
Cellular and Molecular Alterations Underlying Abnormal Bone Growth in X-Linked Hypophosphatemia

 and
and 
Abstract
:1. Introduction
2. X-Linked Hypophosphatemia (XLH)
2.1. Pathogenesis of XLH
2.2. Hyp Mice
3. XLH and Growth
4. Growth Plate Alterations in XLH
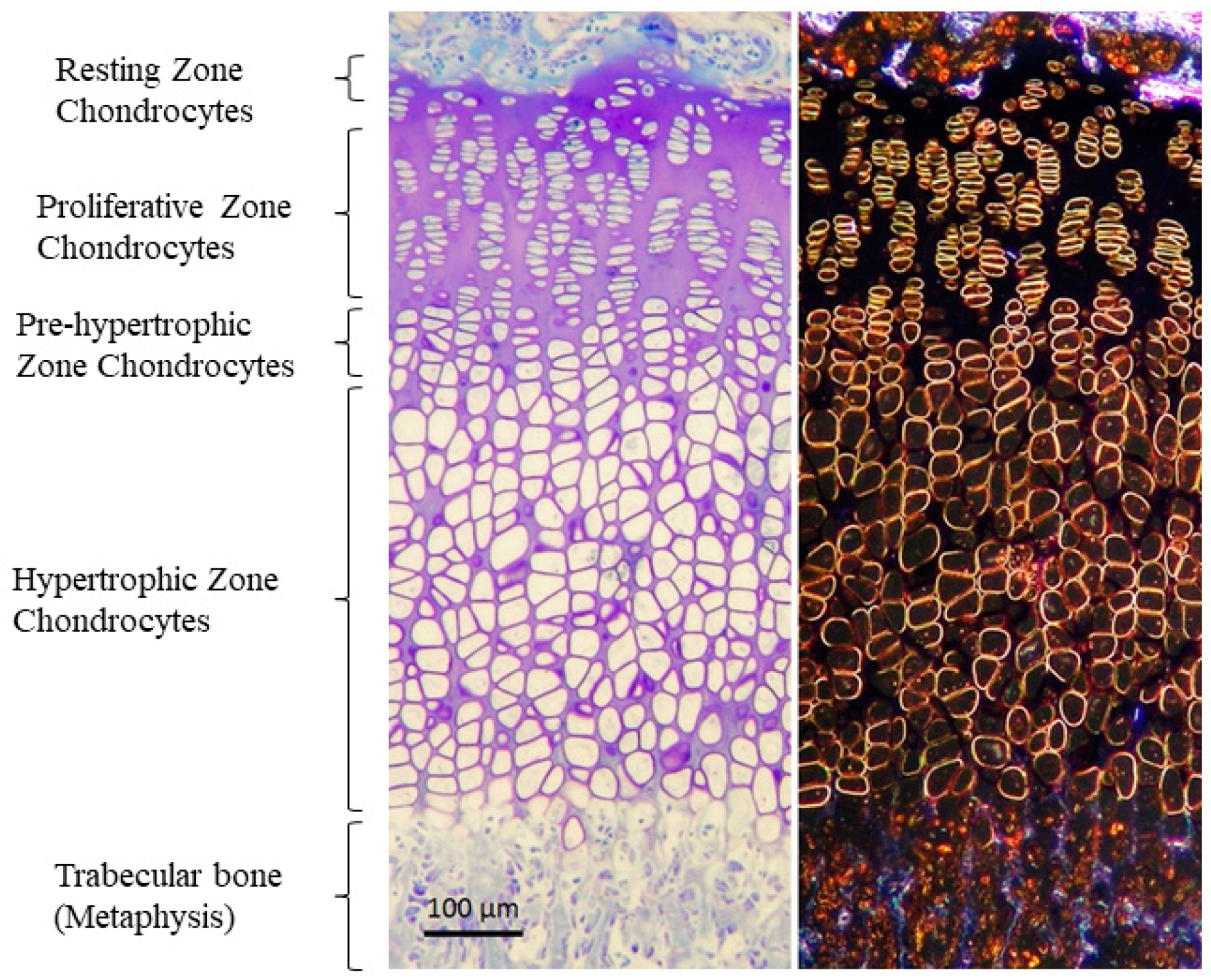
4.1. Changes in the Organization of GP
4.2. Impaired Hypertrophic Chondrocyte Differentiation
4.3. Alteration of Growth Factors and Local Regulators Associated with Growth Plate
5. Treatment Effects in XLH
5.1. Therapeutic Effects of 1,25D and Pi
5.2. Therapeutic Effects of GH
5.3. Therapeutic Effects of FGF23-Neutralizing Antibody (FGF23Ab) or FGF23 Signaling Blockade
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Long, F.; Ornitz, D.M. Development of the endochondral skeleton. Cold Spring Harb. Perspect. Biol. 2013, 5, a028159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holm, I.A.; Huang, X.; Kunkel, L.M. Mutational analysis of the PEX gene in patients with X-linked hypophosphatemic rickets. Am. J. Hum. Genet. 1997, 60, 790–797. [Google Scholar]
- Fuente, R.; Gil-Peña, H.; Claramunt-Taberner, D.; Hernández, O.; Fernández-Iglesias, A.; Alonso-Durán, L.; Rodríguez-Rubio, E.; Santos, F. X-linked hypophosphatemia and growth. Rev. Endocr. Metab. Disord. 2017, 18, 107–115. [Google Scholar] [CrossRef] [Green Version]
- Santos, F.; Fuente, R.; Mejia, N.; Mantecon, L.; Gil-Peña, H.; Ordoñez, F.A. Hypophosphatemia and growth. Pediatr. Nephrol. 2013, 28, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Fuente, R.; Gil-Peña, H.; Claramunt-Taberner, D.; Hernández-Frías, O.; Fernández-Iglesias, A.; Hermida-Prado, F.; Anes-González, G.; Rubio-Aliaga, I.; Lopez, J.M.; Santos, F. Marked alterations in the structure, dynamics and maturation of growth plate likely explain growth retardation and bone deformities of young Hyp mice. Bone 2018, 116, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Razali, N.N.; Hwu, T.T.; Thilakavathy, K. Phosphate homeostasis and genetic mutations of familial hypophosphatemic rickets. J. Pediatr. Endocrinol. Metab. 2015, 28, 1009–1017. [Google Scholar] [CrossRef]
- Francis, F.; Hennig, S.; Korn, B.; Reinhardt, R.; de Jong, P.; Poustka, A.; Lehrach, H.; Rowe, P.S.N.; Goulding, J.N.; Summerfield, T.; et al. A gene (PEX) with homologies to endopeptidases is mutated in patients with X–linked hypophosphatemic rickets. Nat. Genet. 1995, 11, 130–136. [Google Scholar] [CrossRef]
- Kinoshita, Y.; Fukumoto, S. X-linked hypophosphatemia and FGF23-related hypophosphatemic diseases: Prospect for new treatment. Endocr. Rev. 2018, 39, 274–291. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, T.O.; Imel, E.A.; Holm, I.A.; Jan de Beur, S.M.; Insogna, K.L. A clinician’s guide to X-linked hypophosphatemia. J. Bone Miner. Res. 2011, 26, 1381–1388. [Google Scholar] [CrossRef] [Green Version]
- Gaucher, C.; Walrant-Debray, O.; Nguyen, T.M.; Esterle, L.; Garabédian, M.; Jehan, F. PHEX analysis in 118 pedigrees reveals new genetic clues in hypophosphatemic rickets. Hum. Genet. 2009, 125, 401–411. [Google Scholar] [CrossRef]
