Abstract
Protein complexes termed inflammasomes ensure tissue protection from pathogenic and sterile stressors by induction of inflammation. This is mediated by different caspase-1-induced downstream pathways, including activation of the pro-inflammatory cytokines proIL-1β and -18, induction of a lytic type of cell death, and regulation of the release of other pro-inflammatory molecules. Aberrant inflammasome activation underlies the pathology of numerous (auto)inflammatory diseases. Furthermore, inflammasomes support or suppress tumor development in a complex cell-type- and stage-dependent manner. In human keratinocytes and skin, NLRP1 is the central inflammasome sensor activated by cellular perturbation induced, for example, by UVB radiation. UVB represents the main inducer of skin cancer, which is the most common type of malignancy in humans. Recent evidence demonstrates that activation of NLRP1 in human skin supports the development of cutaneous squamous cell carcinomas (cSCCs) by inducing skin inflammation. In contrast, the NLRP1 inflammasome pathway is restrained in established cSCCs, suggesting that, at this stage, the protein complex has a tumor suppressor role. A better understanding of the complex functions of NLRP1 in the development of cSCCs and in general of inflammasomes in cancer might pave the way for novel strategies for cancer prevention and therapy. These strategies might include stage-specific modulation of inflammasome activation or its downstream pathways by mono- or combination therapy.
1. Introduction
Inflammation represents a tissue response induced by many different stress factors [,]. These include PAMPs (pathogen-associated molecular patterns), highly conserved molecules derived from tissue invading bacteria or viruses, and DAMPs (damage-associated molecular patterns), molecules released by impaired endogenous cells after, for example, trauma or injury. PRRs (pattern recognition receptors) are localized on the surface or inside of the cell and ensure the detection of PAMPs and DAMPs. Immune cells are central for inflammation and are attracted and activated by chemokines and cytokines, which are released by immune or other tissue resident cells upon PRR activation. In most cases, acute inflammation is highly beneficial because it is strictly required for efficient defense against pathogens, repair after injury, or induction of anti-tumor responses. However, when chronic, inflammation can also be detrimental, underlying the pathology of inflammatory diseases and tumor development. There is increasing evidence that inflammation is able to support or inhibit cancer development by acting at all stages of tumorigenesis, namely, tumor initiation, promotion, progression, and metastasis [,]. It has been estimated that about 25% of all cancers of epithelial origin might be caused by chronic inflammation, induced by viral or bacterial infection, or associated with other pro-inflammatory conditions []. In contrast, there is increasing evidence for efficient T lymphocyte-based immunotherapy against cancer []. Indeed, tumor cells proliferate with other cell types in the TME (tumor microenvironment), including mesenchymal cells, such as CAFs (cancer-associated fibroblasts), and different types of immune cells. These immune cells can strongly support cancer development by providing factors for tumor cell proliferation or suppression of anti-tumor immunity []. On the other hand, they can also induce anti-tumor immune responses, able to eradicate tumor cells and cure the patient []. Therefore, understanding the different, in part opposing, roles of inflammation and immune cells in the cancer development of the individual patient provides the potential for the establishment of efficient treatment options [].
The IL (interleukin)-1 family consists of 11 members (IL-1β, IL-1α, IL-1Ra, IL-18, IL-36Ra, IL-36α, IL-36β, IL-36γ, IL-37, IL-38, and IL-33) with either pro- or anti-inflammatory activity. IL-1β is a highly conserved, pleiotropic, and very potent pro-inflammatory cytokine that can induce inflammation and, if the dose is high enough, even a septic shock []. IL-1β exerts its biological activity by binding to IL-1RI (IL-1 receptor type I), which is ubiquitously expressed. IL-1 activity is controlled by IL-1Ra (IL-1 receptor antagonist), a secreted protein that binds to IL-1RI, but cannot activate the downstream signaling pathway. IL-1 plays important roles in the induction of inflammation, inflammatory diseases, as well as cancer [,,]. IL-1β is initially synthesized as an inactive 31 kDa precursor (proIL-1β), which cannot bind and activate IL-1RI. The cysteine protease caspase-1 is the principal activator of proIL-1β, cleaving the pro sequence off, and thereby generating the active 17 kDa IL-1β []. In contrast, proIL-1α neither is a substrate for caspase-1 nor requires proteolytic processing for binding and activation of IL-1RI. Furthermore, caspase-1 is the main activator of proIL-18, which binds and stimulates in its mature form IL-18Rα/β []. Unlike IL-1, proIL-18 is ubiquitously expressed and involved in the induction of IFN-γ expression [].
2. Inflammasomes
Caspase-1 is expressed as an enzymatically inactive precursor molecule (pro-caspase-1) and its activation occurs upon assembly of inflammasomes []. Inflammasomes are multiprotein complexes that contain (i) a sensor protein, such as NLRP1 (NLR (NOD-like receptor) family pyrin domain containing 1), NLRP3, NLRC4 (NLR family CARD (caspase activation recruitment domain) domain containing 4), AIM2 (absent in melanoma 2), or pyrin, which names the corresponding type of inflammasome; (ii) the adaptor protein ASC (apoptosis-associated speck-like protein containing a CARD); and (iii) the effector protein pro-caspase-1 (Figure 1). Inflammasomes have been mainly characterized in immune cells, but are also expressed by keratinocytes [,]. They are activated by several different sensor-specific stressors. Once activated, the sensor induces the formation of ASC oligomers, termed ASC specks []. Then, pro-caspase-1 is recruited and activated by proximity-induced dimerization []. All interactions between the inflammasome sensor, ASC, and pro-caspase-1 are homotypic based on the death domain fold, either the CARD (in ASC, pro-caspase-1, NLRP1, and NLRC4) or the pyrin domain (in ASC, NLRP3, AIM2, and pyrin) []. Inflammasomes play key roles in innate immunity protecting from several pathogens []. However, their chronic activation, particularly of NLRP3, also underlies numerous common inflammatory diseases, ranging from Alzheimer’s disease, atherosclerosis, and diabetes to rheumatoid arthritis (for cancer, please see 3) [,]. As NLRP3 seems to be dispensable for immunity, its targeting represents a promising strategy for the treatment of numerous patients suffering from NLRP3-mediated diseases [].
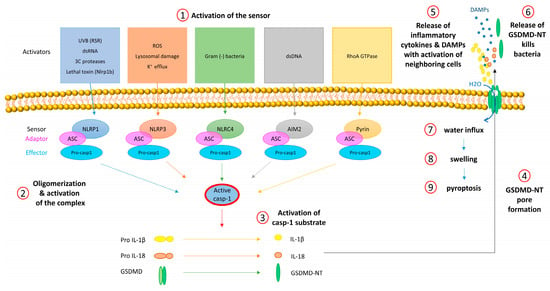
Figure 1.
Activation of inflammasomes and downstream mechanisms. (1) Each inflammasome sensor is activated by specific stressors. (2) Activation is associated with oligomerization of the sensor and of the adaptor protein ASC (ASC speck formation) followed by proteolytic self-activation of pro-caspase-1. (3) Active caspase-1 cleaves and thereby activates the pro-inflammatory cytokines proIL-1β and -18 and GSDMD. (4) GSDM-NT oligomerizes and forms pores in the cell membrane. (5) Via GSDMD pores, IL-1β, IL-18, and other molecules are released and induce inflammation by activation of immune and other neighboring cells. (6) Released GSDMD-NT inserts into the membrane of pathogens and kills them. (7) Through GSDMD-NT pores, water enters the cell (8) causing swelling and (9) rupture of the cell. This lytic type of cell death is termed pyroptosis and supports inflammation.
In most cases, inflammasome activation requires a preceding priming step for transcriptional induction of expression of proIL-1β, NLRP3, or AIM2 expression and posttranslational modifications of NLRP3 [,,]. This is achieved by LPS-induced TLR (toll-like receptor) 4 activation or stimulation of cells with TNFα, IL-1 itself, or IFN-γ.
Inflammasome activation is often equated with proIL-1β activation. Indeed, owing to its fundamental role in inflammation, inflammatory diseases, and cancer (please, see in 1), proIL-1β is the most important substrate of caspase-1. However, proIL-18 also plays crucial roles in immunity, particularly in the intestine, where it seems to be more important for repair than IL-1 [,,]. Importantly, proIL-18 is not only activated by caspase-1, but also by other proteases [].
Inflammasome activation in immune cells leads to pyroptosis, a lytic form of cell death that supports inflammation []. In 2015, GSDMD (gasdermin D) was identified as a substrate of caspase-1 and the inflammatory caspases -4 and -5 (and caspase-11 in mice) [,,]. Caspase-1 activates GSDMD upon cleavage, inducing oligomerization of its aminoterminal (NT) fragment. Then, the GSDMD-NT oligomer inserts into the cell membrane (and into mitochondrial membranes), creating pores [,,]. Consequently, water molecules can enter the cell, causing its rupture. In this way, pyroptosis induced by GSDMD terminates inflammasome activation. In addition, pyroptotic cells release proinflammatory molecules, including DAMPs, which activate neighboring cells []. Moreover, GSDMD-NT can also kill bacteria by membrane insertion, either intracellularly or after GSDMD-NT release by caspase-1 activating cells [].
Most proteins with extracellular function possess a signal peptide for secretion by the canonical ER (endoplasmic reticulum)/Golgi pathway. However, IL-1α, -β, -18, and other members of the IL-1 family lack such a signal peptide []. They are either passively released upon cell lysis and pyroptosis (for instance, IL-1α and IL-33, so-called alarmins []) or by different, partially poorly understood pathways, collectively termed unconventional protein secretion [,]. Several leaderless proteins, including IL-1α, -β, and -18, are released upon pro-caspase-1 activation by living cells by GSDMD-NT pores [,,]. Therefore, pyroptosis is not a prerequisite for IL-1 release, but the cytokine can be additionally unleashed from different living and non-pyroptotic cell types after GSDMD activation [,,,,]. Interestingly, GSDMD pores are dynamically regulated and can be repaired [,]. Furthermore, GSDMD is not essential for pyroptosis and IL-1β release because other GSDM family members can compensate for its role [,,].
