
Complex Relationships between HIV-1 Integrase and Its Cellular Partners
 , ,
, ,
Abstract
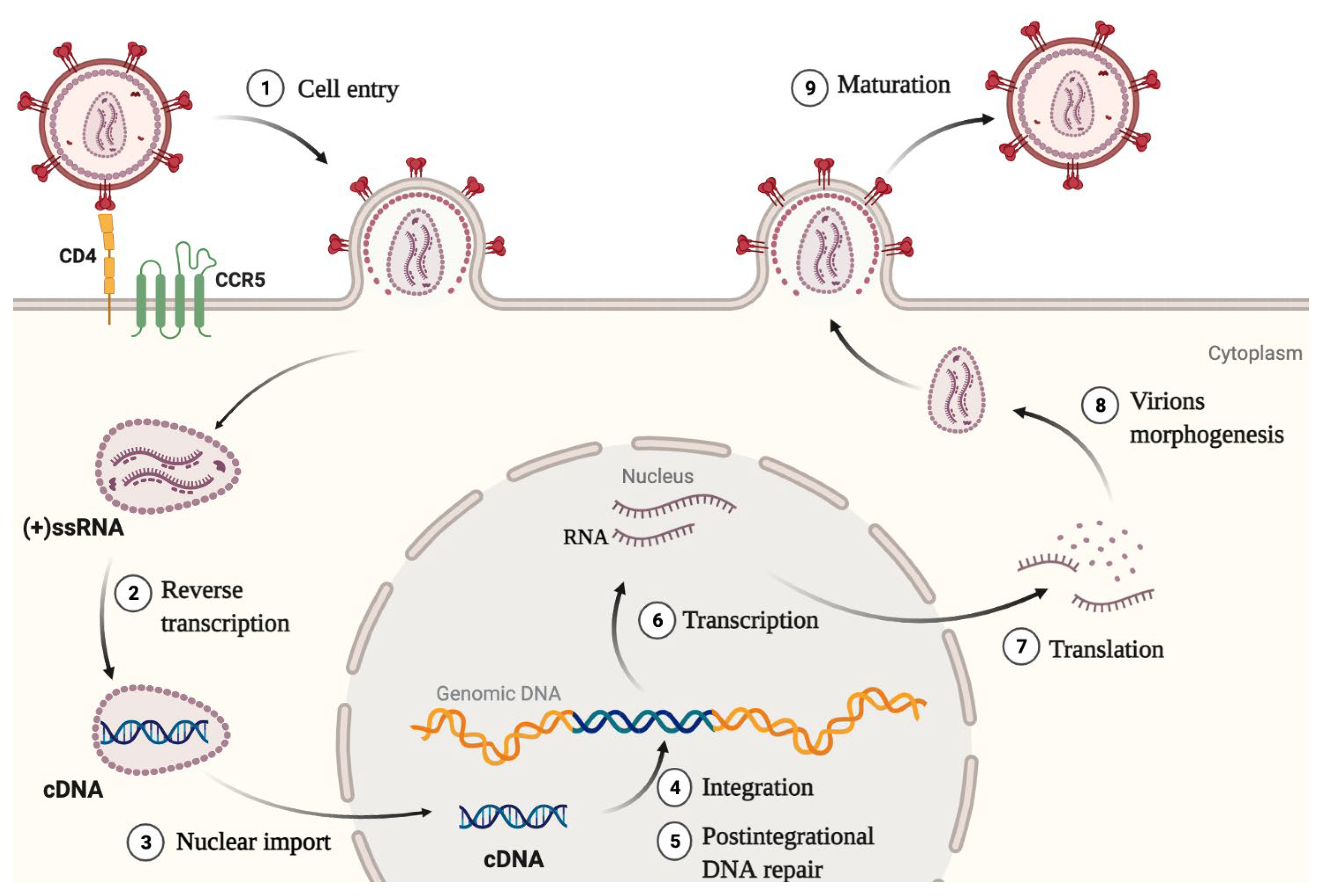
:1. Introduction
- IN was shown to enhance the initiation and elongation of reverse transcription. Also, direct interaction between IN and RT was proved to be necessary for reverse transcription [15].
