
Ectodysplasin A1 Deficiency Leads to Osteopetrosis-like Changes in Bones of the Skull Associated with Diminished Osteoclastic Activity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Osteopetrosis-like Changes of Tabby Calvarial Bone
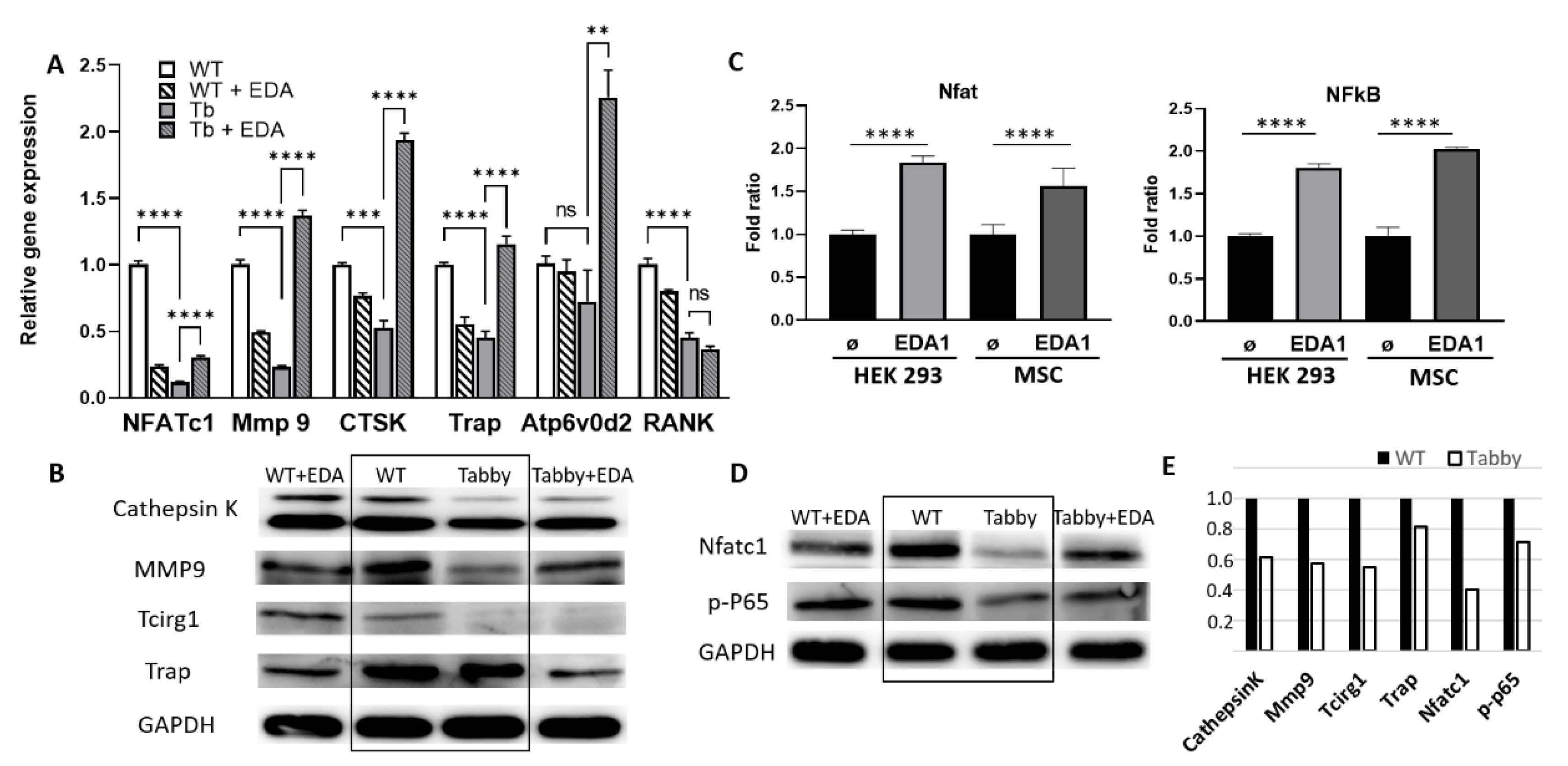
2.2. Diminished Nuclear NFATc1 and Bone-Resorbing Enzymes in Tabby-Derived Osteoclasts and Eda1-Deficient Calvariae
2.3. EDA1 Induces NF-kB and Nfatc1 Activation in Osteoclastic Differentiation
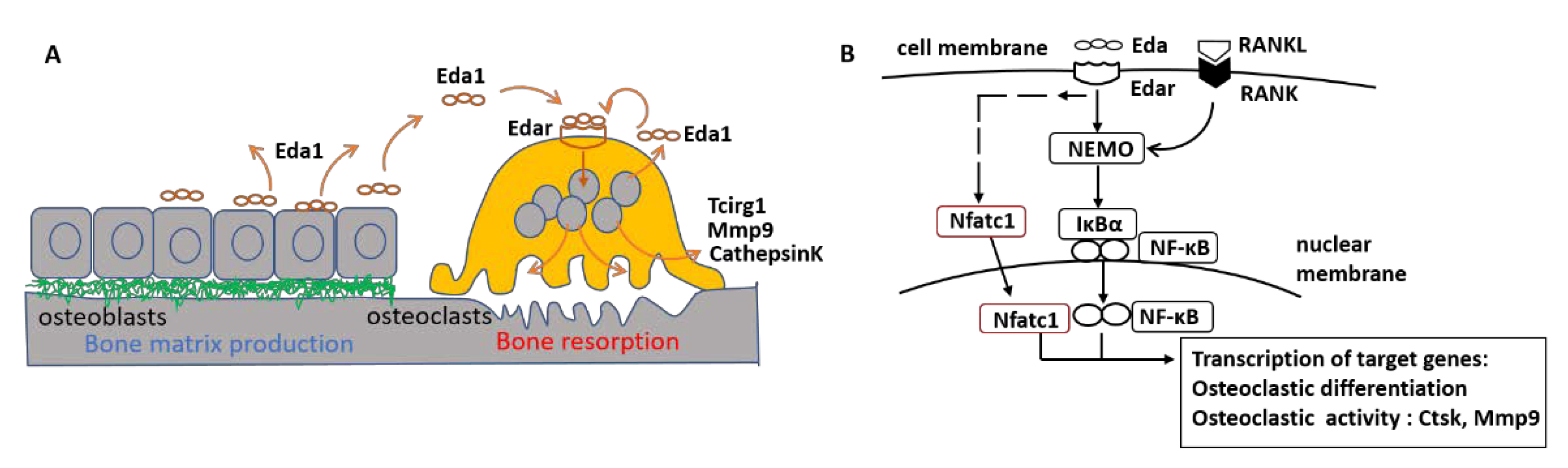
3. Discussion
4. Materials and Methods
4.1. Animal Model
4.2. Histological Assessments and Calvarial Bone Histomorphometry
4.3. In vitro Cell Culture and Osteoclast Differentiation
4.4. Immunostaining and Acquisition of Images
4.5. Quantitative Real-Time PCR
4.6. Western Blot
4.7. Luciferase Assays
4.8. Statistical Analyses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bayes, M.; Hartung, A.J.; Ezer, S.; Pispa, J.; Thesleff, I.; Srivastava, A.K.; Kere, J. The anhidrotic ectodermal dysplasia gene (EDA) undergoes alternative splicing and encodes ectodysplasin-A with deletion mutations in collagenous repeats. Hum. Mol. Genet. 1998, 7, 1661–1669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, M.; Wang, L.C.; Hymowitz, S.G.; Schilbach, S.; Lee, J.; Goddard, A.; de Vos, A.M.; Gao, W.Q.; Dixit, V.M. Two-amino acid molecular switch in an epithelial morphogen that regulates binding to two distinct receptors. Science 2000, 290, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P.; Street, S.L.; Gaide, O.; Hertig, S.; Tardivel, A.; Tschopp, J.; Runkel, L.; Alevizopoulos, K.; Ferguson, B.M.; Zonana, J. Mutations leading to X-linked hypohidrotic ectodermal dysplasia affect three major functional domains in the tumor necrosis factor family member ectodysplasin-A. J. Biol. Chem. 2001, 276, 18819–18827. [Google Scholar] [CrossRef] [Green Version]
- Hymowitz, S.G.; Compaan, D.M.; Yan, M.; Wallweber, H.J.; Dixit, V.M.; Starovasnik, M.A.; de Vos, A.M. The crystal structures of EDA-A1 and EDA-A2: Splice variants with distinct receptor specificity. Structure 2003, 11, 1513–1520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, A. Hypohidrotic ectodermal dysplasia. J. Med. Genet. 1987, 24, 659–663. [Google Scholar] [CrossRef] [Green Version]
- Kere, J.; Srivastava, A.K.; Montonen, O.; Zonana, J.; Thomas, N.; Ferguson, B.; Munoz, F.; Morgan, D.; Clarke, A.; Baybayan, P.; et al. X-linked anhidrotic (hypohidrotic) ectodermal dysplasia is caused by mutation in a novel transmembrane protein. Nat. Genet. 1996, 13, 409–416. [Google Scholar] [CrossRef]
