs and MSCs Derived from hiPSCs with Site-Specific Integration of ITGA2B Promoter-Driven BDDF8 Gene in Hemophilia A



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
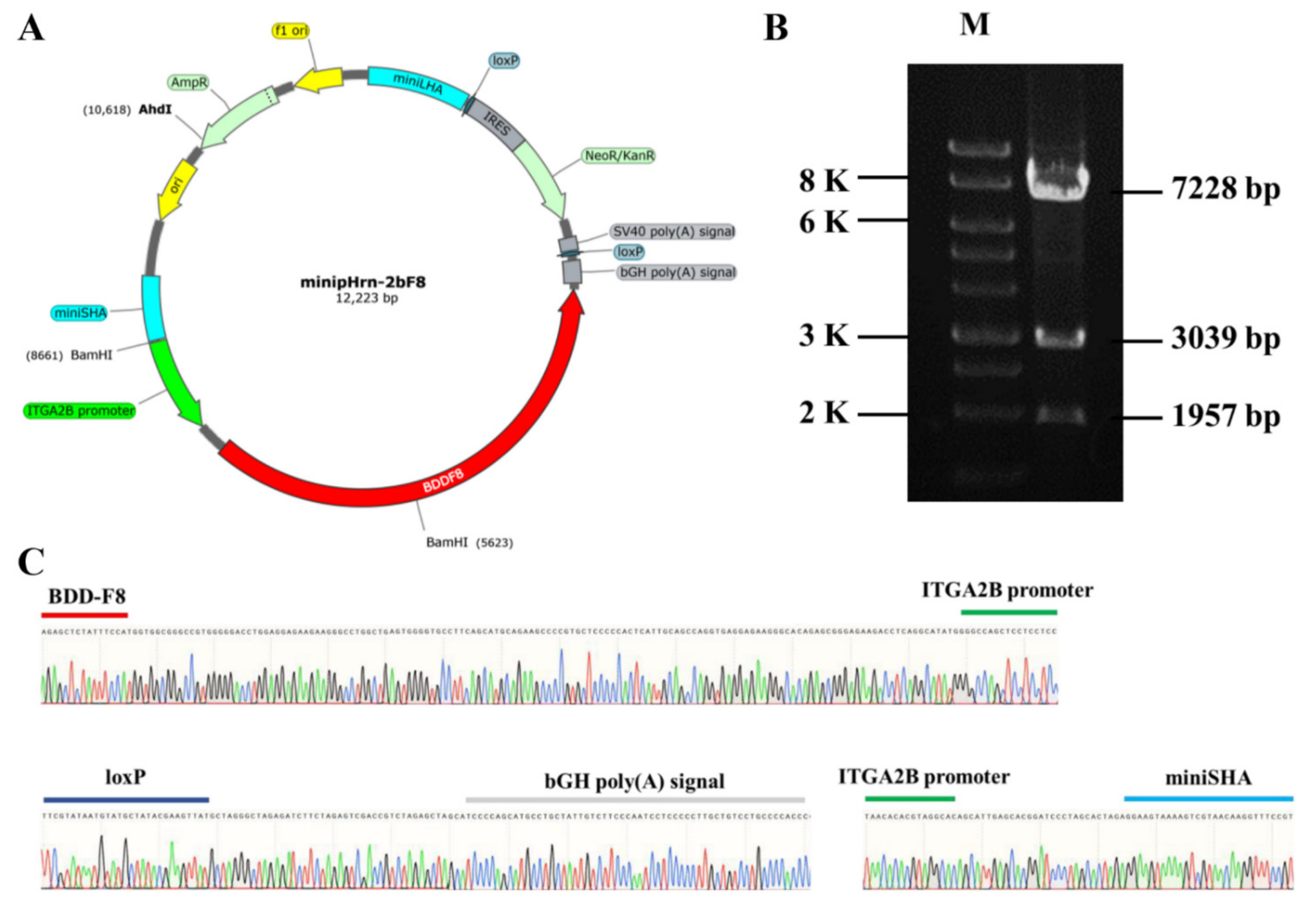
2.1. Construction of Nonviral Targeting Vector minipHrn-2bF8
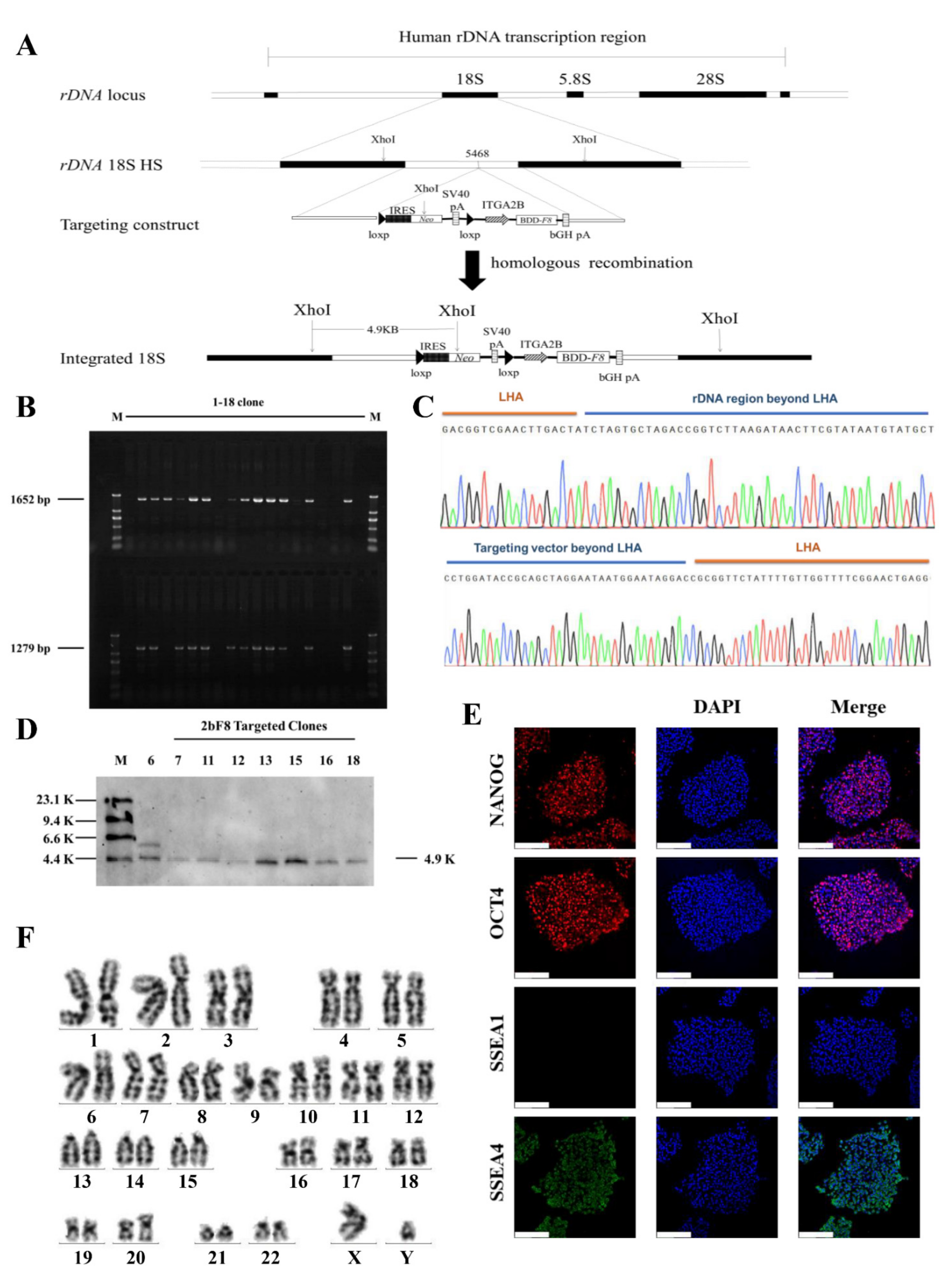
2.2. Gene Targeting of F8 into the rDNA Locus of HA-iPSCs Using TALENickases
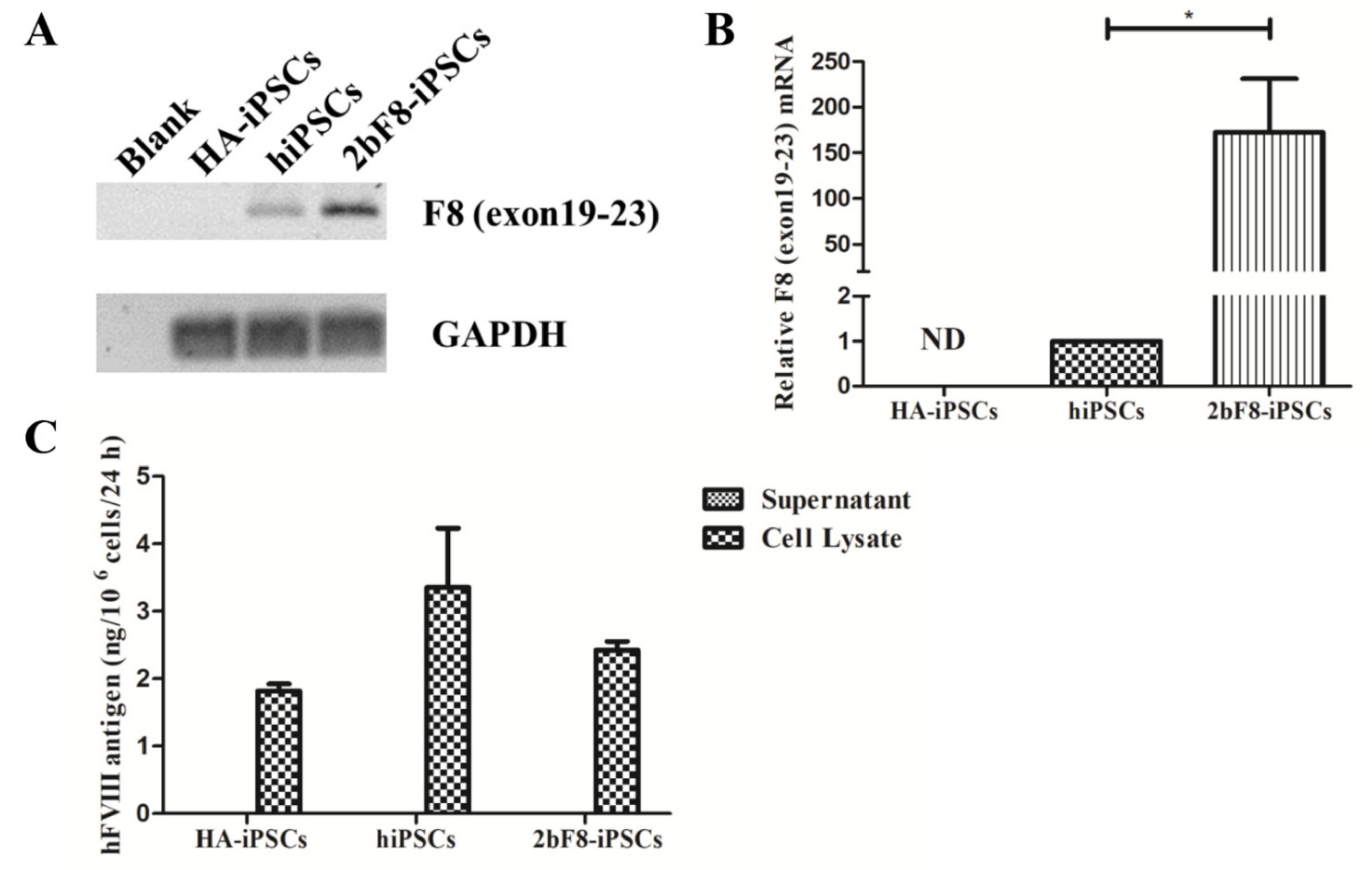
2.3. Verification of F8 Expression in F8-Modified hiPSCs
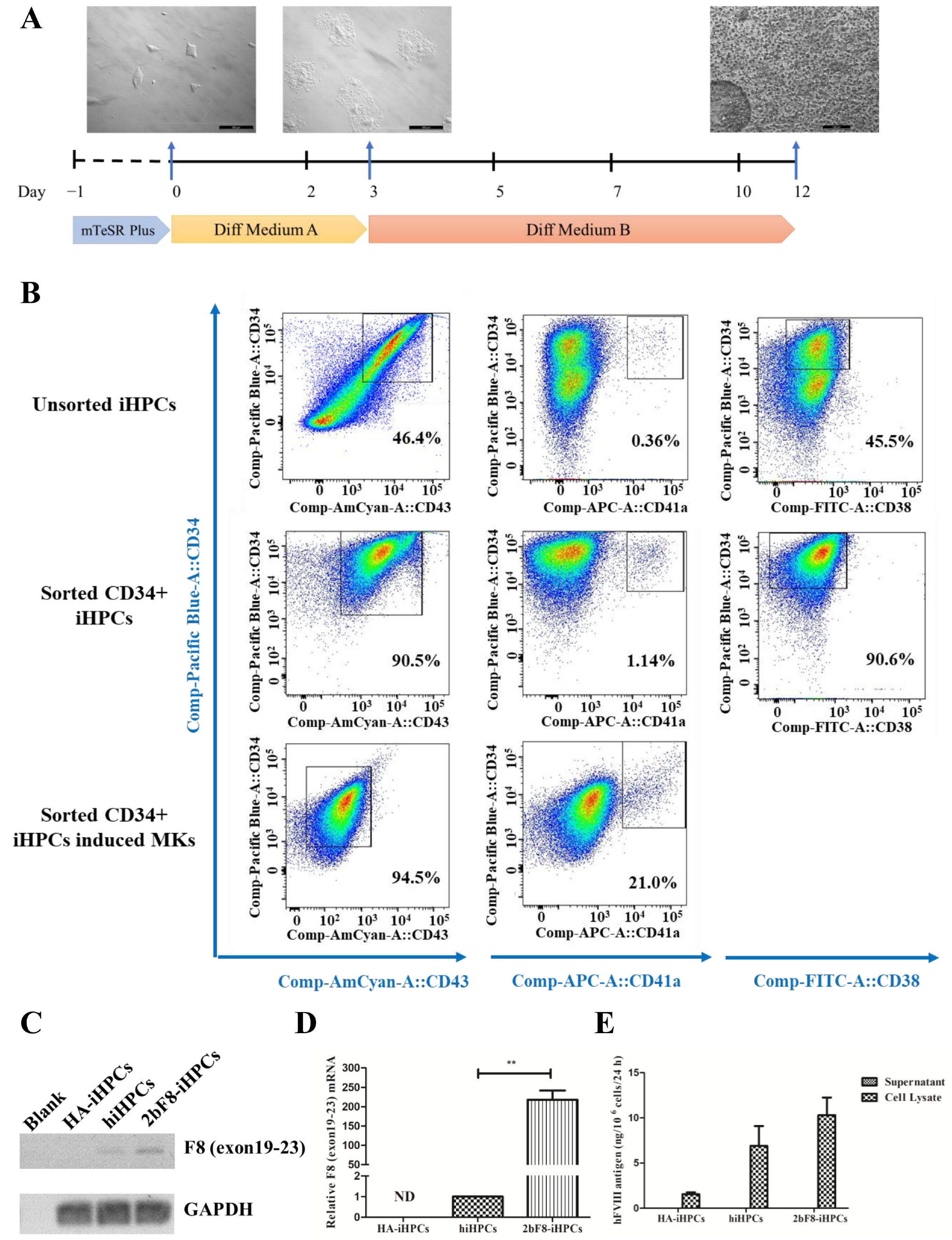
2.4. Generation and Characterization of Induced HPCs (iHPCs) and Induced MKs (iMKs)
2.5. Verification of F8 Expression in F8-Modified iHPCs
2.6. Generation and Characterization of Induced MSCs (iMSCs)
2.7. Verification of FVIII Expression in F8-Modified iMSCs
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Plasmids and Gene Targeting
4.3. RT-PCR and qRT-PCR
4.4. Southern Blotting
4.5. Karyotyping
4.6. Immunofluorescence Staining
4.7. Differentiation of hiPSCs into iHPCs and iMKs
4.8. Characterization of iHPCs and iMKs
4.9. Differentiation of hiPSCs into iMSCs
4.10. Characterization and Identification of Differentiation Potential of iMSCs
4.11. FVIII Assay
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fomin, M.E.; Togarrati, P.P.; Muench, M.O. Progress and challenges in the development of a cell-based therapy for hemophilia A. J. Thromb. Haemost. 