3. Inflammasomes and Cancer
In general, chronic inflammation is believed to support all stages of malignant transformation and represents a hallmark of cancer development [,]. In contrast, acute inflammation can induce anti-tumor immunity, causing tumor regression [,]. Owing to their central roles in inducing inflammation, inflammasomes are generally considered to be tumor promoters [,,,,]. However, this depends on the type of cancer, the type(s) of the activated inflammasome(s) in a given cancer, the cell type(s) where the inflammasome(s) is/are activated, and the time point(s) of activation.
Polymorphisms of inflammasome genes are associated with different types of cancer [,,]. For example, variants of AIM2 contribute to the development of cancer of the small intestine and CRC (colorectal cancer) [,]. Furthermore, single nucleotide polymorphisms (SNPs) of NLRP3 are associated with Crohn’s disease, a risk factor for CRC, and a gain-of-function mutation results in poorer survival of patients suffering from CRC and increases the risk of developing melanoma [,]. In addition, polymorphisms of NLRP3, proIL-1β, caspase-1, and IL-1RN are associated with gastric cancer after Helicobacter pylori infection [,]. In mice, gastric inflammation and, subsequently, gastric cancer can be induced by overexpression of IL-1β [].
ProIL-1β and IL-1Ra variants are associated with non-small-cell lung cancer as well [,,,], and a recent study confirmed the role of IL-1 in the development of lung cancer [,]. The Canakinumab anti-inflammatory thrombosis outcome study (CANTOS) aimed to determine whether patients suffering from coronary artery disease profit from canakinumab, a human neutralizing IL-1β antibody []. Surprisingly, the successful study also revealed protection from lung cancer [].
IL-1β production in tumors, either by tumor cells themselves or by stromal cells, is associated with a worse prognosis [,,,,]. At the molecular and cellular level, IL-1β supports tumor development by different mechanisms. The cytokine directly supports tumor cell proliferation and, consequently, tumor growth []. Furthermore, IL-1β recruits MDSCs (myeloid-derived suppressor cells) to the TME and activates them []. This heterogeneous population of immature myeloid cells plays a key role in the TME by shifting it to the immunosuppressive side upon suppression of NK (natural killer) cells [,] and induction of Treg (regulatory T) cells []. Production of IL-1β for MDSCs’ attraction is NLRP3-driven [] and can induce immunosuppressive CD4+ T cell polarization [], whereas the roles of other types of inflammasomes, such as AIM2, are less well described []. It seems that the effects of IL-1β on the inhibition of anti-tumor responses are associated with its chronic activation by inflammasomes [].
IL-1β, together with IL-18, can also induce epithelial cells to lose their polarity and adherence and to acquire a migratory and mesenchymal cell-like phenotype, a process termed EMT (epithelial-mesenchymal transition) []. This is regulated by transcription factors, including SNAIL, and the downregulation of E-cadherin, required for tight junctions. Specifically, IL-1β induces SNAIL and suppresses E-cadherin expression in gastric cancer cells [] and IL-18 downregulates claudins, which are also tight junction proteins, thereby enhancing breast cancer cell migration []. Furthermore, IL-1β enhances the invasiveness of breast ductal cancer cells [] through induction of MMP9 (matrix metalloproteinase 9) expression, mediated by the transcription factor AP-1 (activator protein 1).
IL-1, by inducing VEGF (vascular endothelial growth factor) expression, also has a role in angiogenesis [,,,]. This process is required for small and localized tumors to progress, enlarge, and metastasize [,]. IL-1α promotes angiogenesis by induction of VEGF expression and, subsequently, by the VEGFR2 pathway in mice []. In a melanoma model, IL-1β expressed by myeloid cells and other pro-inflammatory cytokines induce VEGF expression in endothelial cells, creating a microenvironment favorable for early tumor development []. Furthermore, IL-1 alone or via VEGF also regulates later stages of tumor progression, including metastasis [,,].
Currently, several trials are ongoing, studying the potential of canakinumab and anakinra (recombinant IL-1Ra) in mono- and combination therapy for the treatment of patients suffering from different types of cancer [] and reviewed in [,]).
IL-1α also represents a target for cancer therapy []. Bermekimap, a human IL-1α neutralizing antibody, was tested in patients suffering from different types of cancer, including advanced colorectal cancer, and reduced cancer-associated cachexia [,,].
By the suppression of the tumoricidal function of NK cells through the induction of expression of PD1 (programmed cell death 1), IL-18 can also contribute to tumorigenesis [].
Pro-tumorigenic roles of IL-1 [,] and inflammasomes [] were also demonstrated in several studies with mice. In a murine model of breast cancer, an anti-PD-1 and anti-IL-1β treatment revealed synergistic effects for enhancing anti-tumor immunity [].
A comparison of chemical-induced tumor incidence in mice lacking expression of IL-1RI ligands revealed that IL-1Ra expression strongly protects from tumors, IL-1α had a weak protective effect, whereas IL-1β strongly supports tumor development []. Similarly, the ablation of NLRP3 expression in mice protects from carcinogenesis and metastasis []. In contrast, in a murine tumor model, IL-1β injection resulted in tumor regression [], demonstrating that IL-1 can also have anti-tumor effects, for example, via induction of Th1/Th17 responses [,]. Similarly, the inflammasome pathway seems to have a protective role in murine colitis-associated cancer models [,]. In DSS (dextran sulfate sodium)- and AOM (azoxymethane)-induced colitis models, ablation of NLRP3 expression increases the number of intestinal tumors [,,]. At least in these models, IL-18 rather than IL-1 seems to have a key protective role, as it supports the repair of the colonic epithelium after injury [,,]. Experiments with mice lacking ASC, caspase-1, and IL-18 expression suggest that IL-18 is required for normal intestinal microbiota []. Furthermore, the NLRC4 inflammasome contributes to inflammation-induced colon cancer [].
Other members of the IL-1 family can have roles in tumorigenesis, too. It is believed that IL-33 is biologically active in its pro-form [], although more potent when cleaved [], and has pro-tumorigenic activity [,,], particularly at early stages of cancer development [,]. ProIL-36, on the other hand, requires proteolytic activation by different proteases in order to be active and is associated with the inhibition of tumor development [,].
Finally, inflammasome proteins have, at least in part, inflammasome-independent roles relevant for cancer development, adding another level of complexity. This is especially relevant for ASC. ASC was originally identified as TMS1 (target of methylation-induced silencing-1), whose expression is suppressed in different types of cancer [,]. ASC is not only essential for inflammasome activation, but can also interact with the pro-apoptotic Bcl-2 family member Bax, translocates to mitochondria, and induces apoptosis via the release of cytochrome C []. Therefore, ASC is not only an inflammasome adaptor, but also a pro-apoptotic tumor suppressor, independent of the inflammasome pathway []. Therefore, it might be difficult to estimate which pathway is more relevant when ASC expression is silenced in cancer.
4. The NLRP1 Inflammasome
When NLRP1 was cloned for the first time, it was believed that the protein played a role in apoptosis [,]. Later, NLRP1 was the first inflammasome sensor identified []. Although originally characterized in the monocytic cell line THP-1 [], NLRP1 is expressed at particularly high levels in epithelial cells, such as epidermal keratinocytes, and in the brain [].
The NLRP1 protein consists of 1473 amino acids and five different domains []: an aminoterminal PYD (pyrin domain), followed by a NACHT domain, six leucine-rich repeats (LRRs), a FIIND (function-to-find domain), and a carboxyterminal CARD (Figure 2). The FIIND, consisting of ZU5 (ZO-1 and UNC5) and UPA (UNC5, PIDD, and Ankyrins) subdomains, is a constitutive proteolytic self-activation domain with cleavage occurring at Phe1212 at the border between ZU5 and UPA [,]. The carboxyterminal part (NLRP1-CT) represents the effector fragment, but remains inhibited upon interaction with the aminoterminal inhibitory fragment (NLRP1-NT). In most cases, activation of NLRP1-CT is induced by proteasomal degradation of NLRP1-NT [].
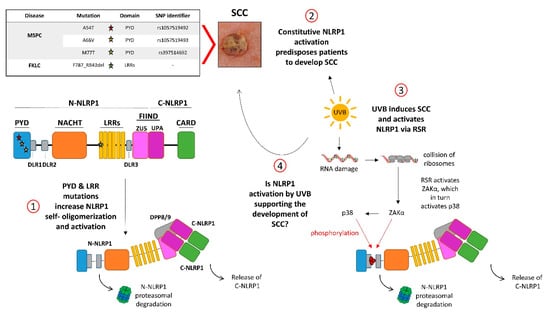
Figure 2.
NLRP1, UVB, and skin cancer. (1) NLRP1 is a five-domain self-processing protein. Self-processing occurs in the FIIND (function-to-find domain) between the ZU5 and UPA subdomains, generating the inhibitory N-NLRP1 and the effector C-NLRP1. Germline gain-of-function mutations of NLRP1 (stars) cause the inflammatory skin syndromes MSPC (multiple self-healing palmoplantar carcinoma) and FKLC (familial keratosis lichenoides chronica), predisposing patients to develop SCCs (2). These mutations induce self-oligomerization and -activation of NLRP1 by release from C-NLRP1 from the dipeptidyl peptidases DPP8/9 and the inhibitory N-NLPR1 (1). (3) The UVB spectrum of the sunlight activates NLRP1 via the ribotoxic stress response (RSR) upon sensing of collision of ribosomes by the kinase ZAKα. Then, ZAKα phosphorylates and activates the stress-induced kinase p38, and both enzymes phosphorylate N-NLRP1 in DLR1 (disordered linker region 1), causing its proteasomal degradation and release of C-NLRP1. (4) It is tempting to speculate that NLRP1 activation either by UVB or by MSPC-causing mutations induces the development of SCCs by overlapping mechanisms.