- Monreal, A.W.; Zonana, J.; Ferguson, B. Identification of a new splice form of the EDA1 gene permits detection of nearly all X-linked hypohidrotic ectodermal dysplasia mutations. Am. J. Hum. Genet. 1998, 63, 380–389. [Google Scholar] [CrossRef] [Green Version]
- Headon, D.J.; Emmal, S.A.; Ferguson, B.M.; Tucker, A.S.; Justice, M.J.; Sharpe, P.T.; Zonana, J.; Overbeek, P.A. Gene defect in ectodermal dysplasia implicates a death domain adapter in development. Nature 2001, 414, 913–916. [Google Scholar] [CrossRef]
- Bluschke, G.; Nusken, K.D.; Schneider, H. Prevalence and prevention of severe complications of hypohidrotic ectodermal dysplasia in infancy. Early Hum. Dev. 2010, 86, 397–399. [Google Scholar] [CrossRef]
- Gaide, O.; Schneider, P. Permanent correction of an inherited ectodermal dysplasia with recombinant EDA. Nat. Med. 2003, 9, 614–618. [Google Scholar] [CrossRef]
- Casal, M.L.; Lewis, J.R.; Mauldin, E.A.; Tardivel, A.; Ingold, K.; Favre, M.; Paradies, F.; Demotz, S.; Gaide, O.; Schneider, P. Significant correction of disease after postnatal administration of recombinant ectodysplasin A in canine X-linked ectodermal dysplasia. Am. J. Hum. Genet. 2007, 81, 1050–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermes, K.; Schneider, P.; Krieg, P.; Dang, A.; Huttner, K.; Schneider, H. Prenatal therapy in developmental disorders: Drug targeting via intra-amniotic injection to treat X-linked hypohidrotic ectodermal dysplasia. J. Investig. Dermatol. 2014, 134, 2985–2987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korber, I.; Klein, O.D.; Morhart, P.; Faschingbauer, F.; Grange, D.K.; Clarke, A.; Bodemer, C.; Maitz, S.; Huttner, K.; Kirby, N.; et al. Safety and immunogenicity of Fc-EDA, a recombinant ectodysplasin A1 replacement protein, in human subjects. Br. J. Clin. Pharm. 2020, 86, 2063–2069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, H.; Faschingbauer, F.; Schuepbach-Mallepell, S.; Korber, I.; Wohlfart, S.; Dick, A.; Wahlbuhl, M.; Kowalczyk-Quintas, C.; Vigolo, M.; Kirby, N.; et al. Prenatal correction of X-Linked hypohidrotic ectodermal dysplasia. N. Engl. J. Med. 2018, 378, 1604–1610. [Google Scholar] [CrossRef]
- Wohlfart, S.; Meiller, R.; Hammersen, J.; Park, J.; Menzel-Severing, J.; Melichar, V.O.; Huttner, K.; Johnson, R.; Porte, F.; Schneider, H. Natural history of X-linked hypohidrotic ectodermal dysplasia: A 5-year follow-up study. Orphanet. J. Rare Dis. 2020, 15, 7. [Google Scholar] [CrossRef]
- DiGiovanna, J.J.; Priolo, M.; Itin, P. Approach towards a new classification for ectodermal dysplasias: Integration of the clinical and molecular knowledge. Am. J. Med. Genet. A 2009, 149A, 2068–2070. [Google Scholar] [CrossRef]