2014, 12, 1954–1965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gouw, S.C.; van der Bom, J.G.; Ljung, R.; Escuriola, C.; Cid, A.R.; Claeyssens-Donadel, S.; van Geet, C.; Kenet, G.; Makipernaa, A.; Molinari, A.C.; et al. Factor VIII products and inhibitor development in severe hemophilia A. N. Engl. J. Med. 2013, 368, 231–239. [Google Scholar] [CrossRef]
- Peyvandi, F.; Kenet, G.; Pekrul, I.; Pruthi, R.K.; Ramge, P.; Spannagl, M. Laboratory testing in hemophilia: Impact of factor and non-factor replacement therapy on coagulation assays. J. Thromb. Haemost. 2020, 18, 1242–1255. [Google Scholar] [CrossRef] [PubMed]
- White, G.C., 2nd; Kempton, C.L.; Grimsley, A.; Nielsen, B.; Roberts, H.R. Cellular immune responses in hemophilia: Why do inhibitors develop in some, but not all hemophiliacs? J. Thromb. Haemost. 2005, 3, 1676–1681. [Google Scholar] [CrossRef]
- Perrin, G.Q.; Herzog, R.W.; Markusic, D.M. Update on clinical gene therapy for hemophilia. Blood 2019, 133, 407–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croteau, S.E.; Wang, M.; Wheeler, A.P. 2021 clinical trials update: Innovations in hemophilia therapy. Am. J. Hematol. 2021, 96, 128–144. [Google Scholar] [CrossRef] [PubMed]
- Gan, S.U.; Calne, R.Y. Gene therapy for hemophilia A. Discov. Med. 2006, 6, 198–202. [Google Scholar]
- Chuah, M.K.; Evens, H.; VandenDriessche, T. Gene therapy for hemophilia. J. Thromb. Haemost. 2013, 11, 99–110. [Google Scholar] [CrossRef] [Green Version]
- Leebeek, F.W.G.; Miesbach, W. Gene therapy for hemophilia: A review on clinical benefit, limitations, and remaining issues. Blood 2021, 138, 923–931. [Google Scholar] [CrossRef]