- Liu, S.; Zhou, J.; Tang, W.; Jiang, X.; Rowe, D.W. Quarles LD. Pathogenic role of Fgf23 in Hyp mice. Am. J. Physiol Endocrinol. Metab. 2006, 291, E38–E49. [Google Scholar] [CrossRef]
- Beck-Nielsen, S.S.; Brusgaard, K.; Rasmussen, L.M.; Brixen, K.; Brock-Jacobsen, B.; Poulsen, M.R.; Vestergaard, P.; Ralston, S.H.; Albagha, O.M.; Poulsen, S.; et al. Phenotype presentation of hypophosphatemic rickets in adults. Calcif. Tissue Int. 2010, 87, 108–119. [Google Scholar] [CrossRef]
- Dhayat, N.A.; Ackermann, D.; Pruijm, M.; Ponte, B.; Ehret, G.; Guessous, I.; Leichtle, A.B.; Paccaud, F.; Mohaupt, M.; Fiedler, G.M.; et al. Fibroblast growth factor 23 and markers of mineral metabolism in individuals with preserved renal function. Kidney Int. 2016, 90, 648–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michigami, T. Skeletal mineralization: Mechanisms and diseases. Ann. Pediatr. Endocrinol. Metab. 2019, 24, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Quarles, L.D. FGF23, PHEX, and MEPE regulation of phosphate homeostasis and skeletal mineralization. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E1–E9. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, Y.; Carpenter, T.O.; Demay, M.B. Hypophosphatemia leads to rickets by impairing caspase-mediated apoptosis of hypertrophic chondrocytes. Proc. Natl. Acad. Sci. USA 2005, 102, 9637–9642. [Google Scholar] [CrossRef] [Green Version]
- Makitie, O.; Doria, A.; Kooh, S.W.; Cole, W.G.; Daneman, A.; Sochett, E. Early treatment improves growth and biochemical and radiographic outcome in X-linked hypophosphatemic rickets. J. Clin. Endocrinol. Metab. 2003, 88, 3591–3597. [Google Scholar] [CrossRef] [Green Version]
- Murali, S.K.; Andrukhova, O.; Clinkenbeard, E.L.; White, K.E.; Erben, R.G. Excessive Osteocytic Fgf23 Secretion Contributes to Pyrophosphate Accumulation and Mineralization Defect in Hyp Mice. PLoS Biol. 2016, 14, e1002427. [Google Scholar] [CrossRef] [Green Version]
- Guo, R.; Quarles, L.D. Cloning and sequencing of human PEX from a bone cDNA library: Evidence for Its Developmental Stage-Specific Regulation in Osteoblasts. J. Bone Miner. Res. 1997, 12, 1009–1017. [Google Scholar] [CrossRef]
- Xiao, Z.; Huang, J.; Cao, L.; Liang, Y.; Han, X.; Quarles, L.D. Osteocyte-specific deletion of Fgfr1 suppresses FGF23. PLoS ONE 2014, 9, e104154. [Google Scholar] [CrossRef] [Green Version]
- Benet-Pagès, A.; Lorenz-Depiereux, B.; Zischka, H.; White, K.E.; Econs, M.J.; Strom, T.M. FGF23 is processed by proprotein convertases but not by PHEX. Bone 2004, 35, 455–462. [Google Scholar] [CrossRef]
- Linglart, A.; Biosse-Duplan, M.; Briot, K.; Chaussain, C.; Esterle, L.; Guillaume-Czitrom, S.; Kamenicky, P.; Nevoux, J.; Prié, D.; Rothenbuhler, A.; et al. Therapeutic management of hypophosphatemic rickets from infancy to adulthood. Endocr. Connect. 2014, 3, R13–R30. [Google Scholar] [CrossRef]
