
Reduced Colonic Mucosal Injury in 2,3,7,8-Tetrachlorodibenzo-p-Dioxin Poly ADP-Ribose Polymerase (TIPARP/PARP7)-Deficient Mice

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
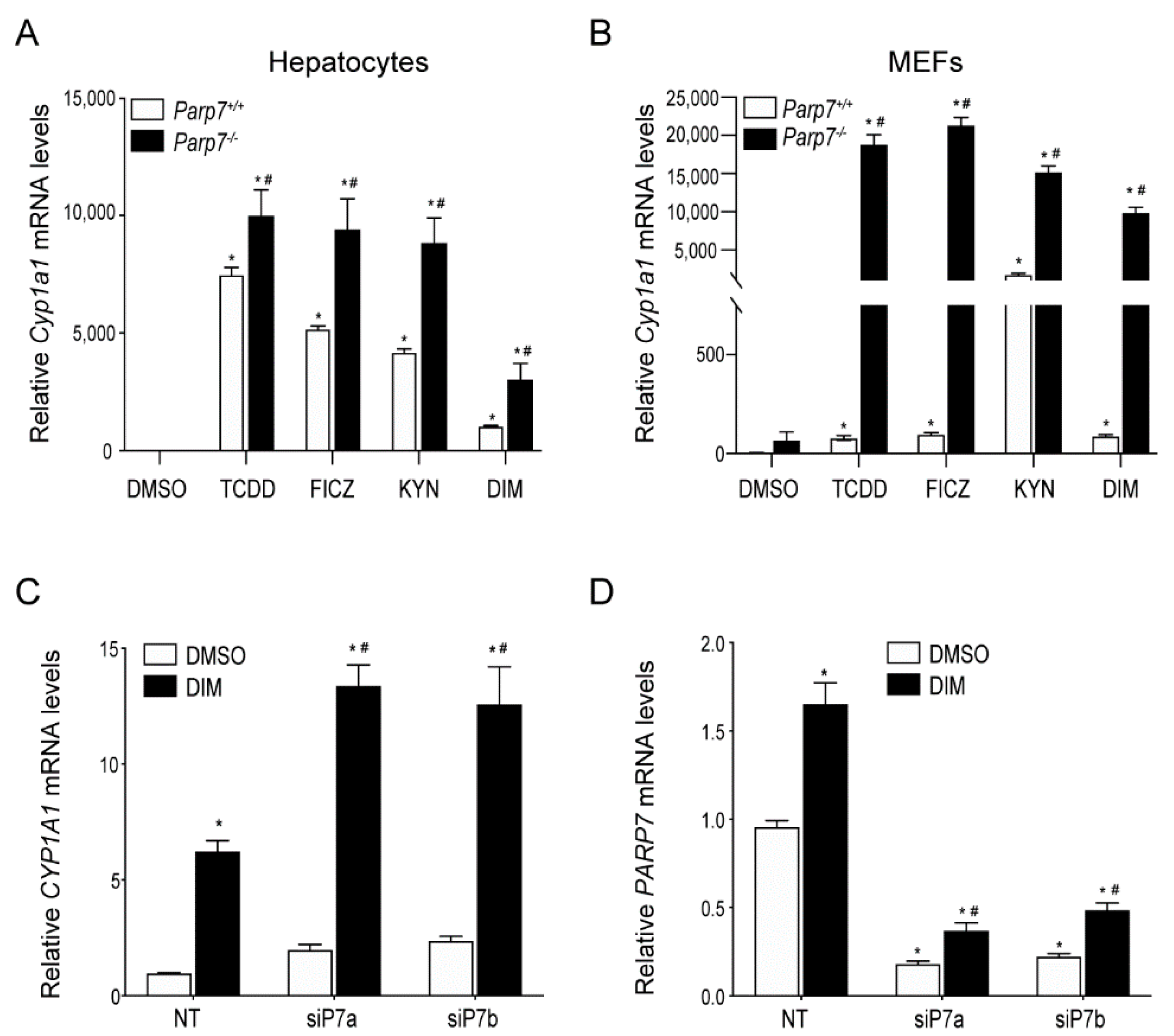
2.1. Increased AHR-Regulated CYP1A1 Levels in Hepatocytes or MEFs Isolated from Parp7−/− Mice
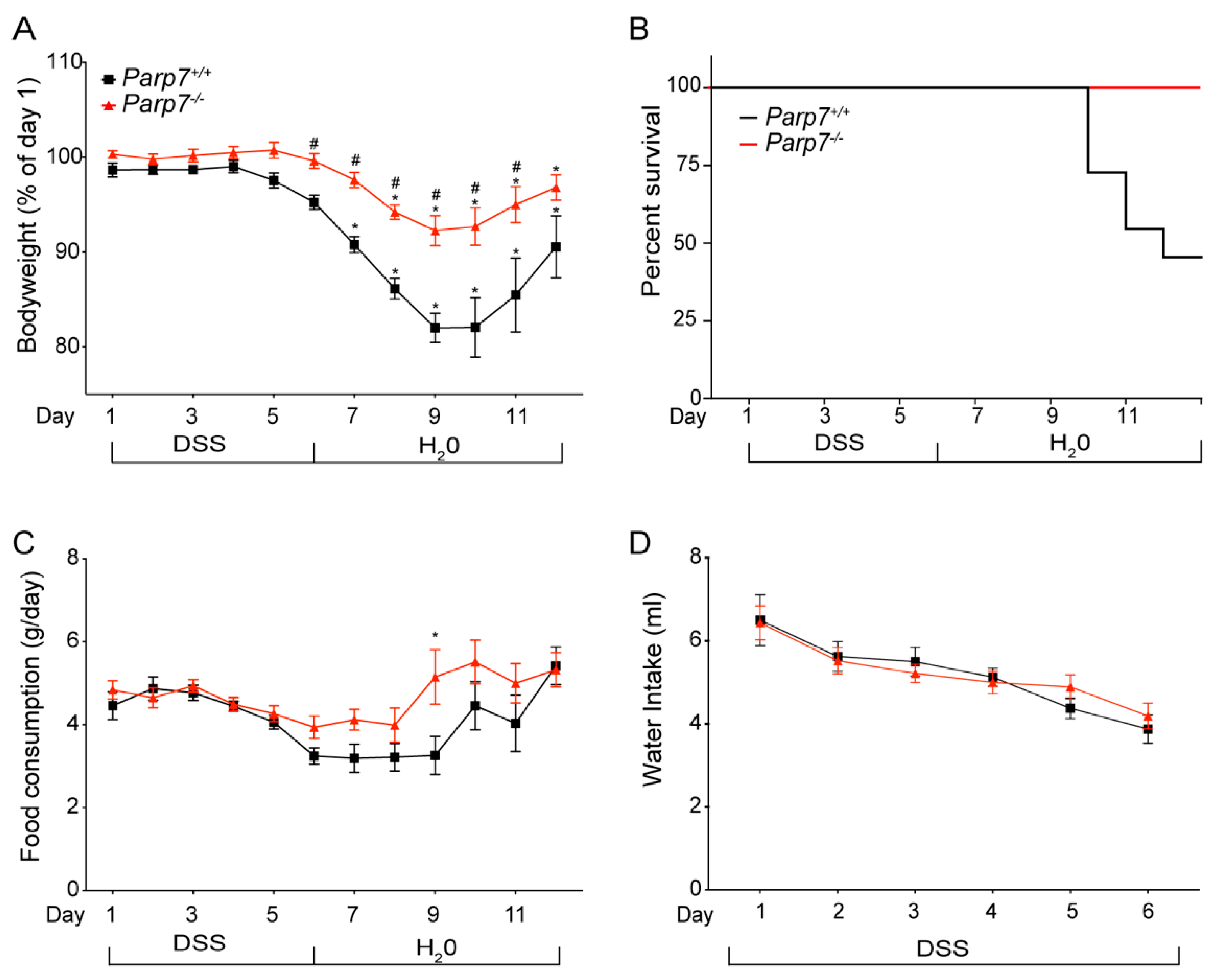
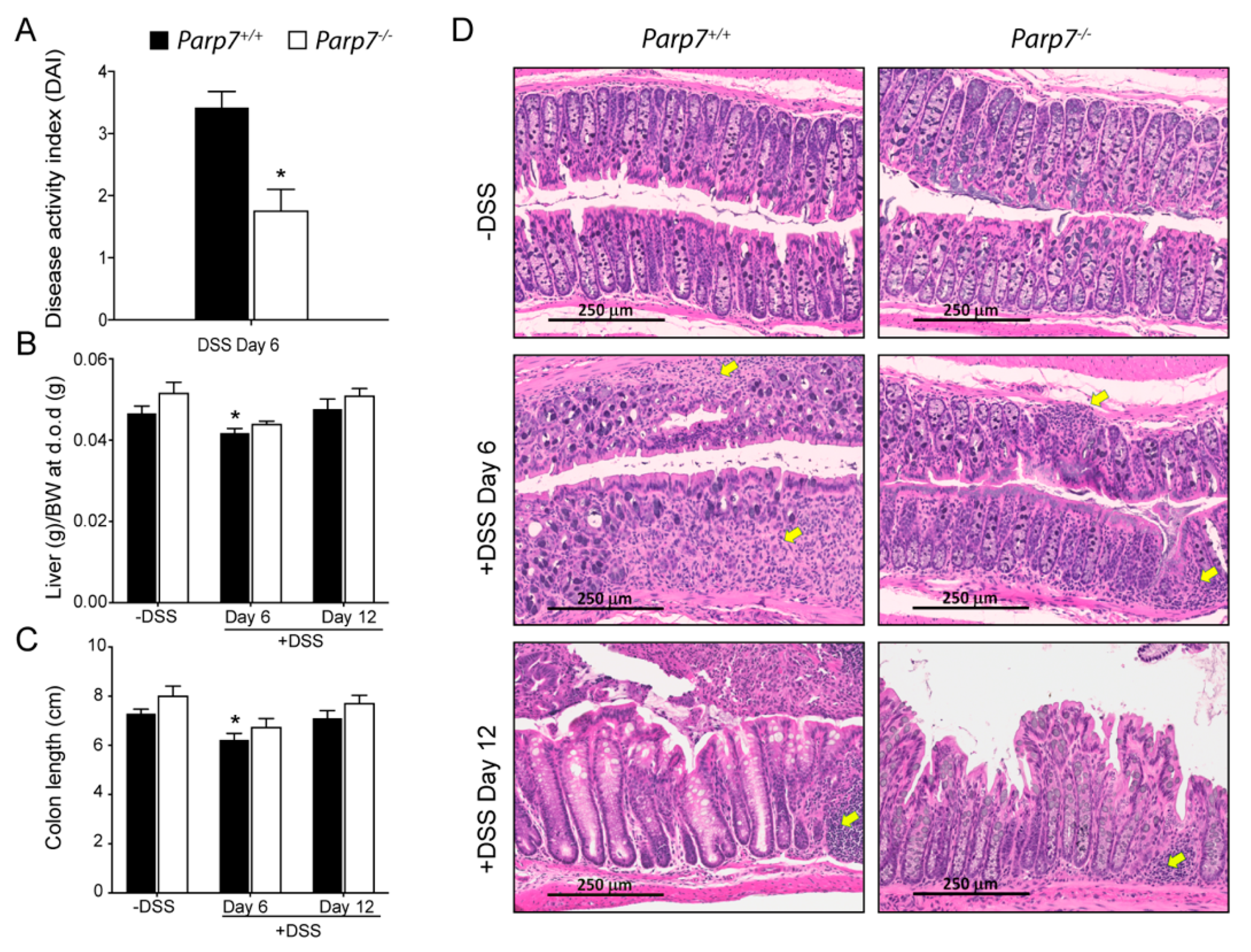
2.2. Reduced Sensitivity of Parp7−/− Mice to DSS-Induced Colitis
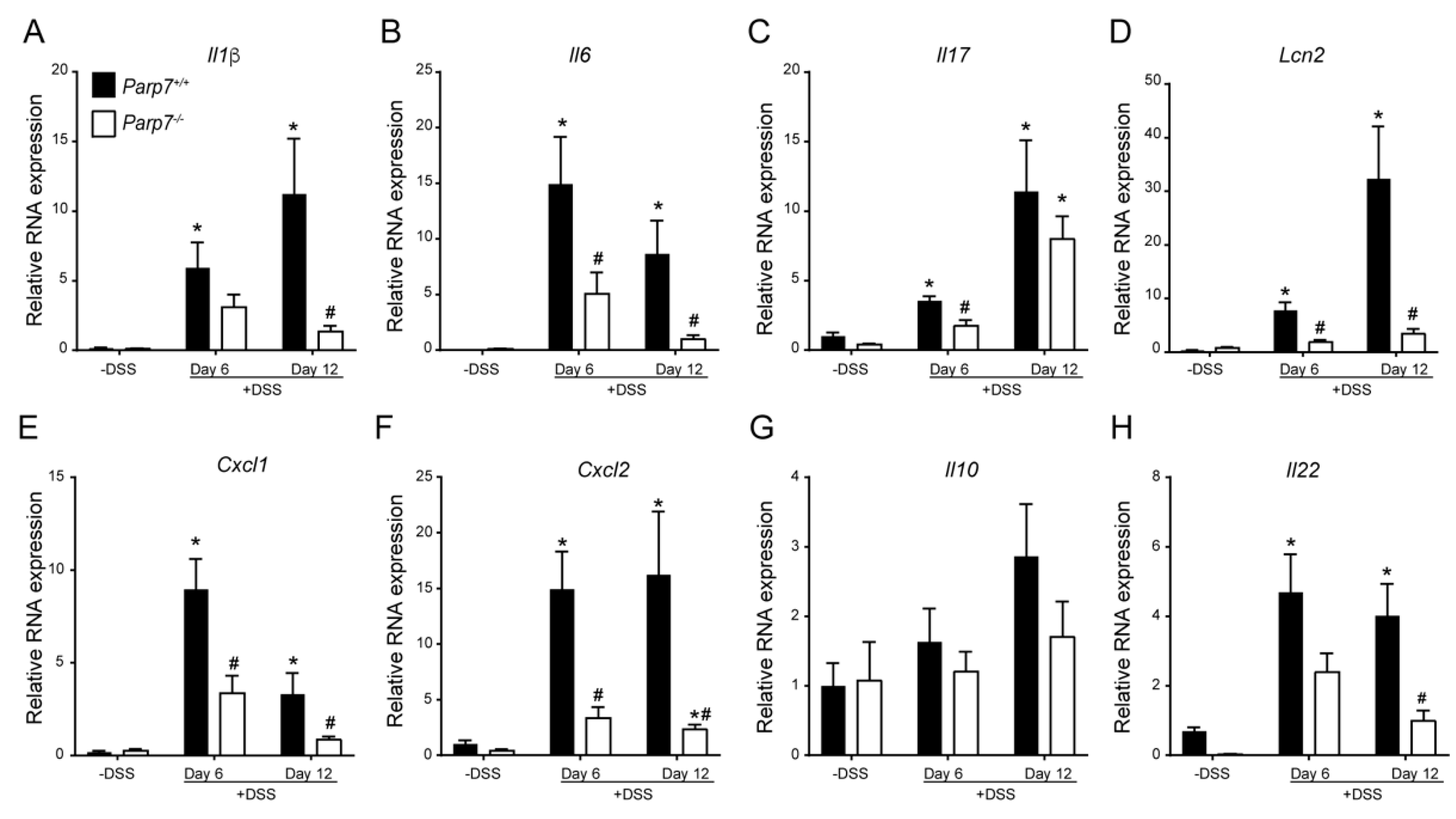
2.3. Increased Expression of Proinflammatory Genes in DSS-Treated Parp7+/+ Compared with Parp7−/− Mice
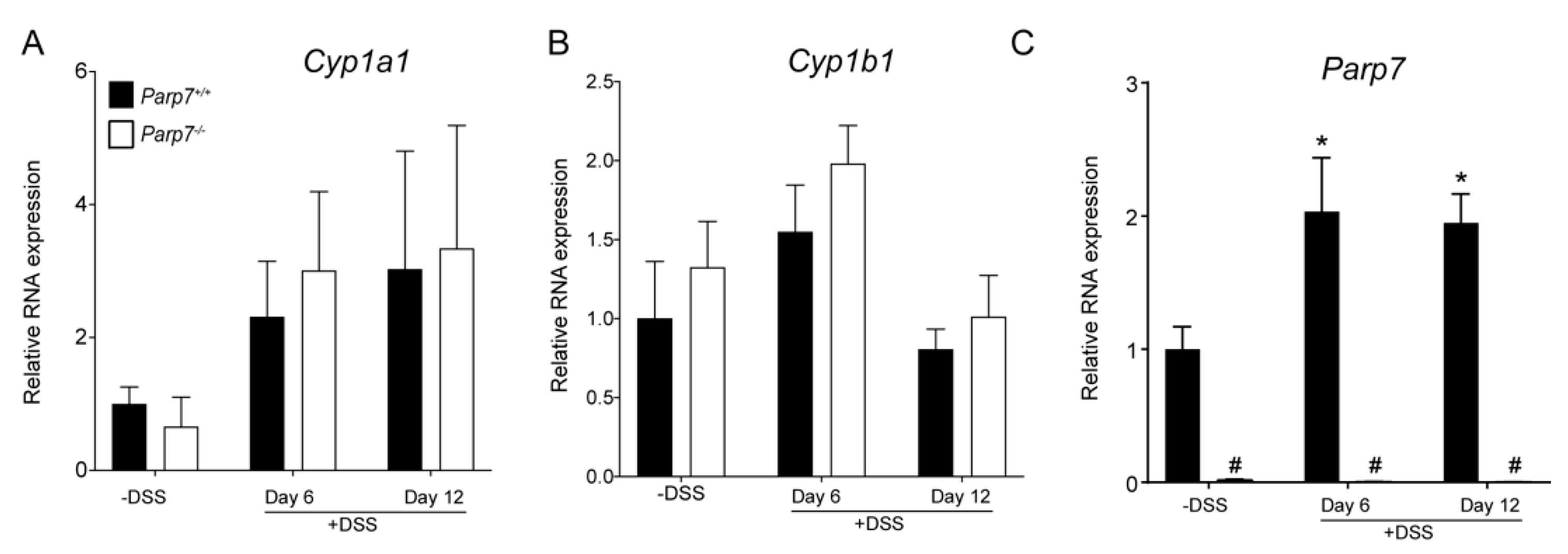
2.4. No Changes in Cyp1a1 and Cyp1b1 Expression Levels in DSS-Treated Parp7+/+ Compared with Parp7−/− Mice
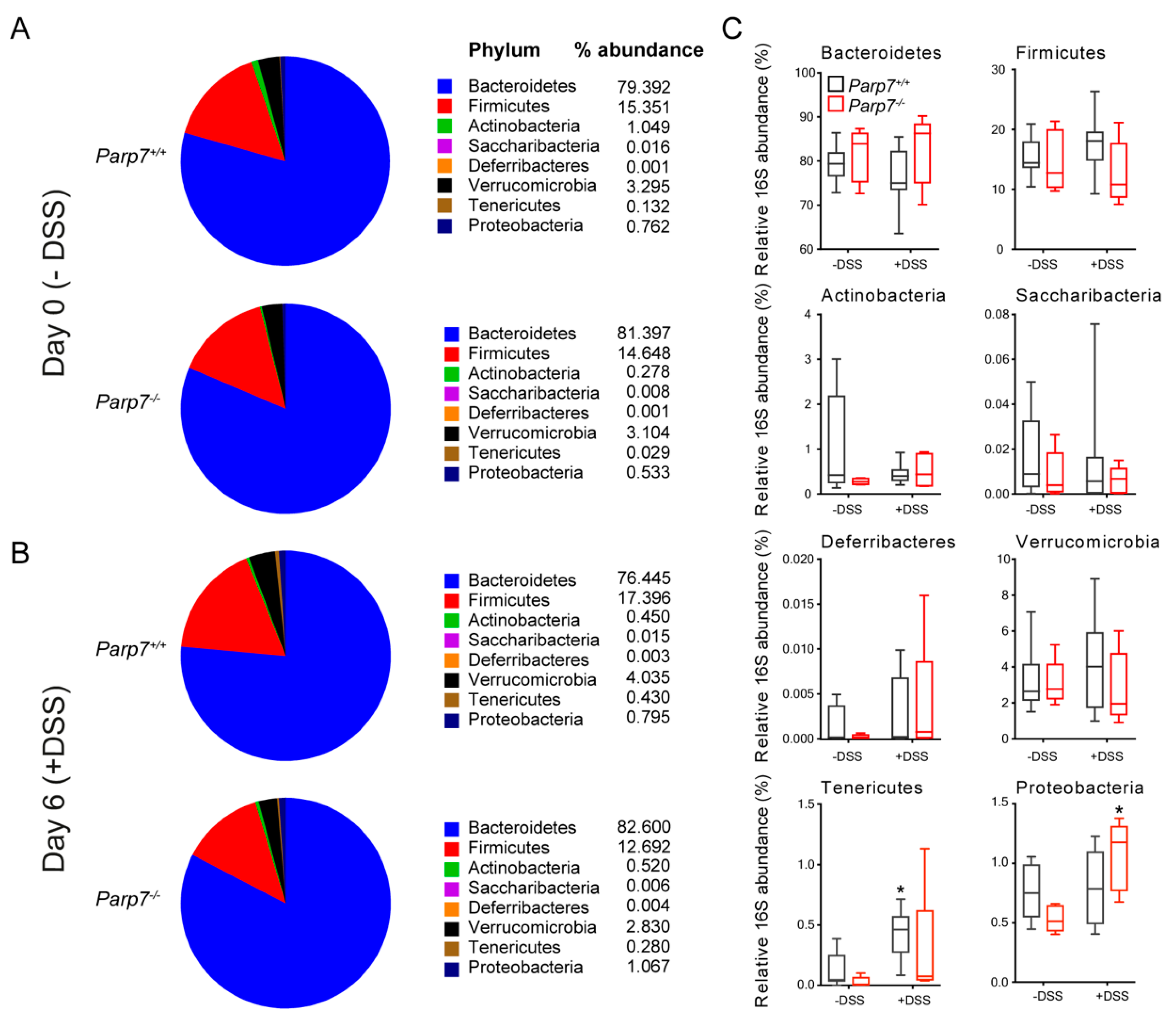
2.5. Influence of PARP7 Loss on the Composition of the Intestinal Microbiota
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Animals and In Vivo Dextran Sulfate Sodium (DSS) Treatment
4.3. Histology
4.4. LiCl Clean-Up Protocol for DNA and RNA
4.5. Hepatocyte and Mouse Embryonic Fibroblast (MEF) Isolation
4.6. RNAi Knockdown Studies
4.7. RNA Extraction and Gene Expression Analysis
4.8. DNA Isolation and Real Time-qPCR Analysis
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Hottiger, M.O.; Hassa, P.O.; Luscher, B.; Schuler, H.; Koch-Nolte, F. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem. Sci. 2010, 35, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Vyas, S.; Matic, I.; Uchima, L.; Rood, J.; Zaja, R.; Hay, R.T.; Ahel, I.; Chang, P. Family-wide analysis of poly(ADP-ribose) polymerase activity. Nat. Commun. 2014, 5, 4426. [Google Scholar] [CrossRef] [Green Version]
- Krishnakumar, R.; Kraus, W.L. The PARP side of the nucleus: Molecular actions, physiological outcomes, and clinical targets. Mol. Cell 2010, 39, 8–24. [Google Scholar] [CrossRef] [Green Version]
- Gibson, B.A.; Kraus, W.L. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 2012, 13, 411–424. [Google Scholar] [CrossRef]
- Luscher, B.; Ahel, I.; Altmeyer, M.; Ashworth, A.; Bai, P.; Chang, P.; Cohen, M.; Corda, D.; Dantzer, F.; Daugherty, M.D.; et al. ADP-ribosyltransferases, an update on function and nomenclature. FEBS J. 2021. [Google Scholar] [CrossRef]
- Yamada, T.; Horimoto, H.; Kameyama, T.; Hayakawa, S.; Yamato, H.; Dazai, M.; Takada, A.; Kida, H.; Bott, D.; Zhou, A.C.; et al. Constitutive aryl hydrocarbon receptor signaling constrains type I interferon-mediated antiviral innate defense. Nat. Immunol. 2016, 17, 687–694. [Google Scholar] [CrossRef] [Green Version]