- IN helps to maintain optimal capsid stability in the cytoplasm by promoting interaction between CA and its cellular cofactor CypA. Lack of IN or a mutation C130S in it impairs the interaction between CA and CypA, which leads to defective reverse transcription and a block in replication [16]. This is an indirect mechanism of IN influence on reverse transcription efficiency.
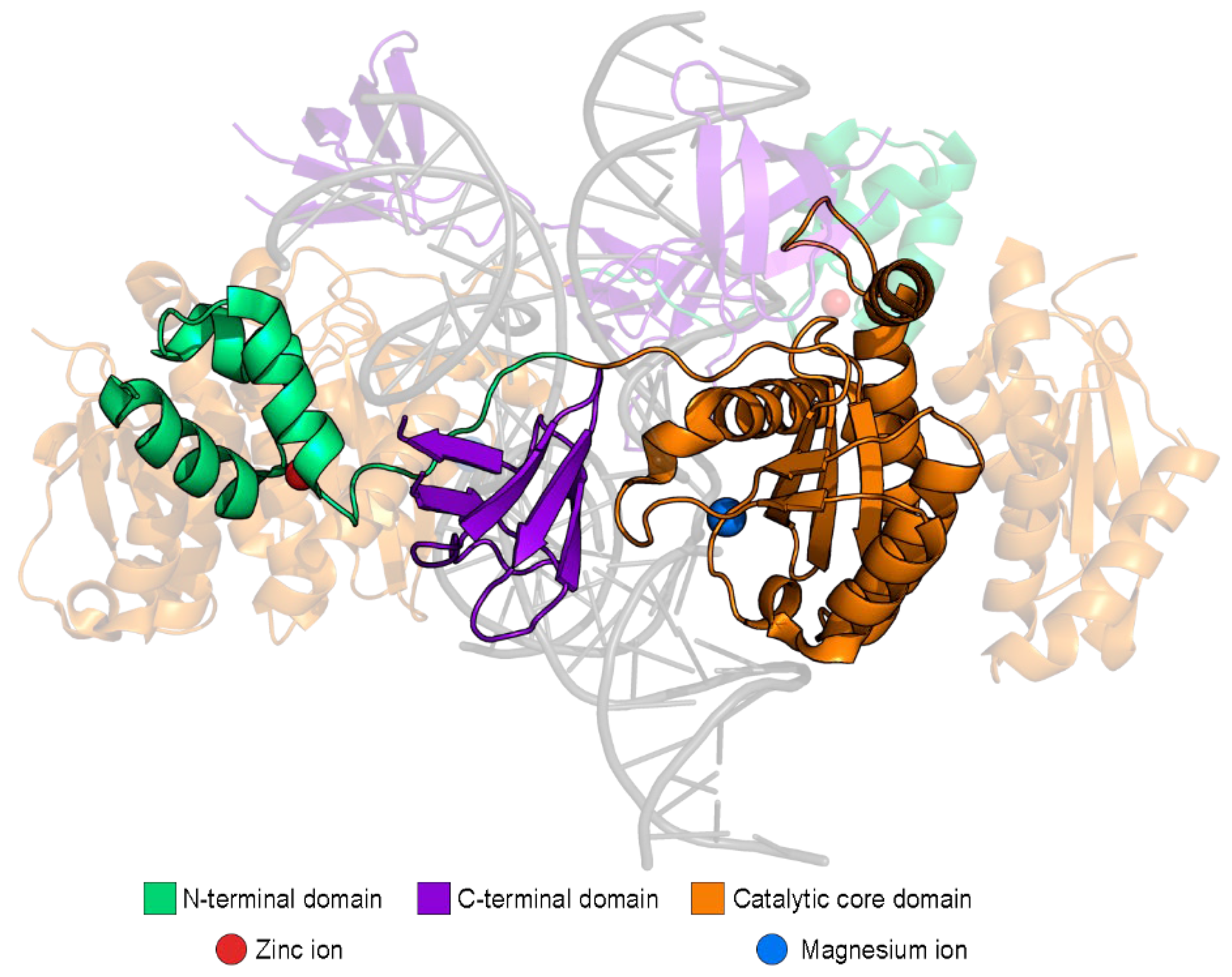
- IN interacts with the HIV-1 RNA genome, and this interaction is necessary for the correct packaging of the genome into viral particles [12]. For the successful interaction, IN must be in a tetramer form. A decreased level of IN, substances that disrupt its multimerization, and mutations that disrupt the RNA—IN interaction lead to the formation of non-infectious virions with eccentric morphology [21].