- Brownstein, C.A.; Zhang, J.; Stillman, A.; Ellis, B.; Troiano, N.; Adams, D.J.; Gundberg, C.M.; Lifton, R.P.; Carpenter, T.O. Increased bone volume and correction of HYP mouse hypophosphatemia in the Klotho/HYP mouse. Endocrinology 2010, 151, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Eicher, E.M.; Southard, J.L.; Scriver, C.R.; Glorieux, F.H. Hypophosphatemia: Mouse model for human familial hypophosphatemic (vitamin D-resistant) rickets. Proc. Natl. Acad. Sci. USA 1976, 73, 4667–4671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tenenhouse, H.S. X-linked hypophosphataemia: A homologous disorder in humans and mice. Nephrol. Dial. Transplant. 1999, 14, 333–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makras, P.; Hamdy, N.A.T.; Kant, S.G.; Papapoulos, S.E. Normal growth and muscle dysfunction in X-linked hypophosphatemic rickets associated with a novel mutation in the PHEX gene. J. Clin. Endocrinol. Metab. 2008, 93, 1386–1389. [Google Scholar] [CrossRef]
- Imel, E.A. Enthesopathy, Osteoarthritis, and mobility in X-linked Hypophosphatemia 1. J. Clin. Endocrinol. Metab. 2020, 105, 2649–2651. [Google Scholar] [CrossRef] [PubMed]
- Cauliez, A.; Zhukouskaya, V.V.; Hilliquin, S.; Sadoine, J.; Slimani, L.; Miceli-Richard, C.; Briot, K.; Linglart, A.; Chaussain, C.; Bardet, C. Impact of early conventional treatment on adult bone and joints in a murine model of X-linked Hypophosphatemia. Front. Cell Dev. Biol. 2021, 8, 591417. [Google Scholar] [CrossRef]
- Santos Rodríguez, F. X-linked hypophosphataemic rickets and growth. Adv. Ther. 2020, 37, 55–61. [Google Scholar] [CrossRef]
- Cagnoli, M.; Richter, R.; Böhm, P.; Knye, K.; Empting, S.; Mohnike, K. Spontaneous growth and effect of early therapy with calcitriol and phosphate in X-linked Hypophosphatemic rickets. Pediatr. Endocrinol. Rev. 2017, 1, 119–122. [Google Scholar] [CrossRef]
- Rodríguez-Rubio, E.; Gil-Peña, H.; Chocron, S.; Madariaga, L.; de la Cerda-Ojeda, F.; Fernández-Fernández, M.; de Lucas-Collantes, C.; Gil, M.; Luis-Yanes, M.I.; Vergara, I.; et al. Phenotypic characterization of X-linked hypophosphatemia in pediatric Spanish population. Orphanet. J. Rare Dis. 2021, 16, 104. [Google Scholar] [CrossRef]
- Zivičnjak, M.; Schnabel, D.; Billing, H.; Staude, H.; Filler, G.; Querfeld, U.; Schumacher, M.; Pyper, A.; Schröder, C.; Brämswig, J.; et al. Age-related stature and linear body segments in children with X-linked hypophosphatemic rickets. Pediatr. Nephrol. 2011, 26, 223–231. [Google Scholar] [CrossRef]
- Polisson, R.P.; Martinez, S.; Khoury, M.; Harrell, R.M.; Lyles, K.W.; Friedman, N.; Harrelson, J.M.; Reisner, E.; Drezner, M.K. Calcification of entheses associated with X-linked Hypophosphatemic Osteomalacia. N. Engl. J. Med. 1985, 313, 1–6. [Google Scholar] [CrossRef]
- Kronenberg, H.M. Developmental regulation of the growth plate. Nature 2003, 15, 332–336. [Google Scholar] [CrossRef]