NLRP1 is the central inflammasome sensor in human skin and is expressed by human keratinocytes []. In addition, human keratinocytes can express and activate the AIM2 inflammasome, which might underlie skin inflammation in psoriasis, as well as the NLRP3 inflammasome [,]. Human keratinocytes in vitro express all NLRP1 inflammasome proteins, including proIL-1α, -β, and -18 [], and irradiation of the cells with a physiological dose of UVB induces NLRP1 inflammasome activation [], which is believed to underlie the induction of sunburn []. UVB-induced NLRP1 activation requires the activity of the stress-induced protein kinases p38 and JNK []. Recently, it was shown that this is induced by the kinase ZAKα (leucine-zipper and sterile-alpha motif kinase) [,] upon activation of the ribotoxic stress response (RSR) pathway [] (Figure 2). The RSR is induced by the collision of ribosomes upon UVB radiation and sensed by ZAKα binding directly to the ribosomes. Then, ZAKα is activated, inducing phosphorylation of p38 and JNK. Finally, activated ZAKα and p38 directly activate NLRP1 by phosphorylation [,]. In non-stressed cells, NLRP1 is inhibited upon binding to DPP8 (dipeptidyl peptidase 8) and 9, partially via interaction of the aminoterminus of NLRP1-CT with the active site of the peptidases [,]. Consequently, treatment of cells with the anti-cancer drug and DPP8/9 active site inhibitor talabostat (Val-boroPro, PT-100) [] induces NLRP1 activation in human keratinocytes and the downstream secretion of high levels of IL-1β [,]. It is not yet known whether long-term treatment of patients with talabostat induces SCCs, because the drug is not yet approved for the treatment of solid cancers (clinical trial NCT04171219 is ongoing). Furthermore, human NLRP1 can be activated by dsRNA [] and viral 3C proteases [,]. Both stimuli result in the degradation of NLRP1-NT and the release of the effector NLRP1-CT [].
A similar mechanism of activation was identified previously for murine Nlrp1b. In contrast to humans, three different Nlrp1 paralogues are expressed in mice, with Nlrp1b possessing the strongest homology to human NLRP1 []. Upon infection with Bacillus anthracis, Nlrp1b but not human NLRP1 is proteolytically activated in the aminoterminal region by the lethal factor, a protease of anthrax lethal toxin [,]. Then, the shortened aminoterminal fragment of Nlrp1b is ubiquitinated and degraded by the proteasome, finally causing Nlrp1b activation [,]. This mechanism is termed functional degradation [].
However, the roles of human NLRP1 are only poorly conserved between humans and mice, demonstrating that the NLRP1 is a relatively young evolutionary pathway []. Mechanistically, only human NLRP1 but not murine Nlrp1b is activated by dsRNA, ORF45, UVB, and the ribotoxic stress response [,,,]. Talabostat also activates Nlrp1b in murine immune cells; however, this occurs independently of ASC expression and induces mainly pyroptosis with very low levels of secreted IL-1β [,]. Furthermore, expression of Nlrp1b in murine keratinocytes is very low and, although sunburn in mice is IL-1β- and caspase-1-dependent, this is not mediated by keratinocytes, but most likely by a currently unknown immune cell type [].
Single nucleotide polymorphisms (SNPs) of the NLRP1 gene are associated with different (auto)inflammatory diseases, which mainly affect the skin, including vitiligo, Addison’s disease, and NAIAD (NLRP1-associated autoinflammation with arthritis and dyskeratosis) [,,,]. Furthermore, germline gain-of-function mutations of NLRP1 cause two rare inflammatory skin diseases, MSPC (multiple self-healing palmoplantar carcinoma) and FKLC (familial keratosis lichenoides chronica) []. These discoveries demonstrate that NLRP1 plays a particularly important role in skin inflammation. Most importantly, patients with these mutations are predisposed to develop cutaneous SCCs, demonstrating a link between NLRP1-induced inflammation and cancer development in human skin [] (Figure 2).
5. Inflammasomes in Skin Cancer and NLRP1 in Cutaneous SCCs
Cutaneous SCCs and BCCs (basal cell carcinomas) originate from keratinocytes and represent the main types (SCC: 20–30%, BCC: 70–80%) of NMSC (non-melanoma skin cancer), the most prevalent cancer worldwide with further increasing incidence rates []. Nearly 90% of NMSC is caused by exposure to UV radiation, either from sunlight exposure or from tanning beds []. Indeed, UVA and particularly UVB can cause skin cancer by inducing DNA damage, inflammation, and immune suppression []. In addition to UV, immunosuppression, infection by human papillomavirus (HPV), light skin, old age, exposure to organic chemicals and ionizing radiation, and genetic predisposition represent other risk factors [,]. Even though invasion and metastasis are rare events in SCC and BCC patients (for SCCs, about 5%, and for BCCs, 0.0028–0.55%), the high number of patients suffering from them creates a significant burden for the public health system [,,]. Excisional surgery is the most efficient gold standard therapy, but it also represents a frequent cosmetic issue, because BCCs and SCCs develop mainly on the sun-exposed body surface, such as the face.
It is believed that short-term activation of NLRP1 in keratinocytes by UVB with the subsequent release of IL-1 underlies sunburn in humans [,,]. In contrast, chronic NLRP1 activation is responsible for persistent inflammation, which can be considered as tumor promoter in the skin [,], which might be mediated by inflammation-associated ROS and IL-1β (Figure 2). Consistently, patients with germline gain-of-function mutations of NLRP1 suffer from chronic inflammation of the skin and have a high risk of developing cutaneous SCCs []. However, the expressions of ASC [], NLRP1, pro-caspase-1, and proIL-1β are suppressed in established human cutaneous SCC tumors and cell lines, most likely by the promoter methylation []. Silencing of ASC expression has been demonstrated in several types of cancer [,,]. It is not known whether SCC cells profit from ASC suppression owing to the pro-apoptotic function of the protein or to its essential role in inflammasome activation. However, reduced expression of the other inflammasome components suggests that inflammasome suppression could have a role in cancer progression and is more than just a bystander effect []. Furthermore, expression of AIM2 is increased in human SCCs [] and SCC cell lines [] and supports the growth and invasion of cutaneous carcinomas [].
The NLRP3 inflammasome contributes to the development of other types of human carcinomas []. In oropharyngeal SCCs, expression of NLRP3, ASC, caspase-1, and proIL-1β/-18 is increased, suggesting a role in tumor development []. In related head and neck SCCs, the NLRP3 inflammasome supports tumorigenesis, survival, and invasiveness [,]. Furthermore, NLRP3 supports the resistance of oral SCCs to 5-fluorouracil in vivo and in vitro []. NLRP3 expression is also increased in human BCCs [] and SCC cell lines [].
Evidence for the roles of inflammasomes in skin cancer development also comes from mouse models. Experiments based on chemically induced skin carcinogenesis revealed an important role for IL-1α derived from keratinocytes/tumor cells []. IL-1α induces NF-κB activation in an autocrine manner, which in turn prevents the expression of differentiation markers. Furthermore, IL-1α signaling is required for pro-inflammatory gene expression []. It is noteworthy that, in contrast to human keratinocytes, which express proIL-1α and -β, murine keratinocytes express mainly proIL-1α []. In the DMBA/TPA model of skin carcinogenesis, expression of IL-1RI, caspase-1, NLRP3, and ASC—the latter in myeloid cells—supports the incidence of papillomas [,]. However, specific deletion of ASC expression in keratinocytes results in more skin lesions, most likely due to an additional pro-apoptotic role of ASC independent of the inflammasome pathway in keratinocytes [,]. Using the same model, Gasparoto et al. reported increased papilloma incidence and volume in mice lacking expression of ASC and caspase-1, suggesting that the inflammasome is required for protective immune responses []. It might be that different housing conditions of mice are responsible for these contradictory results. Furthermore, the IL-1 family member IL-33 expressed by tumor cells supports skin cancer development in mice via an IL-33/TGF-β feedforward loop [].
UV is also the most important risk factor for melanoma, which derive from melanocytes and represent a very dangerous type of skin cancer []. SNPs of NLRP3 and particularly of NLRP1 are associated with the development of melanoma []. As in other types of human cancer, the expression of ASC is downregulated by promoter hypermethylation in melanoma []. However, depending on the stage, ASC has different roles in human melanoma: it acts as a tumor suppressor in primary tumors, but as a tumor promoter in metastatic melanoma, the latter via inflammasome-mediated IL-1β secretion []. Furthermore, it was suggested that expression and activation of the NLRP3 inflammasome in human melanoma cells correlate with malignancy and with spontaneous IL-1β secretion by late-stage melanoma []. Later, it was demonstrated that NLRP1 rather than NLRP3 promotes melanoma growth and suppresses apoptosis []. In mice, the NLRP3 inflammasome, IL-1β, and IL-18 support melanoma growth, migration, and metastasis [,,,].
6. Conclusions and Outlook
It is well accepted that inflammasomes play key roles in cancer development. However, IL-1 and other inflammasome effector pathways seem to have distinct and sometimes even opposite functions in carcinogenesis, dependent on the relevant types of inflammasomes and effector pathways, cell types that express and activate these inflammasomes, and the time points at which the activation occurs. In general, IL-1 and inflammasomes are considered tumor promoters, particularly at the early stages of cancer development []. Consequently, inhibition of IL-1 by anakinra or canakinumab [] or of the NLRP3 inflammasome [] might prevent cancer development or have therapeutic efficacy at the beginning of tumor development, as demonstrated by the CANTOS trial [,]. However, in most cases, cancer is diagnosed not until it causes discomfort, which occurs when cancer has already progressed. At this later stage, IL-1 and inflammasome are often considered to be tumor suppressors and several clinical studies for the treatment of cancer patients by either IL-1 mono- or combination therapy with, for example, immune checkpoint inhibitors [,,] are ongoing.
