
Cellular and Molecular Targets for Non-Invasive, Non-Pharmacological Therapeutic/Rehabilitative Interventions in Acute Ischemic Stroke


 and
and 
Abstract
:1. Introduction
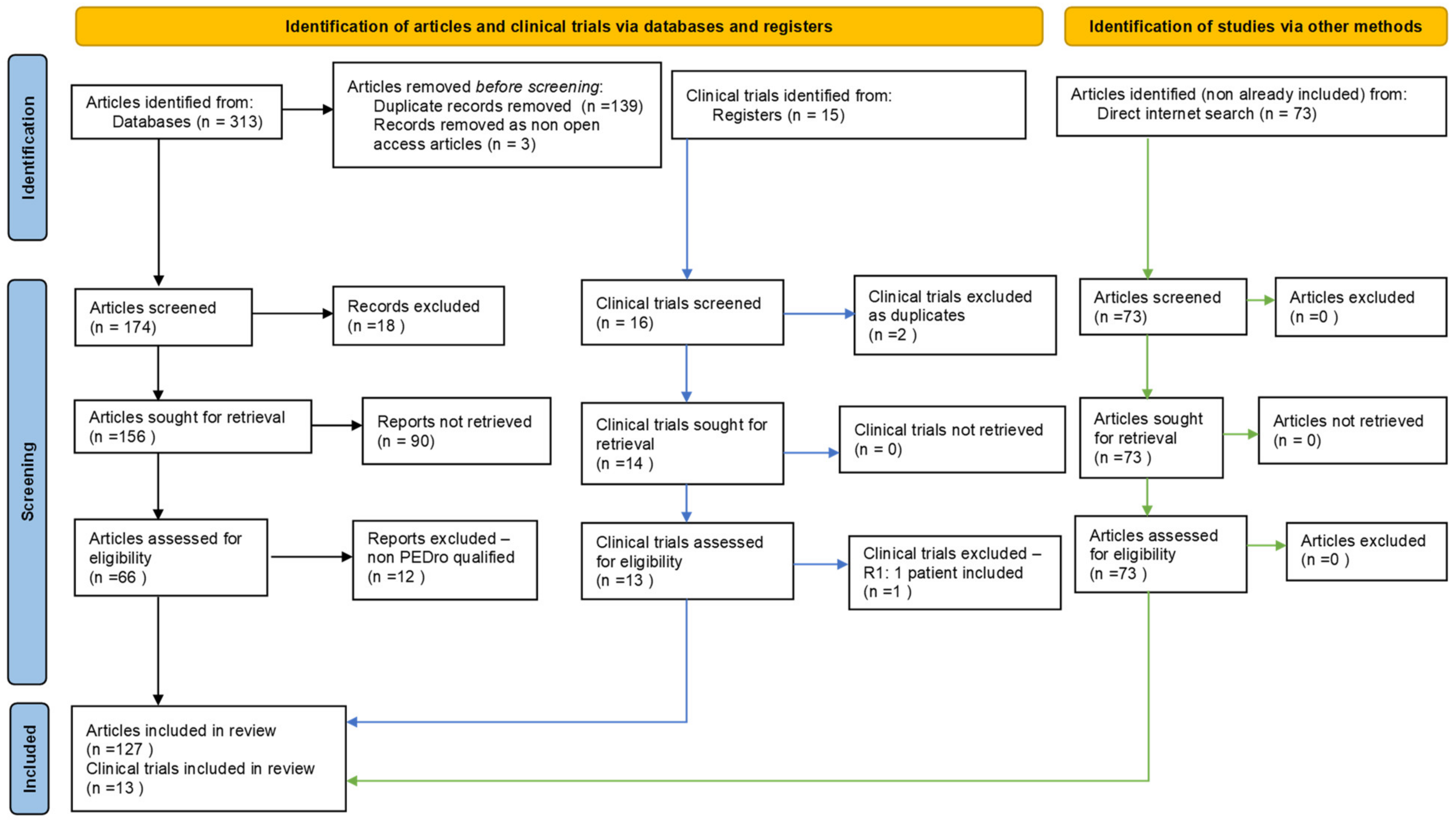
2. Materials and Methods
3. Results
3.1. Cellular and Molecular Mechanisms as Presumptive Therapeutic/Rehabilitative Targets in Acute Ischemic Stroke
3.1.1. Transcriptional Factors
- STAT family members, STAT1-STAT6, found in the cytoplasm, play an essential role in regulating inflammatory responses. Janus kinases (JAKs) and STATs pathways handle dozens of cellular responses such as proliferation, differentiation, migration, apoptosis, and cell survival, depending on the cellular context. The JAK2/STAT3 pathway has been highlighted by in vitro and in vivo experimental stroke models, subsequently activating numerous genes responsible for many cellular functions in neural injury and repair [23].
- NF-κB activates the microglia and transforms these activated cells into the M1 phenotype. It is activated in response to detrimental stresses and induces inflammatory responses. The activated NF-κB is translocated to the nucleus, promoting the secretion of pro-inflammatory cytokines, such as IL18, IL6, and TNF-α [24].
- Nrf2 is a redox-sensitive transcription factor that demonstrates both antioxidant and anti-inflammatory properties, and has been identified as having a protective effect following ischemic stroke [24].
- PPARγ is a ligand-activated transcription factor. In acute ischemic stroke, PPAR γ is also activated and has been shown to reduce tissue damage by directly inhibiting the NF-κB pathway, reducing inflammation, and stimulating the antioxidant response element (Nrf2/ARE) axis to decrease oxidative stress [24].
- Non-coding RNAs (ncRNAs) are a class of functional RNAs that regulate post-transcriptional gene expression [25]. For example, H19 is a long ncRNA (lncRNA) that induces the onset of ischemic stroke and participates in the chronic regeneration process following ischemic stroke. In addition, H19 has been shown to promote M1 microglia polarisation and induce neuroinflammation in ischemic stroke by regulating histone deacetylases. In clinical experiments, H19 levels were identified to be significantly increased in the plasma at 3 h, 7, 30, and 90 days post-ischemic stroke [25].
- Micro RNAs (miRNAs) are small ncRNAs [26] that mediate post-transcriptional gene regulation by controlling the translation of mRNA into protein. In ischemic stroke, miRNAs are involved in multiple cellular functions, such as neuronal development, injured tissue repair, remodeling, and different neuronal activities, and their target genes play a crucial regulatory role in the inflammatory process of post-ischemic reperfusion injury, which explains their potential use as a therapeutic target in ischemic stroke [24].