- Fernández-Iglesias, Á.; López, J.M.; Santos, F. Growth plate alterations in chronic kidney disease. Pediatr. Nephrol. 2020, 35, 367–374. [Google Scholar] [CrossRef]
- Hallett, S.A.; Ono, W.; Ono, N. Growth plate chondrocytes: Skeletal development, growth and beyond. Int. J. Mol. Sci 2019, 20, 6009. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Tsang, K.Y.; Tang, H.C.; Chan, D.; Cheah, K.S.E. Hypertrophic chondrocytes can become osteoblasts and osteocytes in endochondral bone formation. Proc. Natl. Acad. Sci. USA 2014, 111, 12097–12102. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Zhou, S.; Chen, H.; Du, X.; Chen, L. Advances in fibroblast growth factor signaling in growth plate development and disorders. J. Mol. Endocrinol. 2014, 53, T11–T34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miedlich, S.U.; Zalutskaya, A.; Zhu, E.D.; Demay, M.B. Phosphate-induced apoptosis of hypertrophic chondrocytes is associated with a decrease in mitochondrial membrane potential and is dependent upon Erk1/2 phosphorylation. J. Biol. Chem. 2010, 285, 18270–18275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Iglesias, Á.; Fuente, R.; Gil-Peña, H.; Alonso-Duran, L.; García-Bengoa, M.; Santos, F.; López, J.M. A simple method based on confocal microscopy and thick sections recognizes seven subphases in growth plate chondrocytes. Sci. Rep. 2020, 10, 6935. [Google Scholar] [CrossRef] [PubMed]
- Cooper, K.L.; Oh, S.; Sung, Y.; Dasari, R.R.; Kirschner, M.W.; Tabin, C.J. Multiple phases of chondrocyte enlargement underlie differences in skeletal proportions. Nature 2013, 495, 375–378. [Google Scholar] [CrossRef]
- Breur, G.J.; van Enkevort, B.A.; Farnum, C.E.; Wilsman, N.J. Linear relationship between the volume of hypertrophic chondrocytes and the rate of longitudinal bone growth in growth plates. J. Orthop. Res. 1991, 9, 348–359. [Google Scholar] [CrossRef]
- Kuss, P.; Kraft, K.; Stumm, J.; Ibrahim, D.; Vallecillo-Garcia, P.; Mundlos, S.; Stricker, S. Regulation of cell polarity in the cartilage growth plate and perichondrium of metacarpal elements by HOXD13 and WNT5A. Dev. Biol. 2014, 385, 83–93. [Google Scholar] [CrossRef] [Green Version]
- Long, F.; Chung, U.I.; Ohba, S.; McMahon, J.; Kronenberg, H.M.; McMahon, A.P. IHH signaling is directly required for the osteoblast lineage in the endochondral skeleton. Development 2004, 131, 1309–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, T.; Kamekura, S.; Mabuchi, A.; Kou, I.; Seki, S.; Takato, T.; Nakamura, K.; Kawaguchi, H.; Ikegawa, S.; Chung, U.I. The combination of SOX5, SOX6, and SOX9 (the SOX trio) provides signals sufficient for induction of permanent cartilage. Arthritis Rheum. 2004, 50, 3561–3573. [Google Scholar] [CrossRef] [PubMed]
- Miao, D.; Bai, X.; Panda, D.K.; Karaplis, A.C.; Goltzman, D.; McKee, M.D. Cartilage abnormalities are associated with abnormal Phex expression and with altered matrix protein and MMP-9 localization in Hyp mice. Bone 2004, 34, 638–647. [Google Scholar] [CrossRef]