Although inflammasome activation is believed to be mainly associated with immune cells, the NLRP1 inflammasome is highly expressed by epidermal keratinocytes rather than by immune cells []. The fact that patients with germline gain-of-function mutations of NLRP1 are predisposed to develop cSCCs proves that the NLRP1 inflammasome represents a tumor promoter pathway in the development of NMSC []. Most likely, this is mediated by NLRP1 inflammasome activation in keratinocytes, IL-1-induced skin inflammation, and possibly IL-1-dependent immunosuppression. Anti-cancer immunity plays a key role in keeping the development of cSCCs under control, as immunosuppression of organ transplant recipients predisposes them to cSCCs []. Similarly, UVB induces inflammation (sunburn), which is likely mediated by NLRP1 inflammasome activation in keratinocytes, as well as the development of skin cancer [,,]. Furthermore, it is well known that UVB induces immunosuppression in the skin by different mechanisms []. Therefore, it is tempting to speculate that NLRP1-dependent IL-1 production by keratinocytes, when this occurs regularly or even chronically, contributes to keratinocyte proliferation and immunosuppression, supporting the early stages of cSCC development.
Nevertheless, the expression of not only ASC [] but of all inflammasome proteins is suppressed in established cSCC, suggesting a tumor suppressor role of the NLRP1 inflammasome pathway during cSCCs progression []. As the NLRP1 pathway is not conserved in the skin of mice, to prove this hypothesis and to elucidate the underlying molecular mechanisms in vivo will be difficult [].
In conclusion, the NLRP1 inflammasome seems to have opposing roles in the early versus late development of cSCCs. IL-1 blockade or NLRP1 inhibition in the skin might be protective and antagonize the early development of cSCCs. This option might be particularly useful for patients suffering from MSPC and FKLC when they start developing skin lesions as a consequence of NLRP1 activation. Indeed, as NLRP1 is expressed at particularly high levels by keratinocytes in human skin, its pharmacological targeting could be achieved by topical treatment, thus without major side effects.
In contrast, NLRP1 or IL-1RI activation could represent useful novel strategies for patients suffering from advanced cSCCs. Indeed, the TLR agonist AldaraTM, which is also an activator of NLRP1, is already used for patients suffering from BCCs [].
Funding
We would like to thank Wilhelm Sander-Stiftung, Bruno Bloch-Stiftung, Swiss Cancer Research (KFS-3940/5087); Hans Altschüler-Stiftung, Wolfermann-Nägeli-Stiftung, Swiss National Science Foundation (310030_197426); Monique Dornonville de la Cour-Stiftung; and Bangerter-Rhyner-Stiftung for financial support.
Acknowledgments
We are members of the research consortium SKINTEGRITY.CH. P.H., M.D.F., T.K., and M.S. are members of the Life Science Zurich Graduate School.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Rakoff-Nahoum, S.; Medzhitov, R. Toll-like receptors and cancer. Nat. Rev. Cancer 2009, 9, 57–63. [Google Scholar] [CrossRef] [PubMed]
- De Visser, K.E.; Eichten, A.; Coussens, L.M. Paradoxical roles of the immune system during cancer development. Nat. Rev. Cancer 2006, 6, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.R.; Mantovani, A. Cancer-related inflammation: Common themes and therapeutic opportunities. Semin. Cancer Biol. 2012, 22, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- Zheng, S.; Song, Q.; Zhang, P. Metabolic Modifications, Inflammation, and Cancer Immunotherapy. Front. Oncol. 2021, 11, 703681. [Google Scholar] [CrossRef]
- Dinarello, C.A. Immunological and Inflammatory Functions of the Interleukin-1 Family. Annu. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef]
- Gelfo, V.; Romaniello, D.; Mazzeschi, M.; Sgarzi, M.; Grilli, G.; Morselli, A.; Manzan, B.; Rihawi, K.; Lauriola, M. Roles of IL-1 in Cancer: From Tumor Progression to Resistance to Targeted Therapies. Int. J. Mol. Sci. 2020, 21, 6009. [Google Scholar] [CrossRef]
- Mantovani, A.; Dinarello, C.A.; Molgora, M.; Garlanda, C. Interleukin-1 and Related Cytokines in the Regulation of Inflammation and Immunity. Immunity 2019, 50, 778–795. [Google Scholar] [CrossRef]
- Afonina, I.S.; Müller, C.; Martin, S.J.; Beyaert, R. Proteolytic Processing of Interleukin-1 Family Cytokines: Variations on a Common Theme. Immunity 2015, 42, 991–1004. [Google Scholar] [CrossRef]
- Dinarello, C.A.; Novick, D.; Kim, S.; Kaplanski, G. Interleukin-18 and IL-18 Binding Protein. Front. Immunol. 2013, 4, 289. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef]
- Franklin, B.S.; Latz, E.; Schmidt, F.I. The intra- and extracellular functions of ASC specks. Immunol. Rev. 2018, 281, 74–87. [Google Scholar] [CrossRef]
- Boucher, D.; Monteleone, M.; Coll, R.C.; Chen, K.W.; Ross, C.M.; Teo, J.L.; Gomez, G.; Holley, C.L.; Bierschenk, D.; Stacey, K.; et al. Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J. Exp. Med. 2018, 215, 827–840. [Google Scholar] [CrossRef]
- Huoh, Y.; Hur, S. Death domain fold proteins in immune signaling and transcriptional regulation. FEBS J. 2021, 289, 4082–4097. [Google Scholar] [CrossRef]
- Hayward, J.; Mathur, A.; Ngo, C.; Man, S.M. Cytosolic Recognition of Microbes and Pathogens: Inflammasomes in Action. Microbiol. Mol. Biol. Rev. 2018, 82, e00015-18. [Google Scholar] [CrossRef]
- Guo, H.; Callaway, J.B.; Ting, J.P.-Y. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef]
- Mangan, M.S.J.; Olhava, E.J.; Roush, W.R.; Seidel, H.M.; Glick, G.D.; Latz, E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discov. 2018, 17, 688. [Google Scholar] [CrossRef]
- Patel, M.N.; Carroll, R.G.; Galván-Peña, S.; Mills, E.L.; Olden, R.; Triantafilou, M.; Wolf, A.I.; Bryant, C.E.; Triantafilou, K.; Masters, S.L. Inflammasome Priming in Sterile Inflammatory Disease. Trends Mol. Med. 2017, 23, 165–180. [Google Scholar] [CrossRef]
- McKee, C.M.; Coll, R.C. NLRP3 inflammasome priming: A riddle wrapped in a mystery inside an enigma. J. Leukoc. Biol. 2020, 108, 937–952. [Google Scholar] [CrossRef]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D.R.; Latz, E.; Fitzgerald, K.A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009, 458, 514–518. [Google Scholar] [CrossRef]
- Zaki, M.H.; Boyd, K.L.; Vogel, P.; Kastan, M.B.; Lamkanfi, M.; Kanneganti, T.D. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity 2010, 32, 379–391. [Google Scholar] [CrossRef]
- Zaki, H.; Lamkanfi, M.; Kanneganti, T.-D. The Nlrp3 inflammasome: Contributions to intestinal homeostasis. Trends Immunol. 2011, 32, 171–179. [Google Scholar] [CrossRef]
- Dupaul-Chicoine, J.; Yeretssian, G.; Doiron, K.; Bergstrom, K.S.; McIntire, C.R.; LeBlanc, P.M.; Meunier, C.; Turbide, C.; Gros, P.; Beauchemin, N.; et al. Control of Intestinal Homeostasis, Colitis, and Colitis-Associated Colorectal Cancer by the Inflammatory Caspases. Immunity 2010, 32, 367–378. [Google Scholar] [CrossRef]
- Bergsbaken, T.; Fink, S.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- He, W.T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.H.; Zhong, C.Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Pelegrín, P.; Shao, F. The gasdermins, a protein family executing cell death and inflammation. Nat. Rev. Immunol. 2020, 20, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xia, S.; Zhang, Z.; Wu, H.; Lieberman, J. Channelling inflammation: Gasdermins in physiology and disease. Nat. Rev. Drug Discov. 2021, 20, 384–405. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Man, S.M.; Karki, R.; Kanneganti, T.-D. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol. Rev. 2017, 277, 61–75. [Google Scholar] [CrossRef]