- Verdera, H.C.; Kuranda, K.; Mingozzi, F. AAV Vector Immunogenicity in Humans: A Long Journey to Successful Gene Transfer. Mol. Ther. 2020, 28, 723–746. [Google Scholar] [CrossRef]
- Nguyen, G.N.; Everett, J.K.; Kafle, S.; Roche, A.M.; Raymond, H.E.; Leiby, J.; Wood, C.; Assenmacher, C.A.; Merricks, E.P.; Long, C.T.; et al. A long-term study of AAV gene therapy in dogs with hemophilia A identifies clonal expansions of transduced liver cells. Nat. Biotechnol. 2021, 39, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Cicalese, M.P.; Ferrua, F.; Castagnaro, L.; Pajno, R.; Barzaghi, F.; Giannelli, S.; Dionisio, F.; Brigida, I.; Bonopane, M.; Casiraghi, M.; et al. Update on the safety and efficacy of retroviral gene therapy for immunodeficiency due to adenosine deaminase deficiency. Blood 2016, 128, 45–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, K.; Kumar, P.; Cortez-Toledo, E.; Hao, D.; Reynaga, L.; Rose, M.; Wang, C.; Farmer, D.; Nolta, J.; Zhou, J.; et al. Potential long-term treatment of hemophilia A by neonatal co-transplantation of cord blood-derived endothelial colony-forming cells and placental mesenchymal stromal cells. Stem. Cell Res. Ther. 2019, 10, 34. [Google Scholar] [CrossRef] [Green Version]
- Wilcox, D.A.; Shi, Q.; Nurden, P.; Haberichter, S.L.; Rosenberg, J.B.; Johnson, B.D.; Nurden, A.T.; White, G.C., 2nd; Montgomery, R.R. Induction of megakaryocytes to synthesize and store a releasable pool of human factor VIII. J. Thromb. Haemost. 2003, 1, 2477–2489. [Google Scholar] [CrossRef] [PubMed]