- Liu, E.S.; Martins, J.S.; Raimann, A.; Chae, B.T.; Brooks, D.J.; Jorgetti, V.; Bouxsein, M.L.; Demay, M.B. 1,25-dihydroxyvitamin D alone improves skeletal growth, microarchitecture, and strength in a murine model of XLH, despite enhanced FGF23 expression. J. Bone Miner. Res. 2016, 31, 929–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ide, N.; Ye, R.; Courbebaisse, M.; Olauson, H.; Densmore, M.J.; Larsson, T.E.; Hanai, J.I.; Lanske, B. In vivo evidence for an interplay of FGF23/Klotho/PTH axis on the phosphate handling in renal proximal tubules. Am. J. Physiol.-Renal Physiol. 2018, 315, F1261–F1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowe, P.S.N. The molecular background to hypophosphataemic rickets. Arch. Dis. Child. 2000, 83, 192–194. [Google Scholar] [CrossRef] [Green Version]
- Larsson, T.; Marsell, R.; Schipani, E.; Ohlsson, C.; Ljunggren, O.; Tenenhouse, H.S.; Jüppner, H.; Jonsson, K.B. Transgenic mice expressing fibroblast growth factor 23 under the control of the α1(I) collagen promoter exhibit growth retardation, osteomalacia, and disturbed phosphate homeostasis. Endocrinology 2004, 145, 3087–3094. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.S.; Kleeman, C.R.; Felsenfeld, A.J. The journey from Vitamin D-resistant rickets to the regulation of renal phosphate transport lessons from studies of rare genetic disorders. Clin. J. Am. Soc. Nephrol. 2009, 4, 1866–1877. [Google Scholar] [CrossRef] [Green Version]
- Staines, K.A.; Mackenzie, N.C.; Clarkin, C.E.; Zelenchuk, L.; Rowe, P.S.; MacRae, V.E.; Farquharson, C. MEPE is a novel regulator of growth plate cartilage mineralization. Bone 2012, 51, 418–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokarz, D.; Martins, J.S.; Petit, E.T.; Lin, C.P.; Demay, M.B.; Liu, E.S. Hormonal regulation of Osteocyte Perilacunar and canalicular remodeling in the Hyp mouse model of X-linked Hypophosphatemia. J. Bone Miner. Res. 2018, 33, 499–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, E.S.; Zalutskaya, A.; Chae, B.T.; Zhu, E.D.; Gori, F.; Demay, M.B. Phosphate interacts with PTHrP to regulate endochondral bone formation. Endocrinology 2014, 155, 3750–3756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, S.L.; Cohen, A.J.; Lassová, L. Integration of signaling pathways regulating chondrocyte differentiation during endochondral bone formation. J. Cell Physiol. 2007, 213, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Baum, M.; Syal, A.; Quigley, R.; Seikaly, M. Role of prostaglandins in the pathogenesis of X-linked hypophosphatemia. Pediatr. Nephrol. 2006, 21, 1067–1074. [Google Scholar] [CrossRef]
- Wu, S.; Levenson, A.; Kharitonenkov, A.; De Luca, F. Fibroblast growth factor 21 (FGF21) inhibits chondrocyte function and growth hormone action directly at the growth plate. J. Biol. Chem. 2012, 287, 26060–26067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchihashi, K.; Nakatani, T.; Goetz, R.; Mohammadi, M.; He, X.; Razzaque, M.S. FGF23-induced hypophosphatemia persists in Hyp mice deficient in the WNT coreceptor Lrp6. Contrib. Nephrol. 2013, 180, 124–137. [Google Scholar] [CrossRef] [Green Version]