- Itin, P.H. Rationale and background as basis for a new classification of the ectodermal dysplasias. Am. J. Med. Genet. A 2009, 149A, 1973–1976. [Google Scholar] [CrossRef]
- Clauss, F.; Maniere, M.C.; Obry, F.; Waltmann, E.; Hadj-Rabia, S.; Bodemer, C.; Alembik, Y.; Lesot, H.; Schmittbuhl, M. Dento-craniofacial phenotypes and underlying molecular mechanisms in hypohidrotic ectodermal dysplasia (HED): A review. J. Dent. Res. 2008, 87, 1089–1099. [Google Scholar] [CrossRef]
- Srivastava, A.K.; Pispa, J.; Hartung, A.J.; Du, Y.; Ezer, S.; Jenks, T.; Shimada, T.; Pekkanen, M.; Mikkola, M.L.; Ko, M.S.; et al. The Tabby phenotype is caused by mutation in a mouse homologue of the EDA gene that reveals novel mouse and human exons and encodes a protein (ectodysplasin-A) with collagenous domains. Proc. Natl. Acad. Sci. USA 1997, 94, 13069–13074. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, B.M.; Brockdorff, N.; Formstone, E.; Ngyuen, T.; Kronmiller, J.E.; Zonana, J. Cloning of Tabby, the murine homolog of the human EDA gene: Evidence for a membrane-associated protein with a short collagenous domain. Hum. Mol. Genet. 1997, 6, 1589–1594. [Google Scholar] [CrossRef]
- Hill, N.L.; Laib, A.; Duncan, M.K. Mutation of the ectodysplasin-A gene results in bone defects in mice. J. Comp. Pathol. 2002, 126, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Montonen, O.; Ezer, S.; Saarialho-Kere, U.K.; Herva, R.; Karjalainen-Lindsberg, M.L.; Kaitila, I.; Schlessinger, D.; Srivastava, A.K.; Thesleff, I.; Kere, J. The gene defective in anhidrotic ectodermal dysplasia is expressed in the developing epithelium, neuroectoderm, thymus, and bone. J. Histochem. Cytochem. 1998, 46, 281–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iida, Y.; Hibiya, K.; Inohaya, K.; Kudo, A. Eda/Edar signaling guides fin ray formation with preceding osteoblast differentiation, as revealed by analyses of the medaka all-fin less mutant afl. Dev. Dyn. 2014, 243, 765–777. [Google Scholar] [CrossRef] [PubMed]
- Kossel, C.S.; Wahlbuhl, M.; Schuepbach-Mallepell, S.; Park, J.; Kowalczyk-Quintas, C.; Seeling, M.; von der Mark, K.; Schneider, P.; Schneider, H. Correction of vertebral bone development in Ectodysplasin A1-deficient mice by prenatal treatment with a replacement protein. Front. Genet. 2021, 12, 709736. [Google Scholar] [CrossRef]
- Bornert, F.; Choquet, P.; Gros, C.I.; Aubertin, G.; Perrin-Schmitt, F.; Clauss, F.; Lesot, H.; Constantinesco, A.; Schmittbuhl, M. Subtle morphological changes in the mandible of Tabby mice revealed by micro-CT imaging and elliptical Fourier quantification. Front. Physiol. 2011, 2, 15. [Google Scholar] [CrossRef] [Green Version]