- Roper, S.J.; Chrysanthou, S.; Senner, C.E.; Sienerth, A.; Gnan, S.; Murray, A.; Masutani, M.; Latos, P.; Hemberger, M. ADP-ribosyltransferases Parp1 and Parp7 safeguard pluripotency of ES cells. Nucleic Acids Res. 2014, 42, 8914–8927. [Google Scholar] [CrossRef] [Green Version]
- Han, B.; Zhang, Y.; Zhang, Y.; Bai, Y.; Chen, X.; Huang, R.; Wu, F.; Leng, S.; Chao, J.; Zhang, J.H.; et al. Novel insight into circular RNA HECTD1 in astrocyte activation via autophagy by targeting MIR142-TIPARP: Implications for cerebral ischemic stroke. Autophagy 2018, 14, 1164–1184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacPherson, L.; Tamblyn, L.; Rajendra, S.; Bralha, F.; McPherson, J.P.; Matthews, J. 2,3,7,8-tetrachlorodibenzo-p-dioxin poly(ADP-ribose) polymerase (TiPARP, ARTD14) is a mono-ADP-ribosyltransferase and repressor of aryl hydrocarbon receptor transactivation. Nucleic Acids Res. 2013, 41, 1604–1621. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, K.M.; Buch-Larsen, S.C.; Kirby, I.T.; Siordia, I.; Hutin, D.; Rasmussen, M.; Grant, D.M.; David, L.L.; Matthews, J.; Nielsen, M.L.; et al. Chemical genetics and proteome-wide site mapping reveal cysteine MARylation by PARP-7 on immune-relevant protein targets. Elife 2021, 10, e60480. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.V.; Delrow, J.; Corrin, P.D.; Frazier, J.P.; Soriano, P. Identification and validation of PDGF transcriptional targets by microarray-coupled gene-trap mutagenesis. Nat. Genet. 2004, 36, 304–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grunewald, M.E.; Shaban, M.G.; Mackin, S.R.; Fehr, A.R.; Perlman, S. Murine Coronavirus Infection Activates the Aryl Hydrocarbon Receptor in an Indoleamine 2,3-Dioxygenase-Independent Manner, Contributing to Cytokine Modulation and Proviral TCDD-Inducible-PARP Expression. J. Virol. 2020, 94, e01743-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heer, C.D.; Sanderson, D.J.; Voth, L.S.; Alhammad, Y.M.O.; Schmidt, M.S.; Trammell, S.A.J.; Perlman, S.; Cohen, M.S.; Fehr, A.R.; Brenner, C. Coronavirus infection and PARP expression dysregulate the NAD metabolome: An actionable component of innate immunity. J. Biol. Chem. 2020, 295, 17986–17996. [Google Scholar] [CrossRef]