3.1.2. Receptors
- TLR4, predominantly expressed by microglia, is an essential regulator of inflammatory responses. Elevated TLR4 protein levels are associated with poor outcomes in patients with ischemic stroke. Furthermore, in ischemic stroke, TLR 4 has been shown to recognize molecules related to damage, such as lipopolysaccharides (LPS) [27].
- S1PRs: S1PR1-S1PR5 are G-protein-coupled receptors expressed in abundance in the microglia and are demonstrated to regulate inflammatory responses following ischemic stroke. S1P is the ligand protein for S1PR and has been identified to bind to S1PR1, S1PR2, and S1PR3 to trigger neuroinflammatory reactions in ischemic stroke [24].
- TXA2R, another G-protein-coupled receptor, can promote platelet activation and aggregation by regulating thrombosis/hemostasis and inflammatory responses. In ischemia/reperfusion, significant levels of TXA2R expression were detected in microglia/macrophages [24].
3.1.3. Ion and Ionic Channel Proteins
3.1.4. Mitochondrial Involvement in Cerebral Ischemic Stroke
3.1.5. Signaling Pathways Involved in Cerebral Ischemia
ROS Signaling
HIF-1 Signaling
CK2 Signaling
EGFR Signaling
TGF-β Signaling
NF-kB Signaling
Calcium Signaling
ACh and Related Receptors
Nitric Oxide, RNS, and Nitrosative Stress
MAPK Signal Pathway
Dopamine System
3.1.6. Oxidative Stress Role in Cerebral Ischemic Stroke
3.1.7. Excitotoxicity and Apoptosis/Necrosis
3.1.8. Apoptosis Molecular Mechanisms
3.1.9. Autophagy
3.1.10. Angiogenesis
3.1.11. Serum Proteins
3.1.12. Brain-Derived Neurotrophic Factor
3.1.13. Neuroplasticity
3.1.14. Blood–Brain Barrier Dysfunction
3.1.15. Cell Adhesion Molecules and Ischemic Stroke
3.1.16. Anti-Inflammation as a Therapeutic Target for Ischemic Stroke
3.1.17. Microglia Role in Ischemic Stroke
3.2. Non-Invasive, Non-Pharmacological Therapeutic/Rehabilitative Interventions in Acute Ischemic Stroke
3.2.1. Acupuncture/Electro-Acupuncture (EA)
3.2.2. Hyperbaric Oxygen Therapy
3.2.3. Systemic Hypothermia (Selective, Local Brain) Cooling
3.2.4. Photobiomodulation (PBM)
3.2.5. Therapeutic Gases
3.2.6. Transcranial Direct Currents Stimulations (tDCS)
3.2.7. Transcranial Magnetic Stimulation
3.3. Clinical Studies/Trials on Non-Invasive, Non-Pharmacological Therapeutic/Rehabilitative Interventions in Acute Ischemic Stroke
3.3.1. Presentation of Clinical Studies/Trials on Non-Invasive, Non-Pharmacological Therapeutic/Rehabilitative Interventions in Acute Ischemic Stroke
Acupuncture
Hyperbaric Oxygen Therapy
Hypothermia/Cooling
Photobiomodulation
Therapeutic Gases
Transcranial Direct Currents Stimulations
Transcranial Magnetic Stimulation
3.3.2. Meta-Analysis of the Data Obtained from Identified Clinical Studies
| Binary Random-Effects | Model, Metric: Proportion | |||
| Estimate | Lower bound | Upper bound | Std. error | p-Value |
| 0.070 | 0.048 | 0.092 | 0.011 | <0.001 |
| Heterogeneity: tau2: 0.002; Q(df = 13): 782.360; Het. p-Value: <0.001; I2: 98.338. | ||||
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Singh, M.; Pandey, P.K.; Bhasin, A.; Padma, M.V.; Mohanty, S. Application of Stem Cells in Stroke: A Multifactorial Approach. Front. Neurosci. 2020, 14, 473. [Google Scholar] [CrossRef]
- Mestre, H.; Cohen-Minian, Y.; Zajarias-Fainsod, D.; Ibarra, D.Z.-F.A.A. Pharmacological Treatment of Acute Ischemic Stroke. Neurodegener. Dis. 2013, 581–614. [Google Scholar] [CrossRef]
- Dobkin, B.H.; Carmichael, S.T. The Specific Requirements of Neural Repair Trials for Stroke. Neurorehabilit. Neural Repair 2015, 30, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Bernhardt, J.; Hayward, K.; Kwakkel, G.; Ward, N.; Wolf, S.L.; Borschmann, K.; Krakauer, J.W.; Boyd, L.A.; Carmichael, S.T.; Corbett, D.; et al. Agreed definitions and a shared vision for new standards in stroke recovery research: The Stroke Recovery and Rehabilitation Roundtable taskforce. Int. J. Stroke 2017, 12, 444–450. [Google Scholar] [CrossRef]
- Woodruff, T.M.; Thundyil, J.; Tang, S.-C.; Sobey, C.G.; Taylor, S.M.; Arumugam, T.V. Pathophysiology, treatment, and animal and cellular models of human ischemic stroke. Mol. Neurodegener. 2011, 6, 11. [Google Scholar] [CrossRef]
- Zhan, Y.; Li, M.Z.; Yang, L.; Feng, X.F.; Zhang, Q.X.; Zhang, N.; Zhao, H. An MRI study of neurovascular restorative after combination treatment with xiaoshuanenteric-coated capsule and enriched environment in rats after stroke. Front. Neurosci. 2019, 13, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Hentia, C.; Rizzato, A.; Camporesi, E.; Yang, Z.; Muntean, D.M.; Săndesc, D.; Bosco, G. An overview of protective strategies against ischemia/reperfusion injury: The role of hyperbaric oxygen preconditioning. Brain Behav. 2018, 8, e00959. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Chakraborty, D.; Bhowmik, A.; Ghosh, M.K. Cerebral ischemic stroke: Cellular fate and therapeutic opportunities. Front. Biosci. 2019, 24, 435–450. [Google Scholar]
- Stoll, G.; Kleinschnitz, C.; Nieswandt, B. Molecular mechanisms of thrombus formation in ischemic stroke: Novel insights and targets for treatment. Blood 2008, 112, 3555–3562. [Google Scholar] [CrossRef]