- Yarovoi, H.V.; Kufrin, D.; Eslin, D.E.; Thornton, M.A.; Haberichter, S.L.; Shi, Q.; Zhu, H.; Camire, R.; Fakharzadeh, S.S.; Kowalska, M.A.; et al. Factor VIII ectopically expressed in platelets: Efficacy in hemophilia A treatment. Blood 2003, 102, 4006–4013. [Google Scholar] [CrossRef]
- Shi, Q.; Wilcox, D.A.; Fahs, S.A.; Fang, J.; Johnson, B.D.; Du, L.M.; Desai, D.; Montgomery, R.R. Lentivirus-mediated platelet-derived factor VIII gene therapy in murine haemophilia A. J. Thromb. Haemost. 2007, 5, 352–361. [Google Scholar] [CrossRef]
- Takayama, N.; Nishikii, H.; Usui, J.; Tsukui, H.; Sawaguchi, A.; Hiroyama, T.; Eto, K.; Nakauchi, H. Generation of functional platelets from human embryonic stem cells in vitro via ES-sacs, VEGF-promoted structures that concentrate hematopoietic progenitors. Blood 2008, 111, 5298–5306. [Google Scholar] [CrossRef] [Green Version]
- Takayama, N.; Eto, K. In vitro generation of megakaryocytes and platelets from human embryonic stem cells and induced pluripotent stem cells. Methods Mol. Biol. 2012, 788, 205–217. [Google Scholar] [CrossRef]
- Ono-Uruga, Y.; Tozawa, K.; Horiuchi, T.; Murata, M.; Okamoto, S.; Ikeda, Y.; Suda, T.; Matsubara, Y. Human adipose tissue-derived stromal cells can differentiate into megakaryocytes and platelets by secreting endogenous thrombopoietin. J. Thromb. Haemost. 2016, 14, 1285–1297. [Google Scholar] [CrossRef] [Green Version]