- Bertheloot, D.; Latz, E. HMGB1, IL-1α, IL-33 and S100 proteins: Dual-function alarmins. Cell. Mol. Immunol. 2017, 14, 43–64. [Google Scholar] [CrossRef]
- Nickel, W.; Rabouille, C. Mechanisms of regulated unconventional protein secretion. Nat. Rev. Mol. Cell Biol. 2009, 10, 148–155. [Google Scholar] [CrossRef]
- Rabouille, C.; Malhotra, V.; Nickel, W. Diversity in unconventional protein secretion. J. Cell Sci. 2012, 125, 5251–5255. [Google Scholar] [CrossRef]
- Keller, M.; Rüegg, A.; Werner, S.; Beer, H.-D. Active Caspase-1 Is a Regulator of Unconventional Protein Secretion. Cell 2008, 132, 818–831. [Google Scholar] [CrossRef]
- Gaidt, M.M.; Ebert, T.S.; Chauhan, D.; Schmidt, T.; Schmid-Burgk, J.L.; Rapino, F.; Robertson, A.A.; Cooper, M.A.; Graf, T.; Hornung, V. Human Monocytes Engage an Alternative Inflammasome Pathway. Immunity 2016, 44, 833–846. [Google Scholar] [CrossRef]
- Chen, K.W.; Groß, C.J.; Sotomayor, F.V.; Stacey, K.J.; Tschopp, J.; Sweet, M.J.; Schroder, K. The neutrophil NLRC4 inflammasome selectively promotes IL-1β maturation without pyroptosis during acute Salmonella challenge. Cell Rep. 2014, 8, 570–582. [Google Scholar] [CrossRef]
- Conos, S.A.; Lawlor, K.E.; Vaux, D.L.; Vince, J.E.; Lindqvist, L.M. Cell death is not essential for caspase-1-mediated interleukin-1β activation and secretion. Cell Death Differ. 2016, 23, 1827–1838. [Google Scholar] [CrossRef]
- Wolf, A.J.; Reyes, C.N.; Liang, W.; Becker, C.; Shimada, K.; Wheeler, M.L.; Cho, H.C.; Popescu, N.I.; Coggeshall, K.M.; Arditi, M.; et al. Hexokinase Is an Innate Immune Receptor for the Detection of Bacterial Peptidoglycan. Cell 2016, 166, 624–636. [Google Scholar] [CrossRef]
- Zanoni, I.; Tan, Y.; Di Gioia, M.; Broggi, A.; Ruan, J.; Shi, J.; Donado, C.A.; Shao, F.; Wu, H.; Springstead, J.R.; et al. An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science 2016, 352, 1232–1236. [Google Scholar] [CrossRef]
- Santa Cruz Garcia, A.B.; Schnur, K.P.; Malik, A.B.; Mo, G.C.H. Gasdermin D pores are dynamically regulated by local phosphoinositide circuitry. Nat. Commun. 2022, 13, 52. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, W.; Shi, X.; Ding, J.; Liu, W.; He, H.; Wang, K.; Shao, F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017, 547, 99–103. [Google Scholar] [CrossRef]
- Rogers, C.; Fernandes-Alnemri, T.; Mayes, L.; Alnemri, D.; Cingolani, G.; Alnemri, E.S. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat. Commun. 2017, 8, 14128. [Google Scholar] [CrossRef]
- Grossi, S.; Fenini, G.; Kockmann, T.; Hennig, P.; Di Filippo, M.; Beer, H.-D. Inactivation of the Cytoprotective Major Vault Protein by Caspase-1 and -9 in Epithelial Cells during Apoptosis. J. Investig. Dermatol. 2020, 140, 1335–1345.e10. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Pretre, V.; Papadopoulos, D.; Regard, J.; Pelletier, M.; Woo, J. Interleukin-1 (IL-1) and the inflammasome in cancer. Cytokine 2022, 153, 155850. [Google Scholar] [CrossRef]
- North, R.J.; Neubauer, R.H.; Huang, J.J.; Newton, R.; Loveless, S.E. Interleukin 1-induced, T cell-mediated regression of immunogenic murine tumors. Requirement for an adequate level of already acquired host concomitant immunity. J. Exp. Med. 1988, 168, 2031–2043. [Google Scholar] [CrossRef]
- Moossavi, M.; Parsamanesh, N.; Bahrami, A.; Atkin, S.L.; Sahebkar, A. Role of the NLRP3 inflammasome in cancer. Mol. Cancer 2018, 17, 158. [Google Scholar] [CrossRef]
- Kolb, R.; Liu, G.H.; Janowski, A.M.; Sutterwala, F.S.; Zhang, W. Inflammasomes in cancer: A double-edged sword. Protein Cell 2014, 5, 12–20. [Google Scholar] [CrossRef]
- Van Gorp, H.; Lamkanfi, M. The emerging roles of inflammasome-dependent cytokines in cancer development. EMBO Rep. 2019, 20, e47575. [Google Scholar] [CrossRef] [PubMed]
- Karki, R.; Kanneganti, T.-D. Diverging inflammasome signals in tumorigenesis and potential targeting. Nat. Rev. Cancer 2019, 19, 197–214. [Google Scholar] [CrossRef] [PubMed]
- Elaraj, D.M.; Weinreich, D.M.; Varghese, S.; Puhlmann, M.; Hewitt, S.M.; Carroll, N.M.; Feldman, E.D.; Turner, E.M.; Alexander, H.R. The role of interleukin 1 in growth and metastasis of human cancer xenografts. Clin. Cancer Res. 2006, 12, 1088–1096. [Google Scholar] [CrossRef] [PubMed]
- Elkabets, M.; Ribeiro, V.S.; Dinarello, C.A.; Ostrand-Rosenberg, S.; Di Santo, J.P.; Apte, R.N.; Vosshenrich, C.A. IL-1β regulates a novel myeloid-derived suppressor cell subset that impairs NK cell development and function. Eur. J. Immunol. 2010, 40, 3347–3357. [Google Scholar] [CrossRef]
- Ungerbäck, J.; Belenki, D.; ul-Hassan, A.J.; Fredrikson, M.; Fransén, K.; Elander, N.; Verma, D.; Söderkvist, P. Genetic variation and alterations of genes involved in NFkappaB/TNFAIP3- and NLRP3-inflammasome signaling affect susceptibility and outcome of colorectal cancer. Carcinogenesis 2012, 33, 2126–2134. [Google Scholar] [CrossRef]
- Verma, D.; Bivik, C.; Farahani, E.; Synnerstad, I.; Fredrikson, M.; Enerbäck, C.; Rosdahl, I.; Söderkvist, P. Inflammasome polymorphisms confer susceptibility to sporadic malignant melanoma. Pigment. Cell Melanoma Res. 2012, 25, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Castaño-Rodríguez, N.; Kaakoush, N.O.; Goh, K.-L.; Fock, K.M.; Mitchell, H.M. The NOD-Like Receptor Signalling Pathway in Helicobacter pylori Infection and Related Gastric Cancer: A Case-Control Study and Gene Expression Analyses. PLoS ONE 2014, 9, e98899. [Google Scholar] [CrossRef] [PubMed]
- El-Omar, E.M.; Carrington, M.; Chow, W.H.; McColl, K.E.; Bream, J.H.; Young, H.A.; Herrera, J.; Lissowska, J.; Yuan, C.; Rothman, N.; et al. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature 2000, 404, 398–402. [Google Scholar] [CrossRef]
- Faustin, B.; Reed, J.C. Sunburned skin activates inflammasomes. Trends Cell Biol. 2008, 18, 4–8. [Google Scholar] [CrossRef]
- Bhat, I.A.; Naykoo, N.A.; Qasim, I.; Ganie, F.A.; Yousuf, Q.; Bhat, B.A.; Rasool, R.; Aziz, S.; Shah, Z.A. Association of interleukin 1 beta (IL-1β) polymorphism with mRNA expression and risk of non small cell lung cancer. Meta Gene 2014, 2, 123–133. [Google Scholar] [CrossRef]
- Hu, Z.; Shao, M.; Chen, Y.; Zhou, J.; Qian, J.; Xu, L.; Ma, H.; Wang, X.; Xu, Y.; Lu, D.; et al. Allele 2 of the interleukin-1 receptor antagonist gene (IL1RN*2) is associated with a decreased risk of primary lung cancer. Cancer Lett. 2006, 236, 269–275. [Google Scholar] [CrossRef]
- Lind, H.; Zienolddiny, S.; Ryberg, D.; Skaug, V.; Phillips, D.H.; Haugen, A. Interleukin 1 receptor antagonist gene polymorphism and risk of lung cancer: A possible interaction with polymorphisms in the interleukin 1 beta gene. Lung Cancer 2005, 50, 285–290. [Google Scholar] [CrossRef]
- Zienolddiny, S.; Ryberg, D.; Maggini, V.; Skaug, V.; Canzian, F.; Haugen, A. Polymorphisms of the interleukin-1 beta gene are associated with increased risk of non-small cell lung cancer. Int. J. Cancer 2004, 109, 353–356. [Google Scholar] [CrossRef]
- Ridker, P.M.; MacFadyen, J.G.; Thuren, T.; Everett, B.M.; Libby, P.; Glynn, R.J.; CANTOS Trial Group. Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: Exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 2017, 390, 1833–1842. [Google Scholar] [CrossRef]