- Soares, R.O.S.; Losada, D.M.; Jordani, M.C.; Évora, P.; Castro-E-Silva, O. Ischemia/Reperfusion Injury Revisited: An Overview of the Latest Pharmacological Strategies. Int. J. Mol. Sci. 2019, 20, 5034. [Google Scholar] [CrossRef]
- Cheon, S.Y.; Kim, E.J.; Kim, J.M.; Koo, B.-N. Cell Type-Specific Mechanisms in the Pathogenesis of Ischemic Stroke: The Role of Apoptosis Signal-Regulating Kinase 1. Oxid. Med. Cell. Longev. 2018, 2018, 2596043. [Google Scholar] [CrossRef]
- Liu, W.; Wang, X.; O’Connor, M.; Wang, G.; Han, F. Brain-Derived Neurotrophic Factor and Its Potential Therapeutic Role in Stroke Comorbidities. Neural Plast. 2020, 2020, 1969482. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Cheng, J.; Zhong, Y.; Mahgoub, O.K.; Akter, F.; Fan, Y.; Aldughaim, M.; Xie, Q.; Qin, L.; Gu, L.; et al. Mechanism and Treatment Related to Oxidative Stress in Neonatal Hypoxic-Ischemic Encephalopathy. Front. Mol. Neurosci. 2019, 12, 88. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, T.H.; Wider, J.M.; Lee, I.; Reynolds, C.A.; Liu, J.; Lepore, B.; Tousignant, R.; Bukowski, M.J.; Johnston, H.; Fite, A. Inhibitory modulation of cytochrome c oxidase activity with specific near-infrared light wavelengths attenuates brain ischemia/reperfusion injury. Sci. Rep. 2018, 8, 3481. [Google Scholar] [CrossRef] [PubMed]
- Peña, I.D.; Knecht, T.; Borlongan, C. Combination therapy for ischemic stroke: Novel approaches to lengthen therapeutic window of tissue plasminogen activator. Brain Circ. 2018, 4, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Brinjikji, W.; Mereuta, O.M.; Dai, D.; Kallmes, D.F.; Savastano, L.; Liu, Y.; Nimjee, S.M.; Nogueira, R.G.; Abbasi, M.; Kadirvel, R. Mechanisms of fibrinolysis resistance and potential targets for thrombolysis in acute ischaemic stroke: Lessons from retrieved stroke emboli. Stroke Vasc. Neurol. 2021, 6, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Qi, Z.; Luo, Y.; Hinchliffe, T.; Ding, G.; Xia, Y.; Ji, X. Non-pharmaceutical therapies for stroke: Mechanisms and clinical implications. Prog. Neurobiol. 2014, 115, 246–269. [Google Scholar] [CrossRef] [PubMed]
- Munteanu, C. Cell biology considerations in Spinal Cord Injury–Review. Balneo Res. J. 2017, 8, 136–151. [Google Scholar] [CrossRef]
- Mohamadpour, M.; Whitney, K.; Bergold, P.J. The Importance of Therapeutic Time Window in the Treatment of Traumatic Brain Injury. Front. Neurosci. 2019, 13, 07. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Asahina, R.; Fujioka, M.; Matsui, T.K.; Kato, S.; Mori, E.; Hioki, H.; Yamamoto, T.; Kobayashi, K.; Tsuboi, A. Ras-like Gem GTPase induced by Npas4 promotes activity-dependent neuronal tolerance for ischemic stroke. Proc. Natl. Acad. Sci. USA 2021, 118, e2018850118. [Google Scholar] [CrossRef]
- Page, M.J.; Moher, D.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. PRISMA 2020 explanation and elaboration: Updated guidance and exemplars for reporting systematic reviews. BMJ 2021, 372, n160. [Google Scholar] [CrossRef]
- Sadik, N.A.; Rashed, L.A.; Mawla, M.A. Circulating miR-155 and JAK2/STAT3 axis in acute ischemic stroke patients and its relation to post-ischemic inflammation and associated ischemic stroke risk factors. Int. J. Gen. Med. 2021, 14, 1469–1484. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Zhang, M.; Li, W.-B.; Dong, F.; Zhang, F. Mechanisms Involved in the Neuroprotection of Electroacupuncture Therapy for Ischemic Stroke. Front. Neurosci. 2018, 12, 929. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.T.; Wu, W.F.; Deng, Y.H.; Ge, J.W. Modulators of microglia activation and polarization in ischemic stroke (Review). Mol. Med. Rep. 2020, 21, 2006–2018. [Google Scholar] [CrossRef]
- Wang, S.-W.; Liu, Z.; Shi, Z.-S. Non-Coding RNA in Acute Ischemic Stroke: Mechanisms, Biomarkers and Therapeutic Targets. Cell Transplant. 2018, 27, 1763–1777. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Chen, D.; Gao, F.; Lv, H.; Zhang, G.; Sun, X.; Liu, L.; Mo, D.; Ma, N.; Song, L.; et al. Exosomes derived from microRNA-138-5p-overexpressing bone marrow-derived mesenchymal stem cells confer neuroprotection to astrocytes following ischemic stroke via inhibition of LCN2. J. Biol. Eng. 2019, 13, 71. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Liu, J.; Bi, Y.; Chen, J.; Zhao, L. Potential Medications or Compounds Acting on Toll-like Receptors in Cerebral Ischemia. Curr. Neuropharmacol. 2018, 16, 160–175. [Google Scholar] [CrossRef] [PubMed]
- Stankowski, J.N.; Gupta, R. Therapeutic Targets for Neuroprotection in Acute Ischemic Stroke: Lost in Translation? Antioxidants Redox Signal. 2011, 14, 1841–1851. [Google Scholar] [CrossRef]
- Kshatri, A.; González-Hernández, A.J.; Giraldez, T. Physiological Roles and Therapeutic Potential of Ca2+ Activated Potassium Channels in the Nervous System. Front. Mol. Neurosci. 2018, 11, 258. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, N.; Ni, Y.-S.; Yang, J.-M.; Ma, L.; Lan, X.-B.; Wu, J.; Niu, J.-G.; Yu, J.-Q. TRPM2 in ischemic stroke: Structure, molecular mechanisms, and drug intervention. Channels 2021, 15, 136–154. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Lin, P.; Chen, M.; Zhang, Y.; Chen, J.; Zheng, M.; Liu, J.; Du, H.; Chen, R.; Pan, X.; et al. Restoration of L-OPA1 alleviates acute ischemic stroke injury in rats via inhibiting neuronal apoptosis and preserving mitochondrial function. Redox Biol. 2020, 34, 101503. [Google Scholar] [CrossRef] [PubMed]