- Bindesboll, C.; Tan, S.; Bott, D.; Cho, T.; Tamblyn, L.; MacPherson, L.; Gronning-Wang, L.M.; Nebb, H.I.; Matthews, J. TCDD-inducible poly-ADP-ribose polymerase (TIPARP/PARP7) mono-ADP-ribosylates and coactivates liver X receptors. Biochem. J. 2016, 473, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Kamata, T.; Yang, C.S.; Melhuish, T.A.; Frierson, H.F., Jr.; Wotton, D.; Paschal, B.M. Post-Transcriptional Regulation of PARP7 Protein Stability Is Controlled by Androgen Signaling. Cells 2021, 10, 363. [Google Scholar] [CrossRef]
- Zhang, L.; Cao, J.; Dong, L.; Lin, H. TiPARP forms nuclear condensates to degrade HIF-1alpha and suppress tumorigenesis. Proc. Natl. Acad. Sci. USA 2020, 117, 13447–13456. [Google Scholar] [CrossRef]
- Matthews, J. Alternative Negative Feedback Control in the Aryl Hydrocarbon Receptor Signaling Pathway. J. Drug Metab. Toxicol. 2013, 4, 1–2. [Google Scholar] [CrossRef] [Green Version]
- Gomez, A.; Bindesboll, C.; Satheesh, S.V.; Grimaldi, G.; MacPherson, L.; Hutin, D.; Ahmed, S.; Tamblyn, L.; Cho, T.; Nebb, H.I.; et al. Characterization of TCDD-Inducible Poly-ADP-Ribose Polymerase (TIPARP/ARTD14) Catalytic Activity. Biochem. J. 2018, 475, 3827–3846. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, S.; Bott, D.; Gomez, A.; Tamblyn, L.; Rasheed, A.; MacPherson, L.; Sugamori, K.S.; Cho, T.; Yang, Y.; Grant, D.M.; et al. Loss of the Mono-ADP-Ribosyltransferase, TIPARP, Increases Sensitivity to Dioxin-Induced Steatohepatitis and Lethality. J. Biol. Chem. 2015, 290, 16824–16840. [Google Scholar] [CrossRef] [Green Version]
- Hutin, D.; Tamblyn, L.; Gomez, A.; Grimaldi, G.; Soedling, H.; Cho, T.; Ahmed, S.; Lucas, C.; Chakravarthi, K.; Grant, D.M.; et al. Hepatocyte-specific deletion of TIPARP, a negative regulator of the aryl hydrocarbon receptor, is sufficient to increase sensitivity to dioxin-induced wasting syndrome. Toxicol. Sci. 2018, 165, 347–360. [Google Scholar] [CrossRef]
- Hutin, D.; Long, A.; Sugamori, K.; Shao, P.; Hagen, K.A.; Grimaldi, G.; Grant, D.M.; Matthews, J. 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-inducible poly-ADP-ribose polymerase (TIPARP/PARP7) catalytic mutant mice (TiparpH532A) have increased sensitivity to TCDD-induced hepatotoxicity and lethality. Toxicol. Sci. 2021, 183, 154–169. [Google Scholar] [CrossRef]
- Sartor, R.B. Mechanisms of disease: Pathogenesis of Crohn’s disease and ulcerative colitis. Nat. Clin. Pract. Gastroenterol. Hepatol. 2006, 3, 390–407. [Google Scholar] [CrossRef]
- Jijon, H.B.; Churchill, T.; Malfair, D.; Wessler, A.; Jewell, L.D.; Parsons, H.G.; Madsen, K.L. Inhibition of poly(ADP-ribose) polymerase attenuates inflammation in a model of chronic colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 279, G641–G651. [Google Scholar] [CrossRef] [PubMed]