- Smahi, A.; Courtois, G.; Rabia, S.H.; Doffinger, R.; Bodemer, C.; Munnich, A.; Casanova, J.L.; Israel, A. The NF-kappaB signalling pathway in human diseases: From incontinentia pigmenti to ectodermal dysplasias and immune-deficiency syndromes. Hum. Mol. Genet. 2002, 11, 2371–2375. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 Years of NF-kappaB: A blossoming of relevance to human pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef] [Green Version]
- Doffinger, R.; Smahi, A.; Bessia, C.; Geissmann, F.; Feinberg, J.; Durandy, A.; Bodemer, C.; Kenwrick, S.; Dupuis-Girod, S.; Blanche, S.; et al. X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-kappaB signaling. Nat. Genet. 2001, 27, 277–285. [Google Scholar] [CrossRef]
- Wright, J.T.; Fete, M.; Schneider, H.; Zinser, M.; Koster, M.I.; Clarke, A.J.; Hadj-Rabia, S.; Tadini, G.; Pagnan, N.; Visinoni, A.F.; et al. Ectodermal dysplasias: Classification and organization by phenotype, genotype and molecular pathway. Am. J. Med. Genet. A 2019, 179, 442–447. [Google Scholar] [CrossRef]
- Cui, C.Y.; Schlessinger, D. EDA signaling and skin appendage development. Cell Cycle 2006, 5, 2477–2483. [Google Scholar] [CrossRef]
- Koppinen, P.; Pispa, J.; Laurikkala, J.; Thesleff, I.; Mikkola, M.L. Signaling and subcellular localization of the TNF receptor Edar. Exp. Cell Res. 2001, 269, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Ullrich, R.; Tobin, D.J.; Lenhard, D.; Schneider, P.; Paus, R.; Scheidereit, C. NF-kappaB transmits Eda A1/EdaR signalling to activate Shh and cyclin D1 expression, and controls post-initiation hair placode down growth. Development 2006, 133, 1045–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pispa, J.; Pummila, M.; Barker, P.A.; Thesleff, I.; Mikkola, M.L. Edar and Troy signalling pathways act redundantly to regulate initiation of hair follicle development. Hum. Mol. Genet. 2008, 17, 3380–3391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franzoso, G.; Carlson, L.; Xing, L.; Poljak, L.; Shores, E.W.; Brown, K.D.; Leonardi, A.; Tran, T.; Boyce, B.F.; Siebenlist, U. Requirement for NF-kappaB in osteoclast and B-cell development. Genes Dev. 1997, 11, 3482–3496. [Google Scholar] [CrossRef] [Green Version]
- Asagiri, M.; Takayanagi, H. The molecular understanding of osteoclast differentiation. Bone 2007, 40, 251–264. [Google Scholar] [CrossRef]
- Lomaga, M.A.; Yeh, W.C.; Sarosi, I.; Duncan, G.S.; Furlonger, C.; Ho, A.; Morony, S.; Capparelli, C.; Van, G.; Kaufman, S.; et al. TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev. 1999, 13, 1015–1024. [Google Scholar] [CrossRef] [Green Version]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef]