- Guan, R.; Zou, W.; Dai, X.; Yu, X.; Liu, H.; Chen, Q.; Teng, W. Mitophagy, a potential therapeutic target for stroke 06 Biological Sciences 0601 Biochemistry and Cell Biology 11 Medical and Health Sciences 1109 Neurosciences. J. Biomed. Sci. 2018, 25, 87. [Google Scholar] [CrossRef]
- Sun, M.-S.; Jin, H.; Sun, X.; Huang, S.; Zhang, F.-L.; Guo, Z.-N.; Yang, Y. Free Radical Damage in Ischemia-Reperfusion Injury: An Obstacle in Acute Ischemic Stroke after Revascularization Therapy. Oxidative Med. Cell. Longev. 2018, 2018, 3804979. [Google Scholar] [CrossRef]
- Gower, A.; Tiberi, M. The Intersection of Central Dopamine System and Stroke: Potential Avenues Aiming at Enhancement of Motor Recovery. Front. Synaptic Neurosci. 2018, 10, 18. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Huang, L.; Cheng, C.; Li, G.; Xie, J.; Shen, M.; Chen, Q.; Li, W.; He, W.; Qiu, P.; et al. Design, synthesis and biological evaluation of chalcone analogues with novel dual antioxidant mechanisms as potential anti-ischemic stroke agents. Acta Pharm. Sin. B 2019, 9, 335–350. [Google Scholar] [CrossRef]
- Zeng, J.; Zhu, L.; Liu, J.; Zhu, T.; Xie, Z.; Sun, X.; Zhang, H. Metformin Protects against Oxidative Stress Injury Induced by Ischemia/Reperfusion via Regulation of the lncRNA-H19/miR-148a-3p/Rock2 Axis. Oxid. Med. Cell. Longev. 2019, 2019, 8768327. [Google Scholar] [CrossRef] [PubMed]
- George, A.K.; Singh, M.; Homme, R.P.; Majumder, A.; Sandhu, H.S.; Tyagi, S.C. A hypothesis for treating inflammation and oxidative stress with hydrogen sulfide during age-related macular degeneration. Int. J. Ophthalmol. 2018, 11, 881–887. [Google Scholar]
- Xu, D.-S.; Bai, Y.-L.; Gao, B.-Y.; Sun, C.-C.; Xia, G.-H.; Zhou, S.-T.; Zhang, Y.; Mao, Y.-R.; Liu, P.-L.; Zheng, Y.; et al. Paired associated magnetic stimulation promotes neural repair in the rat middle cerebral artery occlusion model of stroke. Neural Regen. Res. 2020, 15, 2047–2056. [Google Scholar] [CrossRef] [PubMed]
- Dilek, N.; Papapetropoulos, A.; Toliver-Kinsky, T.; Szabo, C. Hydrogen sulfide: An endogenous regulator of the immune system. Pharmacol. Res. 2020, 161, 105119. [Google Scholar] [CrossRef]
- Zhao, F.-L.; Fang, F.; Qiao, P.-F.; Yan, N.; Gao, D.; Yan, Y. AP39, a Mitochondria-Targeted Hydrogen Sulfide Donor, Supports Cellular Bioenergetics and Protects against Alzheimer’s Disease by Preserving Mitochondrial Function in APP/PS1 Mice and Neurons. Oxidative Med. Cell. Longev. 2016, 2016, 8360738. [Google Scholar] [CrossRef] [PubMed]
- Carbajo, J.M.; Maraver, F. Salt water and skin interactions: New lines of evidence. Int. J. Biometeorol. 2018, 62, 1345–1360. [Google Scholar] [CrossRef]
- Jeong, N.Y.; Jung, J.; Tabassum, R. Protective effect of hydrogen sulfide on oxidative stress-induced neurodegenerative diseases. Neural Regen. Res. 2020, 15, 232–241. [Google Scholar] [CrossRef]
- Huang, L. Molecular hydrogen: A therapeutic antioxidant and beyond. Med. Gas Res. 2016, 6, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Narne, P.; Pandey, V.; Phanithi, P.B. Role of Nitric Oxide and Hydrogen Sulfide in Ischemic Stroke and the Emergent Epigenetic Underpinnings. Mol. Neurobiol. 2019, 56, 1749–1769. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Y.; Jin, X.-F.; Zhou, X.-H.; Dong, X.-H.; Yu, W.-T.; Gao, W.-J. The Role of Astragaloside IV against Cerebral Ischemia/Reperfusion Injury: Suppression of Apoptosis via Promotion of P62-LC3-Autophagy. Molecules 2019, 24, 1838. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Xiao, Y.; Wang, W.; Qing, L.; Xu, Y.; Song, H.; Zhen, X.; Ao, G.; Alkayed, N.J.; Cheng, J. Differential mechanisms underlying neuroprotection of hydrogen sulfide donors against oxidative stress. Neurochem. Int. 2013, 62, 1072–1078. [Google Scholar] [CrossRef]
- Zhang, M.; Xu, L.; Yang, H. Schisandra chinensis Fructus and Its Active Ingredients as Promising Resources for the Treatment of Neurological Diseases. Int. J. Mol. Sci. 2018, 19, 1970. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Ling, X.; Xi, W.; Wang, P.; Sun, J.; Yang, Q.; Xiao, J. Exogenous hydrogen sulfide reduces atrial remodeling and atrial fibrillation induced by diabetes mellitus via activation of the PI3K/Akt/eNOS pathway. Mol. Med. Rep. 2020, 22, 1759–1766. [Google Scholar] [CrossRef]
- Lee, Y.-S.; Chio, C.-C.; Chang, C.-P.; Wang, L.-C.; Chiang, P.-M.; Niu, K.-C.; Tsai, K.-J. Long Course Hyperbaric Oxygen Stimulates Neurogenesis and Attenuates Inflammation after Ischemic Stroke. Mediat. Inflamm. 2013, 2013, 512978. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.-S.; Song, H.; Kim, E.-H.; Choi, J.-H.; Hong, H.; Han, Y.-M.; Hahm, K.-B. Inhibition of Hydrogen Sulfide-induced Angiogenesis and Inflammation in Vascular Endothelial Cells: Potential Mechanisms of Gastric Cancer Prevention by Korean Red Ginseng. J. Ginseng Res. 2012, 36, 135–145. [Google Scholar] [CrossRef]