- Safe, S.; Cheng, Y.; Jin, U.H. The Aryl Hydrocarbon Receptor (AhR) as a Drug Target for Cancer Chemotherapy. Curr. Opin. Toxicol. 2017, 2, 24–29. [Google Scholar] [CrossRef]
- Stockinger, B.; Shah, K.; Wincent, E. AHR in the intestinal microenvironment: Safeguarding barrier function. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 559–570. [Google Scholar] [CrossRef]
- Denison, M.S.; Nagy, S.R. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 309–334. [Google Scholar] [CrossRef] [PubMed]
- DeGroot, D.E.; Franks, D.G.; Higa, T.; Tanaka, J.; Hahn, M.E.; Denison, M.S. Naturally occurring marine brominated indoles are aryl hydrocarbon receptor ligands/agonists. Chem. Res. Toxicol. 2015, 28, 1176–1185. [Google Scholar] [CrossRef] [Green Version]
- Hubbard, T.D.; Murray, I.A.; Bisson, W.H.; Lahoti, T.S.; Gowda, K.; Amin, S.G.; Patterson, A.D.; Perdew, G.H. Adaptation of the human aryl hydrocarbon receptor to sense microbiota-derived indoles. Sci. Rep. 2015, 5, 12689. [Google Scholar] [CrossRef] [PubMed]
- Hankinson, O. Role of coactivators in transcriptional activation by the aryl hydrocarbon receptor. Arch. Biochem. Biophys. 2005, 433, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Innocentin, S.; Withers, D.R.; Roberts, N.A.; Gallagher, A.R.; Grigorieva, E.F.; Wilhelm, C.; Veldhoen, M. Exogenous stimuli maintain intraepithelial lymphocytes via aryl hydrocarbon receptor activation. Cell 2011, 147, 629–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteleone, I.; Rizzo, A.; Sarra, M.; Sica, G.; Sileri, P.; Biancone, L.; MacDonald, T.T.; Pallone, F.; Monteleone, G. Aryl hydrocarbon receptor-induced signals up-regulate IL-22 production and inhibit inflammation in the gastrointestinal tract. Gastroenterology 2011, 141, 237–248 e231. [Google Scholar] [CrossRef] [PubMed]
- Lamas, B.; Richard, M.L.; Leducq, V.; Pham, H.P.; Michel, M.L.; Da Costa, G.; Bridonneau, C.; Jegou, S.; Hoffmann, T.W.; Natividad, J.M.; et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat. Med. 2016, 22, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Kelsall, B.L. The role of type I interferons in intestinal infection, homeostasis, and inflammation. Immunol. Rev. 2014, 260, 145–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rachmilewitz, D.; Karmeli, F.; Takabayashi, K.; Hayashi, T.; Leider-Trejo, L.; Lee, J.; Leoni, L.M.; Raz, E. Immunostimulatory DNA ameliorates experimental and spontaneous murine colitis. Gastroenterology 2002, 122, 1428–1441. [Google Scholar] [CrossRef] [PubMed]