- Miao, D.; Bai, X.; Panda, D.; McKee, M.D.; Karaplis, A.C.; Goltzman, D. Osteomalacia in Hyp mice is associated with abnormal Phex expression and with altered bone matrix protein expression and deposition. Endocrinology 2001, 142, 926–939. [Google Scholar] [CrossRef]
- De Borst, M.H.; Vervloet, M.G.; Ter Wee, P.M.; Navis, G. Cross talk between the renin-angiotensin-aldosterone system and vitamin D-FGF-23-klotho in chronic kidney disease. J. Am. Soc. Nephrol. 2011, 22, 1603–1609. [Google Scholar] [CrossRef] [Green Version]
- Econs, M.J.; Francis, F. Positional cloning of the PEX gene: New insights into the pathophysiology of X-linked hypophosphatemic rickets. Am. J. Physiol.–Ren. Physiol. 1997, 273, F489–F498. [Google Scholar] [CrossRef]
- Raimann, A.; Mindler, G.T.; Kocijan, R.; Bekes, K.; Zwerina, J.; Haeusler, G.; Ganger, R. Multidisciplinary patient care in X-linked hypophosphatemic rickets: One challenge, many perspectives. Wien. Med. Wochenschr. 2020, 170, 116–123. [Google Scholar] [CrossRef] [Green Version]
- Yorgan, T.; Rendenbach, C.; Jeschke, A.; Amling, M.; Cheah, K.S.E.; Schinke, T. Increased Col10a1 expression is not causative for the phenotype of Phex-deficient Hyp mice. Biochem. Biophys. Res. Commun. 2013, 442, 209–213. [Google Scholar] [CrossRef]
- Carpenter, T.O.; Whyte, M.P.; Imel, E.A.; Boot, A.M.; Högler, W.; Linglart, A.; Padidela, R.; Van't Hoff, W.; Mao, M.; Chen, C.Y.; et al. Burosumab therapy in children with X-linked Hypophosphatemia. N. Engl. J. Med. 2018, 378, 1987–1998. [Google Scholar] [CrossRef] [Green Version]
- González-Meneses López, A. FGF23-related hypophosphataemic bone disease. Adv. Ther. 2020, 37, 25–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haffner, D.; Emma, F.; Eastwood, D.M.; Duplan, M.B.; Bacchetta, J.; Schnabel, D.; Wicart, P.; Bockenhauer, D.; Santos, F.; Levtchenko, E.; et al. Clinical practice recommendations for the diagnosis and management of X-linked hypophosphataemia. Nat. Rev. Nephrol. 2019, 15, 435–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arango Sancho, P. Complications of phosphate and Vitamin D treatment in X-linked Hypophosphataemia. Adv. Ther. 2020, 37, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, I.; Segawa, H.; Ikuta, K.; Hanazaki, A.; Fujii, T.; Tatsumi, S.; Kido, S.; Hasegawa, T.; Amizuka, N.; Saito, H.; et al. Eldecalcitol causes FGF23 resistance for Pi reabsorption and improves rachitic bone phenotypes in the male Hyp Mouse. Endocrinology 2018, 159, 2741–2758. [Google Scholar] [CrossRef]
- Martins, J.S.; Liu, E.S.; Sneddon, W.B.; Friedman, P.A.; Demay, M.B. 1,25-dihydroxyvitamin D maintains brush border membrane NaPi2a and attenuates phosphaturia in Hyp Mice. Endocrinology 2019, 160, 2204–2214. [Google Scholar] [CrossRef]