- Qiu, L.; Xie, M.; Zhou, M.; Liu, X.; Hu, Z.; Wu, L. Restoration of FVIII Function and Phenotypic Rescue in Hemophilia A Mice by Transplantation of MSCs Derived From F8-Modified iPSCs. Front. Cell Dev. Biol. 2021, 9, 630353. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Shaw, G.; Murphy, M.; Barry, F. Induced Pluripotent Stem Cell-Derived Mesenchymal Stromal Cells Are Functionally and Genetically Different From Bone Marrow-Derived Mesenchymal Stromal Cells. Stem. Cells 2019, 37, 754–765. [Google Scholar] [CrossRef] [Green Version]
- Du, L.M.; Nurden, P.; Nurden, A.T.; Nichols, T.C.; Bellinger, D.A.; Jensen, E.S.; Haberichter, S.L.; Merricks, E.; Raymer, R.A.; Fang, J.; et al. Platelet-targeted gene therapy with human factor VIII establishes haemostasis in dogs with haemophilia A. Nat. Commun. 2013, 4, 2773. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef]
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, S. Induced pluripotent stem cell technology: A decade of progress. Nat. Rev. Drug Discov. 2017, 16, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Simpson, E.; Dazzi, F. Bone Marrow Transplantation 1957-2019. Front. Immunol. 2019, 10, 1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.; Wang, F.; Wu, M.; Wang, Z.Z. Development of hematopoietic stem and progenitor cells from human pluripotent stem cells. J. Cell Biochem. 2015, 116, 1179–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Luan, J.; Shen, Y.; Chen, B. Developments in the production of platelets from stem cells (Review). Mol. Med. Rep. 2021, 23, 1. [Google Scholar] [CrossRef]