- Rachmilewitz, D.; Karmeli, F.; Shteingart, S.; Lee, J.; Takabayashi, K.; Raz, E. Immunostimulatory oligonucleotides inhibit colonic proinflammatory cytokine production in ulcerative colitis. Inflamm. Bowel Dis. 2006, 12, 339–345. [Google Scholar] [CrossRef] [PubMed]
- MacPherson, L.; Ahmed, S.; Tamblyn, L.; Krutmann, J.; Forster, I.; Weighardt, H.; Matthews, J. Aryl hydrocarbon receptor repressor and TiPARP (ARTD14) use similar, but also distinct mechanisms to repress aryl hydrocarbon receptor signaling. Int. J. Mol. Sci. 2014, 15, 7939–7957. [Google Scholar] [CrossRef]
- Stockinger, B.; Di Meglio, P.; Gialitakis, M.; Duarte, J.H. The aryl hydrocarbon receptor: Multitasking in the immune system. Annu. Rev. Immunol. 2014, 32, 403–432. [Google Scholar] [CrossRef]
- Chassaing, B.; Aitken, J.D.; Malleshappa, M.; Vijay-Kumar, M. Dextran sulfate sodium (DSS)-induced colitis in mice. Curr. Protoc. Immunol. 2014, 104, 15.25.11–15.25.14. [Google Scholar] [CrossRef]
- Busbee, P.B.; Rouse, M.; Nagarkatti, M.; Nagarkatti, P.S. Use of natural AhR ligands as potential therapeutic modalities against inflammatory disorders. Nutr. Rev. 2013, 71, 353–369. [Google Scholar] [CrossRef] [Green Version]
- Naganuma, M.; Sugimoto, S.; Mitsuyama, K.; Kobayashi, T.; Yoshimura, N.; Ohi, H.; Tanaka, S.; Andoh, A.; Ohmiya, N.; Saigusa, K.; et al. Efficacy of Indigo Naturalis in a Multicenter Randomized Controlled Trial of Patients With Ulcerative Colitis. Gastroenterology 2018, 154, 935–947. [Google Scholar] [CrossRef] [Green Version]
- Furumatsu, K.; Nishiumi, S.; Kawano, Y.; Ooi, M.; Yoshie, T.; Shiomi, Y.; Kutsumi, H.; Ashida, H.; Fujii-Kuriyama, Y.; Azuma, T.; et al. A role of the aryl hydrocarbon receptor in attenuation of colitis. Dig. Dis. Sci. 2011, 56, 2532–2544. [Google Scholar] [CrossRef]
- Rasmussen, M.; Tan, S.; Somisetty, V.S.; Hutin, D.; Olafsen, N.E.; Moen, A.; Anonsen, J.H.; Grant, D.M.; Matthews, J. PARP7 and Mono-ADP-Ribosylation Negatively Regulate Estrogen Receptor α Signaling in Human Breast Cancer Cells. Cells 2021, 10, 623. [Google Scholar] [CrossRef]
- Benson, J.M.; Shepherd, D.M. Aryl hydrocarbon receptor activation by TCDD reduces inflammation associated with Crohn’s disease. Toxicol. Sci. 2011, 120, 68–78. [Google Scholar] [CrossRef]