- Wong, C.C.; Baum, J.; Silvestro, A.; Beste, M.T.; Bharani-Dharan, B.; Xu, S.; Wang, Y.A.; Wang, X.; Prescott, M.F.; Krajkovich, L.; et al. Inhibition of IL1β by Canakinumab May Be Effective against Diverse Molecular Subtypes of Lung Cancer: An Exploratory Analysis of the CANTOS Trial. Cancer Res. 2020, 80, 5597–5605. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Jin, L.; Yuan, R.Q.; Fuchs, A.; Yao, Y.; Joseph, A.; Schwall, R.; Schnitt, S.J.; Guida, A.; Hastings, H.M.; Andres, J.; et al. Expression of interleukin-1β in human breast carcinoma. Cancer 1997, 80, 421–434. [Google Scholar] [CrossRef]
- Dinarello, C.A. Why not treat human cancer with interleukin-1 blockade? Cancer Metastasis Rev. 2010, 29, 317–329. [Google Scholar] [CrossRef]
- Apte, R.N.; Krelin, Y.; Song, X.; Dotan, S.; Recih, E.; Elkabets, M.; Carmi, Y.; Dvorkin, T.; White, R.M.; Gayvoronsky, L.; et al. Effects of micro-environment- and malignant cell-derived interleukin-1 in carcinogenesis, tumour invasiveness and tumour–host interactions. Eur. J. Cancer 2006, 42, 751–759. [Google Scholar] [CrossRef]
- Marvel, D.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the tumor microenvironment: Expect the unexpected. J. Clin. Investig. 2015, 125, 3356–3364. [Google Scholar] [CrossRef]
- Van Deventer, H.W.; Burgents, J.E.; Wu, Q.P.; Woodford, R.M.T.; Brickey, W.J.; Allen, I.C.; McElvania-Tekippe, E.; Serody, J.S.; Ting, J.P.Y. The inflammasome component NLRP3 impairs antitumor vaccine by enhancing the accumulation of tumor-associated myeloid-derived suppressor cells. Cancer Res. 2010, 70, 10161–10169. [Google Scholar] [CrossRef]
- Daley, D.; Mani, V.R.; Mohan, N.; Akkad, N.; Pandian, G.S.B.; Savadkar, S.; Lee, K.B.; Torres-Hernandez, A.; Aykut, B.; Diskin, B.; et al. NLRP3 signaling drives macrophage-induced adaptive immune suppression in pancreatic carcinoma. J. Exp. Med. 2017, 214, 1711–1724. [Google Scholar] [CrossRef]
- Sorrentino, R.; Terlizzi, M.; Di Crescenzo, V.; Popolo, A.; Pecoraro, M.; Perillo, G.; Galderisi, A.; Pinto, A. Human Lung Cancer–Derived Immunosuppressive Plasmacytoid Dendritic Cells Release IL-1α in an AIM2 Inflammasome-Dependent Manner. Am. J. Pathol. 2015, 185, 3115–3124. [Google Scholar] [CrossRef]
- Jee, Y.S.; Jang, T.J.; Jung, K.H. Prostaglandin E(2) and interleukin-1β reduce E-cadherin expression by enhancing snail expression in gastric cancer cells. J. Korean Med. Sci. 2012, 27, 987–992. [Google Scholar] [CrossRef]
- Yang, Y.; Cheon, S.; Jung, M.K.; Song, S.B.; Kim, D.; Kim, H.J.; Park, H.; Bang, S.I.; Cho, D. Interleukin-18 enhances breast cancer cell migration via down-regulation of claudin-12 and induction of the p38 MAPK pathway. Biochem. Biophys. Res. Commun. 2015, 459, 379–386. [Google Scholar] [CrossRef]
- Ohtsuka, T.; Ryu, H.; Minamishima, Y.A.; Macip, S.; Sagara, J.; Nakayama, K.I.; Aaronson, S.A.; Lee, S.W. ASC is a Bax adaptor and regulates the p53–Bax mitochondrial apoptosis pathway. Nat. Cell Biol. 2004, 6, 121–128. [Google Scholar] [CrossRef]
- Chu, Z.-L.; Pio, F.; Xie, Z.; Welsh, K.; Krajewska, M.; Krajewski, S.; Godzik, A.; Reed, J.C. A Novel Enhancer of the Apaf1 Apoptosome Involved in Cytochrome c-dependent Caspase Activation and Apoptosis. J. Biol. Chem. 2001, 276, 9239–9245. [Google Scholar] [CrossRef]
- Hlaing, T.; Guo, R.-F.; Dilley, K.A.; Loussia, J.M.; Morrish, T.A.; Shi, M.M.; Vincenz, C.; Ward, P.A. Molecular Cloning and Characterization of DEFCAP-L and -S, Two Isoforms of a Novel Member of the Mammalian Ced-4 Family of Apoptosis Proteins. J. Biol. Chem. 2001, 276, 9230–9238. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Yu, C.-H.; Moecking, J.; Geyer, M.; Masters, S.L. Mechanisms of NLRP1-Mediated Autoinflammatory Disease in Humans and Mice. J. Mol. Biol. 2018, 430, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Finger, J.N.; Lich, J.D.; Dare, L.C.; Cook, M.N.; Brown, K.K.; Duraiswami, C.; Bertin, J.J.; Gough, P.J. Autolytic proteolysis within the function to find domain (FIIND) is required for NLRP1 inflammasome activity. J. Biol. Chem. 2012, 287, 25030–25037. [Google Scholar] [CrossRef] [PubMed]
- D’Osualdo, A.; Weichenberger, C.X.; Wagner, R.N.; Godzik, A.; Wooley, J.; Reed, J.C. CARD8 and NLRP1 Undergo Autoproteolytic Processing through a ZU5-Like Domain. PLoS ONE 2011, 6, e27396. [Google Scholar] [CrossRef] [PubMed]
- Bauernfried, S.; Hornung, V. Human NLRP1: From the shadows to center stage. J. Exp. Med. 2022, 219, e20211405. [Google Scholar] [CrossRef] [PubMed]
- Briukhovetska, D.; Dörr, J.; Endres, S.; Libby, P.; Dinarello, C.A.; Kobold, S. Interleukins in cancer: From biology to therapy. Nat. Rev. Cancer 2021, 21, 481–499. [Google Scholar] [CrossRef]
- Baker, K.J.; Houston, A.; Brint, E. IL-1 Family Members in Cancer; Two Sides to Every Story. Front. Immunol. 2019, 10, 1197. [Google Scholar] [CrossRef]
- Hong, D.S.; Hui, D.; Bruera, E.; Janku, F.; Naing, A.; Falchook, G.S.; Piha-Paul, S.; Wheler, J.J.; Fu, S.; Tsimberidou, A.M.; et al. MABp1, a first-in-class true human antibody targeting interleukin-1α in refractory cancers: An open-label, phase 1 dose-escalation and expansion study. Lancet Oncol. 2014, 15, 656–666. [Google Scholar] [CrossRef]
- Hickish, T.; Andre, T.; Wyrwicz, L.; Saunders, M.; Sarosiek, T.; Kocsis, J.; Nemecek, R.; Rogowski, W.; Lesniewski-Kmak, K.; Petruzelka, L.; et al. MABp1 as a novel antibody treatment for advanced colorectal cancer: A randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2017, 18, 192–201. [Google Scholar] [CrossRef]
- Kurzrock, R.; Hickish, T.; Wyrwicz, L.; Saunders, M.; Wu, Q.; Stecher, M.; Mohanty, P.; Dinarello, C.A.; Simard, J. Interleukin-1 receptor antagonist levels predict favorable outcome after bermekimab, a first-in-class true human interleukin-1α antibody, in a phase III randomized study of advanced colorectal cancer. OncoImmunology 2019, 8, 1551651. [Google Scholar] [CrossRef]
- Terme, M.; Ullrich, E.; Aymeric, L.; Meinhardt, K.; Desbois, M.; Delahaye, N.; Viaud, S.; Ryffel, B.; Yagita, H.; Kaplanski, G.; et al. IL-18 Induces PD-1–Dependent Immunosuppression in Cancer. Cancer Res. 2011, 71, 5393–5399. [Google Scholar] [CrossRef]
- Kaplanov, I.; Carmi, Y.; Kornetsky, R.; Shemesh, A.; Shurin, G.V.; Shurin, M.R.; Dinarello, C.A.; Voronov, E.; Apte, R.N. Blocking IL-1β reverses the immunosuppression in mouse breast cancer and synergizes with anti-PD-1 for tumor abrogation. Proc. Natl. Acad. Sci. USA 2019, 116, 1361–1369. [Google Scholar] [CrossRef]
- Krelin, Y.; Voronov, E.; Dotan, S.; Elkabets, M.; Reich, E.; Fogel, M.; Huszar, M.; Iwakura, Y.; Segal, S.; Dinarello, C.A.; et al. Interleukin-1β-driven inflammation promotes the development and invasiveness of chemical carcinogen-induced tumors. Cancer Res. 2007, 67, 1062–1071. [Google Scholar] [CrossRef]
- Chow, M.T.; Sceneay, J.; Paget, C.; Wong, C.S.; Duret, H.; Tschopp, J.; Möller, A.; Smyth, M.J. NLRP3 Suppresses NK Cell–Mediated Responses to Carcinogen-Induced Tumors and Metastases. Cancer Res. 2012, 72, 5721–5732. [Google Scholar] [CrossRef]
- Karki, R.; Man, S.M.; Kanneganti, T.-D. Inflammasomes and Cancer. Cancer Immunol. Res. 2017, 5, 94–99. [Google Scholar] [CrossRef]
- Allen, I.C.; TeKippe, E.M.; Woodford, R.-M.T.; Uronis, J.M.; Holl, E.K.; Rogers, A.B.; Herfarth, H.H.; Jobin, C.; Ting, J.P.-Y. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J. Exp. Med. 2010, 207, 1045–1056. [Google Scholar] [CrossRef]
- Salcedo, R.; Worschech, A.; Cardone, M.; Jones, Y.; Gyulai, Z.; Dai, R.-M.; Wang, E.; Ma, W.; Haines, D.; O’Huigin, C.; et al. MyD88-mediated signaling prevents development of adenocarcinomas of the colon: Role of interleukin 18. J. Exp. Med. 2010, 207, 1625–1636. [Google Scholar] [CrossRef]
- Kantono, M.; Guo, B. Inflammasomes and Cancer: The Dynamic Role of the Inflammasome in Tumor Development. Front. Immunol. 2017, 8, 1132. [Google Scholar] [CrossRef]