- Ciccone, V.; Genah, S.; Morbidelli, L. Endothelium as a Source and Target of H2S to Improve Its Trophism and Function. Antioxidants 2021, 10, 486. [Google Scholar] [CrossRef] [PubMed]
- Disdier, C.; Stonestreet, B.S. Hypoxic-ischemic-related cerebrovascular changes and potential therapeutic strategies in the neonatal brain. J. Neurosci. Res. 2020, 98, 1468–1484. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.-M.; Zhang, C.-L.; Chen, A.-Q.; Wang, H.-L.; Zhou, Y.-F.; Li, Y.-N.; Hu, B. Immune Cells in the BBB Disruption After Acute Ischemic Stroke: Targets for Immune Therapy? Front. Immunol. 2021, 12, 678744. [Google Scholar] [CrossRef]
- Xu, Q.; Wei, Y.-T.; Fan, S.-B.; Wang, L.; Zhou, X.-P. Repetitive hyperbaric oxygen treatment increases insulin sensitivity in diabetes patients with acute intracerebral hemorrhage. Neuropsychiatr. Dis. Treat. 2017, 13, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, D.; Gao, X.; Lew, K.; Richards, A.M.; Wang, P. mTORC2 Phosphorylation of Akt1: A Possible Mechanism for Hydrogen Sulfide-Induced Cardioprotection. PLoS ONE 2014, 9, e99665. [Google Scholar] [CrossRef]
- Duan, X.; Yao, G.; Liu, Z.; Cui, R.; Yang, W. Mechanisms of Transcranial Magnetic Stimulation Treating on Post-stroke Depression. Front. Hum. Neurosci. 2018, 12, 215. [Google Scholar] [CrossRef]
- Eyileten, C.; Sharif, L.; Wicik, Z.; Jakubik, D.; Jarosz-Popek, J.; Soplinska, A.; Postula, M.; Czlonkowska, A.; Kaplon-Cieslicka, A.; Mirowska-Guzel, D. The Relation of the Brain-Derived Neurotrophic Factor with MicroRNAs in Neurodegenerative Diseases and Ischemic Stroke. Mol. Neurobiol. 2021, 58, 329–347. [Google Scholar] [CrossRef]
- Rahman, A.A.; Amruta, N.; Pinteaux, E.; Bix, G.J. Neurogenesis After Stroke: A Therapeutic Perspective. Transl. Stroke Res. 2021, 12, 1–14. [Google Scholar] [CrossRef]
- Finch-Edmondson, M.; Morgan, C.; Hunt, R.W.; Novak, I. Emergent Prophylactic, Reparative and Restorative Brain Interventions for Infants Born Preterm With Cerebral Palsy. Front. Physiol. 2019, 10, 15. [Google Scholar] [CrossRef]
- Escobar, I.; Xu, J.; Jackson, C.W.; Perez-Pinzon, M.A. Altered Neural Networks in the Papez Circuit: Implications for Cognitive Dysfunction after Cerebral Ischemia. J. Alzheimer’s Dis. 2019, 67, 425–446. [Google Scholar] [CrossRef]
- Ding, Y.; Huber, C.; Huber, M. Evidence and opportunities of hypothermia in acute ischemic stroke: Clinical trials of systemic versus selective hypothermia. Brain Circ. 2019, 5, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Cramer, S.C. Treatments to Promote Neural Repair after Stroke. J. Stroke 2018, 20, 57–70. [Google Scholar] [CrossRef]
- Szelenberger, R.; Kostka, J.; Saluk-Bijak, J.; Miller, E. Pharmacological Interventions and Rehabilitation Approach for Enhancing Brain Self-repair and Stroke Recovery. Curr. Neuropharmacol. 2019, 18, 51–64. [Google Scholar] [CrossRef]
- Suzuki, Y.; Saito, J.; Munakata, M.; Shibata, Y. Hydrogen sulfide as a novel biomarker of asthma and chronic obstructive pulmonary disease. Allergol. Int. 2021, 70, 181–189. [Google Scholar] [CrossRef]
- Liu, F.; Chen, D.D.; Sun, X.; Xie, H.H.; Yuan, H.; Jia, W.P.; Chen, A.F. Hydrogen sulfide improves wound healing via restoration of endothelial progenitor cell functions and activation of angiopoietin-1 in type 2 diabetes. Diabetes 2014, 63, 1763–1778. [Google Scholar] [CrossRef]
- Liu, H.; Wang, Y.; Xiao, Y.; Hua, Z.; Cheng, J.; Jia, J. Hydrogen Sulfide Attenuates Tissue Plasminogen Activator-Induced Cerebral Hemorrhage Following Experimental Stroke. Transl. Stroke Res. 2016, 7, 209–219. [Google Scholar] [CrossRef]
- Yilmaz, G.; Granger, D.N. Cell adhesion molecules and ischemic stroke. Neurol. Res. 2008, 30, 783–793. [Google Scholar] [CrossRef]
- Carbone, F.; Bonaventura, A.; Montecucco, F. Neutrophil-Related Oxidants Drive Heart and Brain Remodeling after Ischemia/Reperfusion Injury. Front. Physiol. 2020, 10, 1587. [Google Scholar] [CrossRef]
- Liu, Z.; Ran, Y.; Qie, S.; Gong, W.; Gao, F.; Ding, Z.; Xi, J. Melatonin protects against ischemic stroke by modulating microglia/macrophage polarization toward anti-inflammatory phenotype through STAT3 pathway. CNS Neurosci. Ther. 2019, 25, 1353–1362. [Google Scholar] [CrossRef] [PubMed]
- Sha, R.; Zhang, B.; Han, X.; Peng, J.; Zheng, C.; Zhang, F.; Huang, X. Electroacupuncture alleviates ischemic brain injury by inhibiting the miR-223/NLRP3 pathway. Med. Sci. Monit. 2019, 25, 4723–4733. [Google Scholar] [CrossRef] [PubMed]
- Gálvez, I.; Torres-Piles, S.; Ortega, E. Innate/inflammatory bioregulation and clinical effectiveness of whole-body hyperthermia (balneotherapy) in elderly patients with osteoarthritis. Int. J. Hyperth. 2018, 35, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Ushikoshi-Nakayama, R.; Shakya, S.; Omagari, D.; Matsumoto, N.; Nukuzuma, C.; Komatsu, T.; Lee, M.C.-I.; Inoue, H.; Saito, I. The effects of bathing in neutral bicarbonate ion water. Sci. Rep. 2021, 11, 21789. [Google Scholar] [CrossRef]
- Lv, M.; Li, Y.; Ji, M.-H.; Zhuang, M.; Tang, J.-H. Inhibition of invasion and epithelial-mesenchymal transition of human breast cancer cells by hydrogen sulfide through decreased phospho-p38 expression. Mol. Med. Rep. 2014, 10, 341–346. [Google Scholar] [CrossRef]