- Takamura, T.; Harama, D.; Matsuoka, S.; Shimokawa, N.; Nakamura, Y.; Okumura, K.; Ogawa, H.; Kitamura, M.; Nakao, A. Activation of the aryl hydrocarbon receptor pathway may ameliorate dextran sodium sulfate-induced colitis in mice. Immunol. Cell Biol. 2010, 88, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Jin, Y.; Hofseth, A.B.; Pena, E.; Habiger, J.; Chumanevich, A.; Poudyal, D.; Nagarkatti, M.; Nagarkatti, P.S.; Singh, U.P.; et al. Resveratrol suppresses colitis and colon cancer associated with colitis. Cancer Prev. Res. 2010, 3, 549–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, A.R.; Villegas, I.; La Casa, C.; de la Lastra, C.A. Resveratrol, a polyphenol found in grapes, suppresses oxidative damage and stimulates apoptosis during early colonic inflammation in rats. Biochem. Pharmacol. 2004, 67, 1399–1410. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.R.; Villegas, I.; Sanchez-Hidalgo, M.; de la Lastra, C.A. The effects of resveratrol, a phytoalexin derived from red wines, on chronic inflammation induced in an experimentally induced colitis model. Br. J. Pharmacol. 2006, 147, 873–885. [Google Scholar] [CrossRef] [Green Version]
- Schiering, C.; Wincent, E.; Metidji, A.; Iseppon, A.; Li, Y.; Potocnik, A.J.; Omenetti, S.; Henderson, C.J.; Wolf, C.R.; Nebert, D.W.; et al. Feedback control of AHR signalling regulates intestinal immunity. Nature 2017, 542, 242–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosoya, T.; Harada, N.; Mimura, J.; Motohashi, H.; Takahashi, S.; Nakajima, O.; Morita, M.; Kawauchi, S.; Yamamoto, M.; Fujii-Kuriyama, Y. Inducibility of cytochrome P450 1A1 and chemical carcinogenesis by benzo[a]pyrene in AhR repressor-deficient mice. Biochem. Biophys. Res. Commun. 2008, 365, 562–567. [Google Scholar] [CrossRef]
- Brandstatter, O.; Schanz, O.; Vorac, J.; Konig, J.; Mori, T.; Maruyama, T.; Korkowski, M.; Haarmann-Stemmann, T.; von Smolinski, D.; Schultze, J.L.; et al. Balancing intestinal and systemic inflammation through cell type-specific expression of the aryl hydrocarbon receptor repressor. Sci. Rep. 2016, 6, 26091. [Google Scholar] [CrossRef] [PubMed]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Schumm, L.P.; Sharma, Y.; Anderson, C.A.; et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef] [Green Version]
- Kawashima, T.; Kosaka, A.; Yan, H.; Guo, Z.; Uchiyama, R.; Fukui, R.; Kaneko, D.; Kumagai, Y.; You, D.J.; Carreras, J.; et al. Double-stranded RNA of intestinal commensal but not pathogenic bacteria triggers production of protective interferon-beta. Immunity 2013, 38, 1187–1197. [Google Scholar] [CrossRef] [Green Version]