- Teitelbaum, S.L. Bone resorption by osteoclasts. Science 2000, 289, 1504–1508. [Google Scholar] [CrossRef]
- Teitelbaum, S.L.; Ross, F.P. Genetic regulation of osteoclast development and function. Nat. Rev. Genet. 2003, 4, 638–649. [Google Scholar] [CrossRef]
- Takayanagi, H.; Kim, S.; Koga, T.; Nishina, H.; Isshiki, M.; Yoshida, H.; Saiura, A.; Isobe, M.; Yokochi, T.; Inoue, J.; et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev. Cell. 2002, 3, 889–901. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, N. Regulation of NFATc1 in Osteoclast Differentiation. J. Bone Metab. 2014, 21, 233–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, S.Y. Myricetin suppresses LPS-induced MMP expression in human gingival fibroblasts and inhibits osteoclastogenesis by downregulating NFATc1 in RANKL-induced RAW 264.7 cells. Arch. Oral Biol. 2012, 57, 1623–1632. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Kogawa, M.; Wada, S.; Takayanagi, H.; Tsujimoto, M.; Katayama, S.; Hisatake, K.; Nogi, Y. Essential role of p38 mitogen-activated protein kinase in cathepsin K gene expression during osteoclastogenesis through association of NFATc1 and PU.1. J. Biol. Chem. 2004, 279, 45969–45979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, I.; Kim, J.H.; Kim, K.; Jin, H.M.; Youn, B.U.; Kim, N. Regulatory mechanism of NFATc1 in RANKL-induced osteoclast activation. FEBS Lett. 2009, 583, 2435–2440. [Google Scholar] [CrossRef] [Green Version]
- Korn, E.D. Actin polymerization and its regulation by proteins from nonmuscle cells. Physiol. Rev. 1982, 62, 672–737. [Google Scholar] [CrossRef]
- Han, G.; Zuo, J.; Holliday, L.S. Specialized roles for actin in osteoclasts: Unanswered questions and therapeutic opportunities. Biomolecules 2019, 9, 17. [Google Scholar] [CrossRef] [Green Version]
- Taranta, A.; Migliaccio, S.; Recchia, I.; Caniglia, M.; Luciani, M.; De Rossi, G.; Dionisi-Vici, C.; Pinto, R.M.; Francalanci, P.; Boldrini, R.; et al. Genotype-phenotype relationship in human ATP6i-dependent autosomal recessive osteopetrosis. Am. J. Pathol. 2003, 162, 57–68. [Google Scholar] [CrossRef] [Green Version]
- Kornak, U.; Schulz, A.; Friedrich, W.; Uhlhaas, S.; Kremens, B.; Voit, T.; Hasan, C.; Bode, U.; Jentsch, T.J.; Kubisch, C. Mutations in the a3 subunit of the vacuolar H(+)-ATPase cause infantile malignant osteopetrosis. Hum. Mol. Genet. 2000, 9, 2059–2063. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Lin, L.; Yang, B.; Meng, Z.; Zhang, B. Knockdown of Tcirg1 inhibits large-osteoclast generation by down-regulating NFATc1 and IP3R2 expression. PLoS ONE 2020, 15, e0237354. [Google Scholar] [CrossRef]
- Sobacchi, C.; Abinun, M. Osteoclast-poor osteopetrosis. Bone 2022, 164, 116541. [Google Scholar] [CrossRef]