- Staessens, S.; Denorme, F.; Francois, O.; Desender, L.; Dewaele, T.; Vanacker, P.; Deckmyn, H.; Vanhoorelbeke, K.; Andersson, T.; De Meyer, S.F. Structural analysis of ischemic stroke thrombi: Histological indications for therapy resistance. Haematologica 2020, 105, 498–507. [Google Scholar] [CrossRef]
- Peruzzaro, S.T.; Andrews, M.M.; Al-Gharaibeh, A.; Pupiec, O.; Resk, M.; Story, D.; Maiti, P.; Rossignol, J.; Dunbar, G.L. Transplantation of mesenchymal stem cells genetically engineered to overexpress interleukin-10 promotes alternative inflammatory response in rat model of traumatic brain injury 11 Medical and Health Sciences 1109 Neurosciences. J. Neuroinflamm. 2019, 16, 2. [Google Scholar] [CrossRef]
- Liu, R.; Pan, M.-X.; Tang, J.-C.; Zhang, Y.; Liao, H.-B.; Zhuang, Y.; Zhao, D.; Wan, Q. Role of neuroinflammation in ischemic stroke. Neuroimmunol. Neuroinflamm. 2017, 4, 158. [Google Scholar] [CrossRef]
- Onose, G.; Anghelescu, A.; Blendea, C.D.; Ciobanu, V.; Daia, C.O.; Firan, F.C.; Munteanu, C.; Oprea, M.; Spinu, A.; Popescu, C. Non-invasive, non-pharmacological/bio-technological interventions towards neurorestoration upshot after ischemic stroke, in adults—Systematic, synthetic, literature review. Front. Biosci. 2021, 26, 1204. [Google Scholar]
- González-Nieto, D.; Fernández-García, L.; Pérez-Rigueiro, J.; Guinea, G.V.; Panetsos, F. Hydrogels-Assisted Cell Engraftment for Repairing the Stroke-Damaged Brain: Chimera or Reality. Polymers 2018, 10, 184. [Google Scholar] [CrossRef] [PubMed]
- Mei, Z.-G.; Huang, Y.-G.; Feng, Z.-T.; Luo, Y.-N.; Yang, S.-B.; Du, L.-P.; Jiang, K.; Liu, X.-L.; Fu, X.-Y.; Deng, Y.-H.; et al. Electroacupuncture ameliorates cerebral ischemia/reperfusion injury by suppressing autophagy via the SIRT1-FOXO1 signaling pathway. Aging 2020, 12, 13187–13205. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, Q.; Yang, S.; Huang, J.; Feng, X.; Peng, J.; Lin, Z.; Liu, W.; Tao, J.; Chen, L. Electroacupuncture Inhibits Apoptosis of Peri-Ischemic Regions via Modulating p38, Extracellular Signal-Regulated Kinase (ERK1/2), and c-Jun N Terminal Kinases (JNK) in Cerebral Ischemia-Reperfusion-Injured Rats. Med Sci. Monit. 2018, 24, 4395–4404. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.H.; Lee, M.H.; Wu, C.Y.; Silva, A.C.; Possoit, H.E.; Hsieh, T.H.; Minagar, A.; Lin, H.W. Cerebral ischemia and neuroregeneration. Neural Regen. Res. 2018, 13, 373–385. [Google Scholar]
- Hsieh, C.-L.; Chang, Q.-Y.; Lin, Y.-W. Acupuncture and neuroregeneration in ischemic stroke. Neural Regen. Res. 2018, 13, 573–583. [Google Scholar] [CrossRef]
- Oh, S.H.; Kang, H.D.; Jung, S.K.; Choi, S. Implementation of Targeted Temperature Management in a Patient with Cerebral Arterial Gas Embolism. Ther. Hypothermia Temp. Manag. 2018, 8, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.-L.; Feng, H.; Xi, G.-H. Hyperbaric oxygen therapy and preconditioning for ischemic and hemorrhagic stroke. Med Gas Res. 2016, 6, 232–236. [Google Scholar] [CrossRef]
- Shah, V.; Mittal, R.; Shahal, D.; Sinha, P.; Bulut, E.; Mittal, J.; Eshraghi, A.A. Evaluating the Efficacy of Taurodeoxycholic Acid in Providing Otoprotection Using an in vitro Model of Electrode Insertion Trauma. Front. Mol. Neurosci. 2020, 13, 113. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, C.; Gao, C.; Li, Z.; Yang, J.; Liu, X.; Liang, F. Effects of hyperbaric oxygen therapy on RAGE and MCP-1 expression in rats with spinal cord injury. Mol. Med. Rep. 2016, 14, 5619–5625. [Google Scholar] [CrossRef]
- Subadi, I.; Nugraha, B.; Laswati, H.; Josomuljono, H. Pain Relief with Wet Cupping Therapy in Rats is Mediated by Heat Shock Protein 70 and ß-Endorphin. Iran. J. Med Sci. 2017, 42, 384–391. [Google Scholar]
- Raphaeli, S.; Carmon, E.; Bloch, B.; Fruchter, E. The role of hyperbaric oxygen therapy in psychiatry: A review of the current knowledge. Isr. J. Psychiatry 2019, 56, 48–53. [Google Scholar]
- Dao, V.T.-V.; Floeren, M.; Kumpf, S.; Both, C.; Peter, B.; Balz, V.; Suvorava, T.; Kojda, G. Catalase activity prevents exercise-induced up-regulation of vasoprotective proteins in venous tissue. J. Cell. Mol. Med. 2011, 15, 2326–2334. [Google Scholar] [CrossRef]
- Lee, K.-L.; Niu, K.-C.; Lin, M.-T.; Niu, C.-S. Attenuating brain inflammation, ischemia, and oxidative damage by hyperbaric oxygen in diabetic rats after heat stroke. J. Formos. Med Assoc. 2013, 112, 454–462. [Google Scholar] [CrossRef]
- Hadanny, A.; Rittblat, M.; Bitterman, M.; May-Raz, I.; Gil Suzin, G.; Boussi-Gross, R.; Zemel, Y.; Bechor, Y.; Catalogna, M.; Efrati, S. Hyperbaric oxygen therapy improves neurocognitive functions of post-stroke patients–a retrospective analysis. Restor. Neurol. Neurosci. 2020, 38, 93–107. [Google Scholar] [CrossRef]
- Huang, G.; Diao, J.; Yi, H.; Xu, L.; Xu, J.; Xu, W. Signaling pathways involved in HSP32 induction by hyperbaric oxygen in rat spinal neurons. Redox Biol. 2016, 10, 108–118. [Google Scholar] [CrossRef]
- Mattingly, T.K.; Lownie, S.P. Cold blood perfusion for selective hypothermia in acute ischemic stroke. Brain Circ. 2019, 5, 187–194. [Google Scholar] [CrossRef]