- Li, J.Y.; Xiao, J.; Gao, M.; Zhou, H.F.; Fan, H.; Sun, F.; Cui, D.D. IRF/Type I IFN signaling serves as a valuable therapeutic target in the pathogenesis of inflammatory bowel disease. Int. Immunopharmacol. 2021, 92, 107350. [Google Scholar] [CrossRef]
- Rauch, I.; Hainzl, E.; Rosebrock, F.; Heider, S.; Schwab, C.; Berry, D.; Stoiber, D.; Wagner, M.; Schleper, C.; Loy, A.; et al. Type I interferons have opposing effects during the emergence and recovery phases of colitis. Eur. J. Immunol. 2014, 44, 2749–2760. [Google Scholar] [CrossRef]
- Giles, E.M.; Stagg, A.J. Type 1 Interferon in the Human Intestine-A Co-ordinator of the Immune Response to the Microbiota. Inflamm. Bowel Dis. 2017, 23, 524–533. [Google Scholar] [CrossRef]
- Vasbinder, M.M.; Gozgit, J.M.; Abo, R.P.; Kunii, K.; Kuplast-Barr, K.G.; Gui, B.; Lu, A.Z.; Swinger, K.K.; Wigle, T.J.; Blackwell, D.J.; et al. A first-in-class PARP7 inhibitor targeting a newly discovered cancer vulnerability in stress-signaling pathways. In Proceedings of the AACR Annual Meeting, Los Angeles, CA, USA, 27–28 April 2020 and 22–24 June 2020. [Google Scholar]
- Gozgit, J.M.; Vasbinder, M.M.; Abo, R.P.; Kunii, K.; Kuplast-Barr, K.G.; Gui, B.; Lu, A.Z.; Molina, J.R.; Minissale, E.; Swinger, K.K.; et al. PARP7 negatively regulates the type I interferon response in cancer cells and its inhibition triggers antitumor immunity. Cancer Cell 2021, 39, 1214–1226. [Google Scholar] [CrossRef] [PubMed]
- Larmonier, C.B.; Shehab, K.W.; Laubitz, D.; Jamwal, D.R.; Ghishan, F.K.; Kiela, P.R. Transcriptional Reprogramming and Resistance to Colonic Mucosal Injury in Poly(ADP-ribose) Polymerase 1 (PARP1)-deficient Mice. J. Biol. Chem. 2016, 291, 8918–8930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, D.N.; St Amand, A.L.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubbard, T.D.; Murray, I.A.; Nichols, R.G.; Cassel, K.; Podolsky, M.; Kuzu, G.; Tian, Y.; Smith, P.; Kennett, M.J.; Patterson, A.D.; et al. Dietary Broccoli Impacts Microbial Community Structure and Attenuates Chemically Induced Colitis in Mice in an Ah receptor dependent manner. J. Funct. Foods 2017, 37, 685–698. [Google Scholar] [CrossRef]
- Yang, Y.W.; Chen, M.K.; Yang, B.Y.; Huang, X.J.; Zhang, X.R.; He, L.Q.; Zhang, J.; Hua, Z.C. Use of 16S rRNA Gene-Targeted Group-Specific Primers for Real-Time PCR Analysis of Predominant Bacteria in Mouse Feces. Appl. Environ. Microbiol. 2015, 81, 6749–6756. [Google Scholar] [CrossRef] [Green Version]