- Luchin, A.; Purdom, G.; Murphy, K.; Clark, M.Y.; Angel, N.; Cassady, A.I.; Hume, D.A.; Ostrowski, M.C. The microphthalmia transcription factor regulates expression of the tartrate-resistant acid phosphatase gene during terminal differentiation of osteoclasts. J. Bone Miner. Res. 2000, 15, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.Y.; Li, M.; Lin, Y.L. Mitf regulates osteoclastogenesis by modulating NFATc1 activity. Exp. Cell Res. 2014, 15, 32–43. [Google Scholar] [CrossRef] [Green Version]
- Walsh, N.C.; Cahill, M.; Carninci, P.; Kawai, J.; Okazaki, Y.; Hayashizaki, Y.; Hume, D.A.; Cassady, A.I. Multiple tissue-specific promoters control expression of the murine tartrate-resistant acid phosphatase gene. Gene 2003, 27, 111–123. [Google Scholar] [CrossRef]
- Jiang, Y.; Jahagirdar, B.N.; Reinhardt, R.L.; Schwartz, R.E.; Keene, C.D.; Ortiz-Gonzalez, X.R.; Reyes, M.; Lenvik, T.; Lund, T.; Blackstad, M.; et al. Pluripotency of mesenchymal stem cells derived from adult marrow. Nature 2002, 418, 41–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Gelse, K.; Frank, S.; von der Mark, K.; Aigner, T.; Schneider, H. Transgene-activated mesenchymal cells for articular cartilage repair: A comparison of primary bone marrow-, perichondrium/periosteum- and fat-derived cells. J. Gene Med. 2006, 8, 112–125. [Google Scholar] [CrossRef]
- Kowalczyk-Quintas, C.; Willen, L.; Dang, A.T.; Sarrasin, H.; Tardivel, A.; Hermes, K.; Schneider, H.; Gaide, O.; Donze, O.; Kirby, N.; et al. Generation and characterization of function-blocking anti-ectodysplasin A (EDA) monoclonal antibodies that induce ectodermal dysplasia. J. Biol. Chem. 2014, 289, 4273–4285. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schweikl, C.; Maier-Wohlfart, S.; Schneider, H.; Park, J. Ectodysplasin A1 Deficiency Leads to Osteopetrosis-like Changes in Bones of the Skull Associated with Diminished Osteoclastic Activity. Int. J. Mol. Sci. 2022, 23, 12189. https://doi.org/10.3390/ijms232012189
Schweikl C, Maier-Wohlfart S, Schneider H, Park J. Ectodysplasin A1 Deficiency Leads to Osteopetrosis-like Changes in Bones of the Skull Associated with Diminished Osteoclastic Activity. International Journal of Molecular Sciences. 2022; 23(20):12189. https://doi.org/10.3390/ijms232012189
Chicago/Turabian StyleSchweikl, Christine, Sigrun Maier-Wohlfart, Holm Schneider, and Jung Park. 2022. "Ectodysplasin A1 Deficiency Leads to Osteopetrosis-like Changes in Bones of the Skull Associated with Diminished Osteoclastic Activity" International Journal of Molecular Sciences 23, no. 20: 12189. https://doi.org/10.3390/ijms232012189