- Fuente, R.; Gil-Peña, H.; Claramunt-Taberner, D.; Hernández-Frías, O.; Fernández-Iglesias, Á.; Alonso-Durán, L.; Rodríguez-Rubio, E.; Hermida-Prado, F.; Anes-González, G.; Rubio-Aliaga, I.; et al. MAPK inhibition and growth hormone: A promising therapy in XLH. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 8349–8362. [Google Scholar] [CrossRef]
- Meo Burt, P.; Xiao, L.; Hurley, M.M. FGF23 regulates Wnt/β-catenin signaling-mediated osteoarthritis in mice overexpressing high-molecular-weight FGF2. Endocrinology 2018, 159, 2386–2396. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, A.; Guibert, M.; Cailotto, F.; Gasser, A.; Presle, N.; Mainard, D.; Netter, P.; Kempf, H.; Jouzeau, J.Y.; Reboul, P. Fibroblast growth factor 23 drives MMP13 expression in human osteoarthritic chondrocytes in a Klotho-independent manner. Osteoarthr. Cartil. 2016, 24, 1961–1969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Insogna, K.L.; Rauch, F.; Kamenický, P.; Ito, N.; Kubota, T.; Nakamura, A.; Zhang, L.; Mealiffe, M.; San Martin, J.; Portale, A.A. Burosumab improved histomorphometric measures of osteomalacia in adults with X-linked Hypophosphatemia: A phase 3, single-arm, international trial. J. Bone Miner. Res. 2019, 34, 2183–2191. [Google Scholar] [CrossRef] [Green Version]
- Goetz, R.; Nakada, Y.; Hu, M.C.; Kurosu, H.; Wang, L.; Nakatani, T.; Shi, M.; Eliseenkova, A.V.; Razzaque, M.S.; Moe, O.W.; et al. Isolated C-terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23-FGFR-Klotho complex formation. Proc. Natl. Acad. Sci. USA 2010, 107, 407–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
| Skeletal manifestations |
| Short stature |
| Rickets in children |
| Osteomalacia in adults (less severe in females) |
| Osteoarthritis (common in the ankles, wrists, knees, feet, and sacroiliac joints) |
| Joint and bone pain (in adults) |
| Impaired mobility |
| Bowed legs (valgus or varus deformities) |
| Enthesophaty or calcification of tendons, ligaments, and joint capsules (in adults) |
| Premature cranial synostosis and increased antero-posterior head length |
| Dental abnormalities (abscesses, enamel and dentin defects) such as oral pain, delayed eruption, enlarged pulp chambers, and taurodontism of permanent molars. |
| Spinal cord stenosis |
| Hearing loss (in adults) |
| Renal disorders |
| Impaired renal tubular reabsorption of phosphate |
| Renal phosphate wasting |
| Biochemical abnormalities |
| Hypophosphatemia |
| Elevated circulating FGF23 concentrations |
| Low or normal levels of 1.25(OH)2D or calcitriol |
| Normal or slightly increased levels of serum PTH |
| Increased levels of circulating α-Klotho |
| Elevated levels of serum alkaline phosphatase |
| Normal calcemia |
| FGF23: Fibroblast growth factor 23; 1.25(OH)2D: 1.25-dihydroxyvitamin D; PTH: Parathyroid hormone. |
| Syndromes | Cause |
|---|---|
| XLH | PHEX mutations |
| ADHR | FGF23 mutations |
| ARHR1 | DMP1 mutations |
| ARHR2 | ENPP1 mutations |
| ARHR3 or Raine syndrome | FAM20C mutations |