- Liu, H.; Liu, J.; Wang, L.; Zhu, F. In vitro Generation of Megakaryocytes and Platelets. Front. Cell Dev. Biol. 2021, 9, 713434. [Google Scholar] [CrossRef] [PubMed]
- Ono-Uruga, Y.; Ikeda, Y.; Matsubara, Y. Platelet production using adipose-derived mesenchymal stem cells: Mechanistic studies and clinical application. J. Thromb. Haemost. 2021, 19, 342–350. [Google Scholar] [CrossRef]
- Hollmann, J.; Brecht, J.; Goetzke, R.; Franzen, J.; Selich, A.; Schmidt, M.; Eipel, M.; Ostrowska, A.; Hapala, J.; Fernandez-Rebollo, E.; et al. Genetic barcoding reveals clonal dominance in iPSC-derived mesenchymal stromal cells. Stem. Cell Res. Ther. 2020, 11, 105. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Hu, Z.; Li, Z.; Pang, J.; Feng, M.; Hu, X.; Wang, X.; Lin-Peng, S.; Liu, B.; Chen, F.; et al. In situ genetic correction of F8 intron 22 inversion in hemophilia A patient-specific iPSCs. Sci. Rep. 2016, 6, 18865. [Google Scholar] [CrossRef]
- Pang, J.; Wu, Y.; Li, Z.; Hu, Z.; Wang, X.; Hu, X.; Wang, X.; Liu, X.; Zhou, M.; Liu, B.; et al. Targeting of the human F8 at the multicopy rDNA locus in Hemophilia A patient-derived iPSCs using TALENickases. Biochem. Biophys. Res. Commun. 2016, 472, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Chen, F.; Wu, Y.; Wang, X.; Feng, M.; Li, Z.; Zhou, M.; Wang, Y.; Wu, L.; Liu, X.; et al. Enhanced tumor growth inhibition by mesenchymal stem cells derived from iPSCs with targeted integration of interleukin24 into rDNA loci. Oncotarget 2017, 8, 