2. Factors Affecting Integrase Stability
2.1. TRIM33
2.2. CRL2VHL Complex
2.3. hRAD18
2.4. LEDGF/p75
2.5. Ku70
3. Factors Affecting Integrase Acetylation
3.1. p300
3.2. GCN5
3.3. HDAC1
3.4. KAP1, Link with HDAC1
3.5. HDAC10
4. Factors Affecting Integrase Phosphorylation
4.1. JNK and Pin1
4.2. GCN2
5. Factors Affecting Integrase Sumoylation
6. Factors Affecting Nuclear Import of Integrase
6.1. Importin α
6.2. Importin 7
6.3. Transportin-SR2 (TNPO3)
6.4. NUP153
7. Factors Affecting Integration Site Selection
7.1. LEDGF/p75
7.2. HRP2
7.3. INI1
8. DNA-Repair Factors Interacting with Integrase
8.1. hRAD51 and hRAD18
8.2. Ku70
8.3. FANCI-D2 Complex
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Hamid, F.B.; Kim, J.; Shin, C.G. Distribution and fate of HIV-1 unintegrated DNA species: A comprehensive update. AIDS Res. Ther. 2017, 14, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freed, E.O. HIV-1 assembly, release and maturation. Nat. Rev. Microbiol. 2015, 13, 484. [Google Scholar] [CrossRef] [PubMed]
- Spearman, P.; Freed, E.O. HIV Interactions with Host Cell Proteins; Springer: Berlin/Heidelberg, Germany, 2009; 204p. [Google Scholar]
- Dyda, F.; Hickman, A.B.; Jenkins, T.M.; Engelman, A.; Craigie, R.; Davies, D.R. Crystal structure of the catalytic domain of HIV-1 integrase: Similarity to other polynucleotidyl transferases. Science 1994, 266, 1981–1986. [Google Scholar] [CrossRef] [PubMed]
- Lesbats, P.; Engelman, A.N.; Cherepanov, P. Retroviral DNA Integration. Chem. Rev. 2016, 116, 12730–12757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposito, D.; Craigie, R. HIV integrase structure and function. Adv. Virus Res. 1999, 52, 319–333. [Google Scholar]
- Jaskolski, M.; Alexandratos, J.N.; Bujacz, G.; Wlodawer, A. Piecing together the structure of retroviral integrase, an important target in AIDS therapy. FEBS J. 2009, 276, 2926–2946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhargava, A.; Lahaye, X.; Manel, N. Let me in: Control of HIV nuclear entry at the nuclear envelope. Cytokine Growth Factor Rev. 2018, 40, 59–67. [Google Scholar] [CrossRef]
- Hamid, F.B.; Kim, J.; Shin, C.G. Cellular and viral determinants of retroviral nuclear entry. Can. J. Microbiol. 2016, 62, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Zheng, R.; Jenkins, T.M.; Craigie, R. Zinc folds the N-terminal domain of HIV-1 integrase, promotes multimerization, and enhances catalytic activity. Proc. Natl. Acad. Sci. USA 1996, 93, 13659–13664. [Google Scholar] [CrossRef] [Green Version]
- Craigie, R. HIV integrase, a brief overview from chemistry to therapeutics. J. Biol. Chem. 2001, 276, 23213–23216. [Google Scholar] [CrossRef] [Green Version]