| Osteoglophonic dysplasia * | FGFR1 mutations |
| Jansen-type metaphyseal chondrodysplasia | PTHRP1 mutations |
| Epidermal nevus syndrome | FGFR3 mutations |
| DAKGM | KL mutations |
| McCune–Albright syndrome | GNAS |
| Epidermal nevus syndrome |
| Regulators | Expression/Effect at the GP | X-Linked Hypophosphatemia | |
|---|---|---|---|
| Systemic regulators | |||
| GH | Resting and Proliferative zones | Normal/Low levels | |
| Serum Calcium | Hypertrophic | Normal levels | |
| Urine calcium | − | Low levels | |
| Circulating phosphate | − | Increased | |
| Urine phosphate | − | Increased | |
| 1.25(OH)2D3 | Proliferative and hypertrophic | Normal/Low levels | |
| PTH | Hypertrophic | Normal/high levels | |
| Local regulators | |||
| FGF23 | Prehypertrophic | Increased | |
| FGFR1 | Prehypertrophic and hypertrophic | Increased | |
| FGFR3 | Prehypertrophic and hypertrophic | Increased | |
| PTHrP | Prehypertrophic | Increased levels | |
| MEPE | Hypertrophic | Increased levels | |
| AQP1 | Hypertrophic | Decreased levels | |
| NKCC1 | Hypertrophic | Decreased levels | |
| Col10a1 | Hypertrophic | Increased expression in bone | |
| MMP13 | Hypertrophic | Increased levels | |
| MMP9 | Hypertrophic | Decreased levels | |
| VEGF | Hypertrophic | Decreased levels | |
| IGF1 | Hypertrophic | Decreased levels | |
| pERK1/2 | Proliferative and prehypertrophic | Increased levels | |
| ALP | Hypertrophic | Increased levels | |
| OCN | Hypertrophic | Decreased levels | |
| BSP | Hypertrophic | Decreased levels | |
| ON | Hypertrophic | Increased levels | |
| TRAP | Late hypertrophic | Increased levels | |
| CLCN7 | Hypertrophic | Increased levels | |
| ATPase H+ | − | Increased levels | |
| NHEDC2 | − | Increased levels |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fuente, R.; García-Bengoa, M.; Fernández-Iglesias, Á.; Gil-Peña, H.; Santos, F.; López, J.M. Cellular and Molecular Alterations Underlying Abnormal Bone Growth in X-Linked Hypophosphatemia. Int. J. Mol. Sci. 2022, 23, 934. https://doi.org/10.3390/ijms23020934
Fuente R, García-Bengoa M, Fernández-Iglesias Á, Gil-Peña H, Santos F, López JM. Cellular and Molecular Alterations Underlying Abnormal Bone Growth in X-Linked Hypophosphatemia. International Journal of Molecular Sciences. 2022; 23(2):934. https://doi.org/10.3390/ijms23020934
Chicago/Turabian StyleFuente, Rocío, María García-Bengoa, Ángela Fernández-Iglesias, Helena Gil-Peña, Fernando Santos, and José Manuel López. 2022. "Cellular and Molecular Alterations Underlying Abnormal Bone Growth in X-Linked Hypophosphatemia" International Journal of Molecular Sciences 23, no. 2: 934. https://doi.org/10.3390/ijms23020934
APA StyleFuente, R., García-Bengoa, M., Fernández-Iglesias, Á., Gil-Peña, H., Santos, F., & López, J. M. (2022). Cellular and Molecular Alterations Underlying Abnormal Bone Growth in X-Linked Hypophosphatemia. International Journal of Molecular Sciences, 23(2), 934. https://doi.org/10.3390/ijms23020934