- Hu, B.; Elinav, E.; Huber, S.; Booth, C.J.; Strowig, T.; Jin, C.; Eisenbarth, S.C.; Flavell, R.A. Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4. Proc. Natl. Acad. Sci. USA 2010, 107, 21635–21640. [Google Scholar] [CrossRef]
- Fenini, G.; Karakaya, T.; Hennig, P.; Di Filippo, M.; Slaufova, M.; Beer, H.-D. NLRP1 Inflammasome Activation in Keratinocytes: Increasing Evidence of Important Roles in Inflammatory Skin Diseases and Immunity. J. Investig. Dermatol. 2022, 142, 2313–2322. [Google Scholar] [CrossRef]
- Bauernfried, S.; Scherr, M.J.; Pichlmair, A.; Duderstadt, K.E.; Hornung, V. Human NLRP1 is a sensor for double-stranded RNA. Science 2021, 371, eabd0811. [Google Scholar] [CrossRef]
- Taniguchi, S.; Elhance, A.; Van Duzer, A.; Kumar, S.; Leitenberger, J.J.; Oshimori, N. Tumor-initiating cells establish an IL-33-TGF-β niche signaling loop to promote cancer progression. Science 2020, 369, eaay1813. [Google Scholar] [CrossRef]
- Andersson, P.; Yang, Y.; Hosaka, K.; Zhang, Y.; Fischer, C.; Braun, H.; Liu, S.; Yu, G.; Liu, S.; Beyaert, R.; et al. Molecular mechanisms of IL-33–mediated stromal interactions in cancer metastasis. JCI Insight 2018, 3, e122375. [Google Scholar] [CrossRef]
- Conway, K.E.; McConnell, B.B.; Bowring, C.; Donald, C.D.; Warren, S.T.; Vertino, P.M. TMS1, a novel proapoptotic caspase recruitment domain protein, is a target of methylation-induced gene silencing in human breast cancers. Cancer Res. 2000, 60, 6236–6242. [Google Scholar]
- Protti, M.P.; De Monte, L. Dual Role of Inflammasome Adaptor ASC in Cancer. Front. Cell Dev. Biol. 2020, 8, 40. [Google Scholar] [CrossRef]
- Fenini, G.; Karakaya, T.; Hennig, P.; Di Filippo, M.; Beer, H.-D. The NLRP1 Inflammasome in Human Skin and Beyond. Int. J. Mol. Sci. 2020, 21, 4788. [Google Scholar] [CrossRef]
- Zhong, F.L.; Mamaï, O.; Sborgi, L.; Boussofara, L.; Hopkins, R.; Robinson, K.; Szeverényi, I.; Takeichi, T.; Balaji, R.; Lau, A.; et al. Germline NLRP1 Mutations Cause Skin Inflammatory and Cancer Susceptibility Syndromes via Inflammasome Activation. Cell 2016, 167, 187–202.e17. [Google Scholar] [CrossRef]
- Dombrowski, Y.; Peric, M.; Koglin, S.; Kammerbauer, C.; Göß, C.; Anz, D.; Simanski, M.; Gläser, R.; Harder, J.; Hornung, V.; et al. Cytosolic DNA Triggers Inflammasome Activation in Keratinocytes in Psoriatic Lesions. Sci. Transl. Med. 2011, 3, 82ra38. [Google Scholar] [CrossRef]
- Dai, X.; Tohyama, M.; Murakami, M.; Sayama, K. Epidermal keratinocytes sense dsRNA via the NLRP3 inflammasome, mediating interleukin (IL)-1β and IL-18 release. Exp. Dermatol. 2017, 26, 904–911. [Google Scholar] [CrossRef]
- Feldmeyer, L.; Keller, M.; Niklaus, G.; Hohl, D.; Werner, S.; Beer, H.D. The inflammasome mediates UVB-induced activation and secretion of interleukin-1β by keratinocytes. Curr. Biol. 2007, 17, 1140–1145. [Google Scholar] [CrossRef] [PubMed]
- Fenini, G.; Grossi, S.; Contassot, E.; Biedermann, T.; Reichmann, E.; French, L.E.; Beer, H.-D. Genome Editing of Human Primary Keratinocytes by CRISPR/Cas9 Reveals an Essential Role of the NLRP1 Inflammasome in UVB Sensing. J. Investig. Dermatol. 2018, 138, 2644–2652. [Google Scholar] [CrossRef] [PubMed]
- Fenini, G.; Grossi, S.; Gehrke, S.; Beer, H.-D.; Satoh, T.K.; Contassot, E.; French, L.E. The p38 Mitogen-Activated Protein Kinase Critically Regulates Human Keratinocyte Inflammasome Activation. J. Investig. Dermatol. 2018, 138, 1380–1390. [Google Scholar] [CrossRef] [PubMed]
- Robinson, K.S.; Toh, G.A.; Rozario, P.; Bayat, S.; Sun, Z.; Bauernfried, S.; Nadkarni, R.; Harapas, C.R.; Lim, C.K.; Chu, W.; et al. Human NLRP1 is activated by ZAKɑ-driven ribotoxic stress response. bioRxiv 2022. bioRxiv:2022.01.24.477516. [Google Scholar]
- Jenster, L.-M.; Lange, K.-E.; Normann, S.; vom Hemdt, A.; Wuerth, J.D.; Schiffelers, L.D.J.; Tesfamariam, Y.M.; Gohr, F.N.; Klein, L.; Kaltheuner, I.H.; et al. P38 kinases mediate NLRP1 inflammasome activation after ribotoxic stress response and virus infection. bioRxiv 2022. bioRxiv:2022.01.24.477423. [Google Scholar]
- Vind, A.C.; Genzor, A.V.; Bekker-Jensen, S. Ribosomal stress-surveillance: Three pathways is a magic number. Nucleic Acids Res. 2020, 48, 10648–10661. [Google Scholar] [CrossRef]
- Hollingsworth, L.R.; Sharif, H.; Griswold, A.R.; Fontana, P.; Mintseris, J.; Dagbay, K.B.; Paulo, J.A.; Gygi, S.P.; Bachovchin, D.A.; Wu, H. DPP9 sequesters the C terminus of NLRP1 to repress inflammasome activation. Nature 2021, 592, 778–783. [Google Scholar] [CrossRef]
- Huang, M.; Zhang, X.; Toh, G.A.; Gong, Q.; Wang, J.; Han, Z.; Wu, B.; Zhong, F.; Chai, J. Structural and biochemical mechanisms of NLRP1 inhibition by DPP9. Nature 2021, 592, 773–777. [Google Scholar] [CrossRef]
- Adams, S.; Miller, G.T.; Jesson, M.I.; Watanabe, T.; Jones, B.; Wallner, B.P. PT-100, a Small Molecule Dipeptidyl Peptidase Inhibitor, Has Potent Antitumor Effects and Augments Antibody-Mediated Cytotoxicity via a Novel Immune Mechanism. Cancer Res. 2004, 64, 5471–5480. [Google Scholar] [CrossRef]
- Zhong, F.L.; Robinson, K.; Teo, D.E.T.; Tan, K.Y.; Lim, C.; Harapas, C.R.; Yu, C.-H.; Xie, W.H.; Sobota, R.M.; Au, V.B.; et al. Human DPP9 represses NLRP1 inflammasome and protects against auto-inflammatory diseases via both peptidase activity and FIIND domain binding. J. Biol. Chem. 2018, 293, 18864–18878. [Google Scholar] [CrossRef]
- Chavarria-Smith, J.; Vance, R.E. The NLRP1 inflammasomes. Immunol. Rev. 2015, 265, 22–34. [Google Scholar] [CrossRef]
- Sandstrom, A.; Mitchell, P.S.; Goers, L.; Mu, E.W.; Lesser, C.F.; Vance, R.E. Functional degradation: A mechanism of NLRP1 inflammasome activation by diverse pathogen enzymes. Science 2019, 364, eaau1330. [Google Scholar] [CrossRef]
- Chui, A.J.; Okondo, M.C.; Rao, S.D.; Gai, K.; Griswold, A.R.; Johnson, D.C.; Ball, D.P.; Taabazuing, C.Y.; Orth, E.L.; Vittimberga, B.A.; et al. N-terminal degradation activates the NLRP1B inflammasome. Science 2019, 364, 82–85. [Google Scholar] [CrossRef]
- Chavarría-Smith, J.; Vance, R.E. Direct Proteolytic Cleavage of NLRP1B Is Necessary and Sufficient for Inflammasome Activation by Anthrax Lethal Factor. PLOS Pathog. 2013, 9, e1003452. [Google Scholar] [CrossRef]
- Yang, X.; Zhou, J.; Liu, C.; Qu, Y.; Wang, W.; Xiao, M.Z.X.; Zhu, F.; Liu, Z.; Liang, Q. KSHV-encoded ORF45 activates human NLRP1 inflammasome. Nat. Immunol. 2022, 23, 916–926. [Google Scholar] [CrossRef]
- de Vasconcelos, N.M.; Vliegen, G.; Gonçalves, A.; De Hert, E.; Martín-Pérez, R.; Van Opdenbosch, N.; Jallapally, A.; Geiss-Friedlander, R.; Lambeir, A.-M.; Augustyns, K.; et al. DPP8/DPP9 inhibition elicits canonical Nlrp1b inflammasome hallmarks in murine macrophages. Life Sci. Alliance 2019, 2, e201900313. [Google Scholar] [CrossRef]
- Okondo, M.C.; Rao, S.D.; Taabazuing, C.Y.; Chui, A.J.; Poplawski, S.E.; Johnson, D.C.; Bachovchin, D.A. Inhibition of Dpp8/9 Activates the Nlrp1b Inflammasome. Cell Chem. Biol. 2018, 25, 262–267.e5. [Google Scholar] [CrossRef]
- Sand, J.; Haertel, E.; Biedermann, T.; Contassot, E.; Reichmann, E.; French, L.E.; Werner, S.; Beer, H.-D. Expression of inflammasome proteins and inflammasome activation occurs in human, but not in murine keratinocytes. Cell Death Dis. 2018, 9, 24. [Google Scholar] [CrossRef]
- Jin, Y.; Mailloux, C.M.; Gowan, K.; Riccardi, S.L.; Laberge, G.; Bennett, D.C.; Fain, P.R.; Spritz, R.A. NALP1 in Vitiligo-Associated Multiple Autoimmune Disease. N. Engl. J. Med. 2007, 356, 1216–1225. [Google Scholar] [CrossRef]
- Żurawek, M.; Fichna, M.; Januszkiewicz-Lewandowska, D.; Gryczyńska, M.; Fichna, P.; Nowak, J. A coding variant in NLRP1 is associated with autoimmune Addison’s disease. Hum. Immunol. 2010, 71, 530–534. [Google Scholar] [CrossRef]