- Stojanov, S.; Berlec, A.; Strukelj, B. The Influence of Probiotics on the Firmicutes/Bacteroidetes Ratio in the Treatment of Obesity and Inflammatory Bowel disease. Microorganisms 2020, 8, 1715. [Google Scholar] [CrossRef]
- Martin-Gallausiaux, C.; Marinelli, L.; Blottiere, H.M.; Larraufie, P.; Lapaque, N. SCFA: Mechanisms and functional importance in the gut. Proc. Nutr. Soc. 2020, 80, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Marinelli, L.; Martin-Gallausiaux, C.; Bourhis, J.M.; Beguet-Crespel, F.; Blottiere, H.M.; Lapaque, N. Identification of the novel role of butyrate as AhR ligand in human intestinal epithelial cells. Sci. Rep. 2019, 9, 643. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, A. Animal models of inflammatory bowel disease. Prog. Mol. Biol. Transl. Sci. 2012, 105, 263–320. [Google Scholar] [CrossRef] [PubMed]
- Viennois, E.; Chen, F.; Laroui, H.; Baker, M.T.; Merlin, D. Dextran sodium sulfate inhibits the activities of both polymerase and reverse transcriptase: Lithium chloride purification, a rapid and efficient technique to purify RNA. BMC Res. Notes 2013, 6, 360. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hutin, D.; Hagen, K.A.; Shao, P.; Sugamori, K.; Grant, D.M.; Matthews, J. Reduced Colonic Mucosal Injury in 2,3,7,8-Tetrachlorodibenzo-p-Dioxin Poly ADP-Ribose Polymerase (TIPARP/PARP7)-Deficient Mice. Int. J. Mol. Sci. 2022, 23, 920. https://doi.org/10.3390/ijms23020920
Hutin D, Hagen KA, Shao P, Sugamori K, Grant DM, Matthews J. Reduced Colonic Mucosal Injury in 2,3,7,8-Tetrachlorodibenzo-p-Dioxin Poly ADP-Ribose Polymerase (TIPARP/PARP7)-Deficient Mice. International Journal of Molecular Sciences. 2022; 23(2):920. https://doi.org/10.3390/ijms23020920
Chicago/Turabian StyleHutin, David, Karoline Alvik Hagen, Peng Shao, Kim Sugamori, Denis M. Grant, and Jason Matthews. 2022. "Reduced Colonic Mucosal Injury in 2,3,7,8-Tetrachlorodibenzo-p-Dioxin Poly ADP-Ribose Polymerase (TIPARP/PARP7)-Deficient Mice" International Journal of Molecular Sciences 23, no. 2: 920. https://doi.org/10.3390/ijms23020920
APA StyleHutin, D., Hagen, K. A., Shao, P., Sugamori, K., Grant, D. M., & Matthews, J. (2022). Reduced Colonic Mucosal Injury in 2,3,7,8-Tetrachlorodibenzo-p-Dioxin Poly ADP-Ribose Polymerase (TIPARP/PARP7)-Deficient Mice. International Journal of Molecular Sciences, 23(2), 920. https://doi.org/10.3390/ijms23020920