- Tang, Y.-N.; Zhang, G.-F.; Chen, H.-L.; Sun, X.-P.; Qin, W.-W.; Shi, F.; Sun, L.-X.; Xu, X.-N.; Wang, M.-S. Selective brain hypothermia-induced neuroprotection against focal cerebral ischemia/reperfusion injury is associated with Fis1 inhibition. Neural Regen. Res. 2020, 15, 903–911. [Google Scholar]
- Ceausu, M.; Hostiuc, S.; Dermengiu, D.; Curca, G.C. Morphological diagnosis of hyperthermia-related deaths. Romanian J. Leg. Med. 2010, 18, 239–246. [Google Scholar] [CrossRef]
- Raichle, M.E. Cerebral blood flow and metabolism. Outcome Sev. Damage Cent. Nerv. Syst. 2008, 2001, 85–96. [Google Scholar]
- Harris, E.; Seelye, E.R.; Squire, A. Oxygen consumption during cardiopulmonary bypass with moderate hypothermia in man. Br. J. Anaesth. 1971, 43, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.-Y.; Shen, Z.; He, P.; Jiang, L.; Hou, W.-W.; Shen, Y.; Zhang, X.-N.; Chen, Z. A Novel Neuroprotective Strategy for Ischemic Stroke: Transient Mild Acidosis Treatment by CO2 Inhalation at Reperfusion. Br. J. Pharmacol. 2014, 34, 275–283. [Google Scholar] [CrossRef]
- Jackson, T.C.; Kochanek, P.M. A New Vision for Therapeutic Hypothermia in the Era of Targeted Temperature Management: A Speculative Synthesis. Ther. Hypothermia Temp. Manag. 2019, 9, 13–47. [Google Scholar] [CrossRef] [PubMed]
- A Almekhlafi, M.; Kuczynski, A.M.; Demchuk, A.M. Therapeutic hypothermia: Applications in adults with acute ischemic stroke. Brain Circ. 2019, 5, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, L.; Ding, Y.; Han, Z.; Ji, X. Effects of Therapeutic Hypothermia Combined with Other Neuroprotective Strategies on Ischemic Stroke: Review of Evidence. Aging Dis. 2018, 9, 507–522. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, M.; Hamblin, M.R. Photobiomodulation and the brain: A new paradigm. J. Opt. 2017, 19, 013003. [Google Scholar] [CrossRef]
- Yang, M.; Yang, Z.; Yuan, T.; Feng, W.; Wang, P. A Systemic Review of Functional Near-Infrared Spectroscopy for Stroke: Current Application and Future Directions. Front. Neurol. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Raychaudhuri, S. Introduction to monte carlo simulation. In Proceedings of the 2008 Winter Simulation Confnference, Miami, FL, USA, 7–10 December 2008; pp. 91–100. [Google Scholar]
- Iglesias-Rey, R.; Castillo, J. New strategies for ischemic stroke: Internal photobiomodulation therapy. Neural Regen. Res. 2020, 15, 1658. [Google Scholar] [CrossRef]
- Munteanu, C.; Dogaru, G.; Rotariu, M.; Onose, G. Therapeutic gases used in balneotherapy and rehabilitation medicine–scientific relevance in the last ten years (2011–2020)-Synthetic literature review. Balneo PRM Res. J. 2021, 12, 111–122. [Google Scholar] [CrossRef]
- Munteanu, C.; Munteanu, D.; Onose, G. Hydrogen sulfide (H2S)–therapeutic relevance in rehabilitation and balneotherapy Systematic literature review and meta-analysis based on the PRISMA paradig. Balneo PRM Res. J. 2021, 12, 176–195. [Google Scholar] [CrossRef]
- Bornheim, S.; Croisier, J.-L.; Maquet, P.; Kaux, J.-F. Transcranial direct current stimulation associated with physical-therapy in acute stroke patients-A randomized, triple blind, sham-controlled study. Brain Stimul. 2020, 13, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Su, F.; Xu, W. Enhancing Brain Plasticity to Promote Stroke Recovery. Front. Neurol. 2020, 11, 554089. [Google Scholar] [CrossRef] [PubMed]
- Koo, W.R.; Jang, B.H.; Kim, C.R. Effects of Anodal Transcranial Direct Current Stimulation on Somatosensory Recovery after Stroke. Am. J. Phys. Med. Rehabil. 2018, 97, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.-Y.; Rui, G.; Zhang, J.-P.; Guo, L.; An, G.-Z.; Lin, J.-J.; He, W.; Ding, G.-R. Cathodal tDCS exerts neuroprotective effect in rat brain after acute ischemic stroke. BMC Neurosci. 2020, 21, 1–13. [Google Scholar] [CrossRef]
- Pruvost-Robieux, E.; Calvet, D.; Ben Hassen, W.; Turc, G.; Marchi, A.; Mélé, N.; Seners, P.; Oppenheim, C.; Baron, J.-C.; Mas, J.-L.; et al. Design and Methodology of a Pilot Randomized Controlled Trial of Transcranial Direct Current Stimulation in Acute Middle Cerebral Artery Stroke (STICA). Front. Neurol. 2018, 9, 816. [Google Scholar] [CrossRef]
- Huang, J.; Zhao, Z.; Qu, Q.K.Z. Neuroprotection by Transcranial Direct Current Stimulation in Rodent Models of Focal Ischemic Stroke: A Meta-analysis. Front. Neurosci. 2021, 15, 761971. [Google Scholar] [CrossRef]
- Choi, E.H.; Blasiak, A.; Lee, J.; Yang, I.H. Modulation of Neural Activity for Myelination in the Central Nervous System. Front. Neurosci. 2019, 13, 952. [Google Scholar] [CrossRef] [PubMed]
- Pruvost-Robieux, E.; Benzakoun, J.; Turc, G.; Marchi, A.; Mancusi, R.L.; Lamy, C.; Domigo, V.; Oppenheim, C.; Calvet, D.; Baron, J.-C.; et al. Cathodal Transcranial Direct Current Stimulation in Acute Ischemic Stroke: Pilot Randomized Controlled Trial. Stroke 2021, 52, 1951–1960. [Google Scholar] [CrossRef] [PubMed]