- Grandemange, S.; Sanchez, E.; Louis-Plence, P.; Mau-Them, F.T.; Bessis, D.; Coubes, C.; Frouin, E.; Seyger, M.; Girard, M.; Puechberty, J.; et al. A new autoinflammatory and autoimmune syndrome associated with NLRP1 mutations: NAIAD (NLRP1-associated autoinflammation with arthritis and dyskeratosis). Ann. Rheum. Dis. 2017, 76, 1191–1198. [Google Scholar] [CrossRef]
- Leiter, U.; Eigentler, T.; Garbe, C. Epidemiology of skin cancer. Adv. Exp. Med. Biol. 2014, 810, 120–140. [Google Scholar]
- D’Orazio, J.; Jarrett, S.; Amaro-Ortiz, A.; Scott, T. UV Radiation and the Skin. Int. J. Mol. Sci. 2013, 14, 12222–12248. [Google Scholar] [CrossRef]
- Bernard, J.J.; Gallo, R.L.; Krutmann, J. Photoimmunology: How ultraviolet radiation affects the immune system. Nat. Rev. Immunol. 2019, 19, 688–701. [Google Scholar] [CrossRef]
- Brandt, M.G.; Moore, C.C. Nonmelanoma Skin Cancer. Facial Plast. Surg. Clin. 2019, 27, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Mushtaq, S. The Immunogenetics of Non-melanoma Skin Cancer. In The Immunogenetics of Dermatologic Diseases; Springer: Cham, Switzerland, 2022; pp. 397–409. [Google Scholar]
- Kallini, J.R.; Hamed, N.; Khachemoune, A. Squamous cell carcinoma of the skin: Epidemiology, classification, management, and novel trends. Int. J. Dermatol. 2015, 54, 130–140. [Google Scholar] [CrossRef]
- Molho-Pessach, V.; Lotem, M. Ultraviolet Radiation and Cutaneous Carcinogenesis. Environ. Factors Ski. Dis. 2007, 35, 14–27. [Google Scholar] [CrossRef]
- Tampa, M.; Georgescu, S.R.; Mitran, C.I.; Mitran, M.I.; Matei, C.; Scheau, C.; Constantin, C.; Neagu, M. Recent Advances in Signaling Pathways Comprehension as Carcinogenesis Triggers in Basal Cell Carcinoma. J. Clin. Med. 2020, 9, 3010. [Google Scholar] [CrossRef] [PubMed]
- Meier, K.; Drexler, S.K.; Eberle, F.C.; Lefort, K.; Yazdi, A.S. Silencing of ASC in Cutaneous Squamous Cell Carcinoma. PLoS ONE 2016, 11, e0164742. [Google Scholar] [CrossRef] [PubMed]
- Sand, J.; Fenini, G.; Grossi, S.; Hennig, P.; Di Filippo, M.; Levesque, M.; Werner, S.; French, L.E.; Beer, H.-D. The NLRP1 Inflammasome Pathway Is Silenced in Cutaneous Squamous Cell Carcinoma. J. Investig. Dermatol. 2019, 139, 1788–1797.e6. [Google Scholar] [CrossRef]
- Guan, X.; Sagara, J.; Yokoyama, T.; Koganehira, Y.; Oguchi, M.; Saida, T.; Taniguchi, S.I. ASC/TMS1, a caspase-1 activating adaptor, is downregulated by aberrant methylation in human melanoma. Int. J. Cancer 2003, 107, 202–208. [Google Scholar] [CrossRef]
- Liu, W.; Luo, Y.; Dunn, J.H.; Norris, D.A.; Dinarello, C.A.; Fujita, M. Dual Role of Apoptosis-Associated Speck-Like Protein Containing a CARD (ASC) in Tumorigenesis of Human Melanoma. J. Investig. Dermatol. 2013, 133, 518–527. [Google Scholar] [CrossRef]
- Farshchian, M.; Nissinen, L.; Siljamäki, E.; Riihilä, P.; Piipponen, M.; Kivisaari, A.; Kallajoki, M.; Grénman, R.; Peltonen, J.; Peltonen, S.; et al. Tumor cell-specific AIM2 regulates growth and invasion of cutaneous squamous cell carcinoma. Oncotarget 2017, 8, 45825–45836. [Google Scholar] [CrossRef]
- Takano, K.; Kondo, A.; Kurose, M.; Yamashita, K.; Nomura, K.; Obata, K.; Murayama, K.; Ito, F.; Himi, T. Expression of Inflammasome-Associated Proteins in Human Oropharyngeal Squamous Cell Carcinoma. In Excellence in Otolaryngology; Karger Publishers: Basel, Switzerland, 2016; pp. 98–104. [Google Scholar] [CrossRef]
- Bae, J.Y.; Lee, S.-W.; Shin, Y.-H.; Lee, J.-H.; Jahng, J.W.; Park, K. P2X7 receptor and NLRP3 inflammasome activation in head and neck cancer. Oncotarget 2017, 8, 48972–48982. [Google Scholar] [CrossRef]
- Huang, C.-F.; Chen, L.; Li, Y.-C.; Wu, L.; Yu, G.-T.; Zhang, W.-F.; Sun, Z.-J. NLRP3 inflammasome activation promotes inflammation-induced carcinogenesis in head and neck squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2017, 36, 116. [Google Scholar] [CrossRef]
- Feng, X.; Luo, Q.; Zhang, H.; Wang, H.; Chen, W.; Meng, G.; Chen, F. The role of NLRP3 inflammasome in 5-fluorouracil resistance of oral squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2017, 36, 1–14. [Google Scholar] [CrossRef]
- Ahmad, I.; Muneer, K.M.; Chang, M.E.; Nasr, H.M.; Clay, J.M.; Huang, C.C.; Yusuf, N. Ultraviolet Radiation-Induced Downregulation of SERCA2 Mediates Activation of NLRP3 Inflammasome in Basal Cell Carcinoma. Photochem. Photobiol. 2017, 93, 1025–1033. [Google Scholar] [CrossRef]
- Cataisson, C.; Salcedo, R.; Hakim, S.; Moffitt, B.A.; Wright, L.; Yi, M.; Stephens, R.; Dai, R.M.; Lyakh, L.; Schenten, D.; et al. IL-1R-MyD88 signaling in keratinocyte transformation and carcinogenesis. J. Exp. Med. 2012, 209, 1689–1702. [Google Scholar] [CrossRef]
- Chow, M.T.; Tschopp, J.; Möller, A.; Smyth, M.J. NLRP3 promotes inflammation-induced skin cancer but is dispensable for asbestos-induced mesothelioma. Immunol. Cell Biol. 2012, 90, 983–986. [Google Scholar] [CrossRef]
- Drexler, S.K.; Bonsignore, L.; Masin, M.; Tardivel, A.; Jackstadt, R.; Hermeking, H.; Schneider, P.; Gross, O.; Tschopp, J.; Yazdi, A.S. Tissue-specific opposing functions of the inflammasome adaptor ASC in the regulation of epithelial skin carcinogenesis. Proc. Natl. Acad. Sci. USA 2012, 109, 18384–18389. [Google Scholar] [CrossRef]
- Gasparoto, T.H.; De Oliveira, C.E.; De Freitas, L.T.; Pinheiro, C.R.; Hori, J.; Garlet, G.; Cavassani, K.A.; Schillaci, R.; Da Silva, J.S.; Zamboni, D.; et al. Inflammasome Activation Is Critical to the Protective Immune Response during Chemically Induced Squamous Cell Carcinoma. PLoS ONE 2014, 9, e107170. [Google Scholar] [CrossRef]
- Apalla, Z.; Lallas, A.; Sotiriou, E.; Lazaridou, E.; Ioannides, D. Epidemiological trends in skin cancer. Dermatol. Pract. Concept. 2017, 7, 1–6. [Google Scholar] [CrossRef]
- Okamoto, M.; Liu, W.; Luo, Y.; Tanaka, A.; Cai, X.; Norris, D.A.; Dinarello, C.A.; Fujita, M. Constitutively active inflammasome in human melanoma cells mediating autoinflammation via caspase-1 processing and secretion of interleukin-1β. J. Biol. Chem. 2010, 285, 6477–6488. [Google Scholar] [CrossRef]
- Zhai, Z.; Liu, W.; Kaur, M.; Luo, Y.; Domenico, J.; Samson, J.M.; Shellman, Y.G.; Norris, D.A.; Dinarello, C.A.; Spritz, R.A.; et al. NLRP1 promotes tumor growth by enhancing inflammasome activation and suppressing apoptosis in metastatic melanoma. Oncogene 2017, 36, 3820–3830. [Google Scholar] [CrossRef] [PubMed]
- Voronov, E.; Shouval, D.S.; Krelin, Y.; Cagnano, E.; Benharroch, D.; Iwakura, Y.; Dinarello, C.A.; Apte, R.N. IL-1 is required for tumor invasiveness and angiogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 2645–2650. [Google Scholar] [CrossRef]
- Carrascal, M.T.; Mendoza, L.; Valcárcel, M.; Salado, C.; Egilegor, E.; Tellería, N.; Vidal-Vanaclocha, F.; Dinarello, C.A. Interleukin-18 binding protein reduces b16 melanoma hepatic metastasis by neutralizing adhesiveness and growth factors of sinusoidal endothelium. Cancer Res. 2003, 63, 491–497. [Google Scholar]
- Dziunycz, P.J.; Lefort, K.; Wu, X.; Freiberger, S.N.; Neu, J.; Djerbi, N.; Iotzowa-Weiss, G.; French, L.E.; Dotto, G.-P.; Hofbauer, G.F. The Oncogene ATF3 Is Potentiated by Cyclosporine A and Ultraviolet Light A. J. Investig. Dermatol. 2014, 134, 1998–2004. [Google Scholar] [CrossRef] [PubMed]
- Walter, A.; Schäfer, M.; Cecconi, V.; Matter, C.; Urosevic-Maiwald, M.; Belloni, B.; Schönewolf, N.; Dummer, R.; Bloch, W.; Werner, S.; et al. Aldara activates TLR7-independent immune defence. Nat. Commun. 2013, 4, 1560. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).