40791–40803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Gao, T.; Wang, X.; Hu, Y.; Hu, X.; Hu, Z.; Pang, J.; Li, Z.; Xue, J.; Feng, M.; et al. TALE nickase mediates high efficient targeted transgene integration at the human multi-copy ribosomal DNA locus. Biochem. Biophys. Res. Commun. 2014, 446, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Liu, M.; Xue, Z.; Pan, Q.; Wu, L.; Long, Z.; Xia, K.; Liang, D.; Xia, J. Non-viral ex vivo transduction of human hepatocyte cells to express factor VIII using a human ribosomal DNA-targeting vector. J. Thromb. Haemost. 2007, 5, 347–351. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, J.; Zhou, M.; Wang, Z.; Wu, L.; Hu, Z.; Liang, D. Ectopic Expression of FVIII in HPCs and MSCs Derived from hiPSCs with Site-Specific Integration of ITGA2B Promoter-Driven BDDF8 Gene in Hemophilia A. Int. J. Mol. Sci. 2022, 23, 623. https://doi.org/10.3390/ijms23020623
Zhao J, Zhou M, Wang Z, Wu L, Hu Z, Liang D. Ectopic Expression of FVIII in HPCs and MSCs Derived from hiPSCs with Site-Specific Integration of ITGA2B Promoter-Driven BDDF8 Gene in Hemophilia A. International Journal of Molecular Sciences. 2022; 23(2):623. https://doi.org/10.3390/ijms23020623
Chicago/Turabian StyleZhao, Junya, Miaojin Zhou, Zujia Wang, Lingqian Wu, Zhiqing Hu, and Desheng Liang. 2022. "Ectopic Expression of FVIII in HPCs and MSCs Derived from hiPSCs with Site-Specific Integration of ITGA2B Promoter-Driven BDDF8 Gene in Hemophilia A" International Journal of Molecular Sciences 23, no. 2: 623. https://doi.org/10.3390/ijms23020623