- Caglayan, A.B.; Beker, M.; Caglayan, B.; Yalcin, E.; Caglayan, A.; Yulug, B.; Hanoglu, L.; Kutlu, S.; Doeppner, T.R.; Hermann, D.M.; et al. Acute and Post-acute Neuromodulation Induces Stroke Recovery by Promoting Survival Signaling, Neurogenesis, and Pyramidal Tract Plasticity. Front. Cell. Neurosci. 2019, 13, 144. [Google Scholar] [CrossRef] [PubMed]
- Blesneag, A.V.; Slăvoacă, D.F.; Popa, L.; Stan, A.D.; Jemna, N.; Moldovan, F.I.; Mureșanu, D.F. Low-frequency rTMS in patients with subacute ischemic stroke: Clinical evaluation of short and long-term outcomes and neurophysiological assessment of cortical excitability. J. Med. Life 2015, 8, 378–387. [Google Scholar] [PubMed]
- Fletcher, J. What is heterogeneity and is it important? BMJ 2007, 334, 94–96. [Google Scholar] [CrossRef] [PubMed]
- Zhaoa, L.-R.; Willing, A. Enhancing endogenous capacity to repair a stroke-damaged brain: An evolving field for stroke research. Prog. Neurobiol. 2018, 163–164, 5–26. [Google Scholar] [CrossRef]
- Gennaro, M.; Mattiello, A.; Pizzorusso, T. Rodent Models of Developmental Ischemic Stroke for Translational Research: Strengths and Weaknesses. Neural Plast. 2019, 2019, 5089321. [Google Scholar] [CrossRef]
- Wolska, M.; Jarosz-Popek, J.; Junger, E.; Wicik, Z.; Porshoor, T.; Sharif, L.; Czajka, P.; Postula, M.; Mirowska-Guzel, D.; Czlonkowska, A.; et al. Long Non-coding RNAs as Promising Therapeutic Approach in Ischemic Stroke: A Comprehensive Review. Mol. Neurobiol. 2021, 58, 1664–1682. [Google Scholar] [CrossRef]
- Kho, A.; Hong, D.; Kang, B.; Park, W.-J.; Choi, K.; Park, K.-H.; Suh, S. The Effects of Atorvastatin on Global Cerebral Ischemia-Induced Neuronal Death. Int. J. Mol. Sci. 2021, 22, 4385. [Google Scholar] [CrossRef] [PubMed]
- Lees, T.; Shad-Kaneez, F.; Simpson, A.M.; Nassif, N.; Lin, Y.; Lal, S. Heart Rate Variability as a Biomarker for Predicting Stroke, Post-stroke Complications and Functionality. Biomark. Insights 2018, 13, 20–22. [Google Scholar] [CrossRef] [PubMed]
- Hussain, I.; Park, S.J. Big-ECG: Cardiographic Predictive Cyber-Physical System for Stroke Management. IEEE Access 2021, 9, 123146–123164. [Google Scholar] [CrossRef]
- Hussain, I.; Park, S.-J. Quantitative Evaluation of Task-Induced Neurological Outcome after Stroke. Brain Sci. 2021, 11, 900. [Google Scholar] [CrossRef] [PubMed]
- Hussain, I.; Park, S.-J. Prediction of Myoelectric Biomarkers in Post-Stroke Gait. Sensors 2021, 21, 5334. [Google Scholar] [CrossRef] [PubMed]
- Hussain, I.; Park, S.J. HealthSOS: Real-Time Health Monitoring System for Stroke Prognostics. IEEE Access 2020, 8, 213574–213586. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Study | Start Year | END Year | N—Total Patients | Acupuncture | Hyperbaric Oxygen Therapy | Hypothermia/Cooling | Therapeutic Gases (CO2) | Transcranial Direct Current Stimulation (tDCS) | CONTROL |
|---|---|---|---|---|---|---|---|---|---|---|
| 0 | AXIS LEGEND | 1 | 2 | 3 | 4 | 5 | 6 | 7 | ||
| 1 | NCT03097055 | 2017 | 2019 | 50 | 50 | |||||
| 2 | NCT03431402 | 2018 | 2020 | 60 | 60 | |||||
| 3 | ChiCTR2000040026 | 2020 | 2022 | 60 | 30 | 30 | ||||
| 4 | ChiCTR2000035074 | 2020 | 2022 | 100 | 50 | 50 | ||||
| 5 | NCT04554797 | 2020 | 2020 | 60 | 60 | |||||
| 6 | NCT04695236 | 2020 | 2021 | 80 | 80 | |||||
| 7 | NCT03804060 | 2019 | 2020 | 120 | 120 | |||||
| 8 | ChiCTR2000038972 | 2020 | 2021 | 8 | 8 | |||||
| 9 | NCT04839224 | 2021 | 2022 | 40 | 40 | |||||
| 10 | NCT04061577 | 2019 | 2025 | 24 | 24 | |||||
| 11 | NCT04938076 | 2020 | 2022 | 172 | 172 | |||||
| 12 | NCT04287231 | 2019 | 2020 | 24 | 24 | |||||
| 13 | NCT04687033 | 2020 | 2021 | 40 | 40 | |||||
| TOTAL | 838 | 50 | 140 | 268 | 40 | 260 | 80 | |||
| % | 100 | 5.96 | 16.69 | 31.94 | 4.77 | 31.11 | 9.54 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Onose, G.; Anghelescu, A.; Blendea, D.; Ciobanu, V.; Daia, C.; Firan, F.C.; Oprea, M.; Spinu, A.; Popescu, C.; Ionescu, A.; et al. Cellular and Molecular Targets for Non-Invasive, Non-Pharmacological Therapeutic/Rehabilitative Interventions in Acute Ischemic Stroke. Int. J. Mol. Sci. 2022, 23, 907. https://doi.org/10.3390/ijms23020907
Onose G, Anghelescu A, Blendea D, Ciobanu V, Daia C, Firan FC, Oprea M, Spinu A, Popescu C, Ionescu A, et al. Cellular and Molecular Targets for Non-Invasive, Non-Pharmacological Therapeutic/Rehabilitative Interventions in Acute Ischemic Stroke. International Journal of Molecular Sciences. 2022; 23(2):907. https://doi.org/10.3390/ijms23020907
Chicago/Turabian StyleOnose, Gelu, Aurelian Anghelescu, Dan Blendea, Vlad Ciobanu, Cristina Daia, Florentina Carmen Firan, Mihaela Oprea, Aura Spinu, Cristina Popescu, Anca Ionescu, and et al. 2022. "Cellular and Molecular Targets for Non-Invasive, Non-Pharmacological Therapeutic/Rehabilitative Interventions in Acute Ischemic Stroke" International Journal of Molecular Sciences 23, no. 2: 907. https://doi.org/10.3390/ijms23020907
APA StyleOnose, G., Anghelescu, A., Blendea, D., Ciobanu, V., Daia, C., Firan, F. C., Oprea, M., Spinu, A., Popescu, C., Ionescu, A., Busnatu, Ș., & Munteanu, C. (2022). Cellular and Molecular Targets for Non-Invasive, Non-Pharmacological Therapeutic/Rehabilitative Interventions in Acute Ischemic Stroke. International Journal of Molecular Sciences, 23(2), 907. https://doi.org/10.3390/ijms23020907