- Kessl, J.J.; Kutluay, S.B.; Townsend, D.; Rebensburg, S.; Slaughter, A.; Larue, R.C.; Shkriabai, N.; Bakouche, N.; Fuchs, J.R.; Bieniasz, P.D.; et al. HIV-1 Integrase Binds the Viral RNA Genome and Is Essential during Virion Morphogenesis. Cell 2016, 166, 1257–1268.e12. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Dobard, C.; Chow, S.A. Requirement for integrase during reverse transcription of human immunodeficiency virus type 1 and the effect of cysteine mutations of integrase on its interactions with reverse transcriptase. J. Virol. 2004, 78, 5045–5055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hehl, E.A.; Joshi, P.; Kalpana, G.V.; Prasad, V.R. Interaction between human immunodeficiency virus type 1 reverse transcriptase and integrase proteins. J. Virol. 2004, 78, 5056–5067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tekeste, S.S.; Wilkinson, T.A.; Weiner, E.M.; Xu, X.; Miller, J.T.; Le Grice, S.F.J.; Clubb, R.T.; Chow, S.A. Interaction between Reverse Transcriptase and Integrase Is Required for Reverse Transcription during HIV-1 Replication. J. Virol. 2015, 89, 12058–12069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briones, M.S.; Dobard, C.W.; Chow, S.A. Role of human immunodeficiency virus type 1 integrase in uncoating of the viral core. J. Virol. 2010, 84, 5181–5190. [Google Scholar] [CrossRef] [Green Version]
- Levin, A.; Armon-Omer, A.; Rosenbluh, J.; Melamed-Book, N.; Graessmann, A.; Waigmann, E.; Loyter, A. Inhibition of HIV-1 integrase nuclear import and replication by a peptide bearing integrase putative nuclear localization signal. Retrovirology 2009, 6, 112. [Google Scholar] [CrossRef] [Green Version]
- Bouyac-Bertoia, M.; Dvorin, J.D.; Fouchier, R.A.M.; Jenkins, Y.; Meyer, B.E.; Wu, L.I.; Emerman, M.; Malim, M.H. HIV-1 Infection Requires a Functional Integrase NLS. Mol. Cell 2001, 7, 1025–1035. [Google Scholar] [CrossRef]
- Gallay, P.; Hope, T.; Chin, D.; Trono, D. HIV-1 infection of nondividing cells through the recognition of integrase by the importin/karyopherin pathway. Proc. Natl. Acad. Sci. USA 1997, 94, 9825–9830. [Google Scholar] [CrossRef] [Green Version]
- Ao, Z.; Danappa Jayappa, K.; Wang, B.; Zheng, Y.; Kung, S.; Rassart, E.; Depping, R.; Kohler, M.; Cohen, E.A.; Yao, X. Importin α3 Interacts with HIV-1 Integrase and Contributes to HIV-1 Nuclear Import and Replication. J. Virol. 2010, 84, 8650–8663. [Google Scholar] [CrossRef] [Green Version]
- Elliott, J.L.; Kutluay, S.B. Going beyond Integration: The Emerging Role of HIV-1 Integrase in Virion Morphogenesis. Viruses 2020, 12, 1005. [Google Scholar] [CrossRef]
- Anisenko, A.N.; Knyazhanskaya, E.S.; Zalevsky, A.O.; Agapkina, J.Y.; Sizov, A.I.; Zatsepin, T.S.; Gottikh, M.B. Characterization of HIV-1 integrase interaction with human Ku70 protein and initial implications for drug targeting. Sci. Rep. 2017, 7, 5649. [Google Scholar] [CrossRef] [PubMed]
- Knyazhanskaya, E.; Anisenko, A.; Shadrina, O.; Kalinina, A.; Zatsepin, T.; Zalevsky, A.; Mazurov, D.; Gottikh, M. NHEJ pathway is involved in post-integrational DNA repair due to Ku70 binding to HIV-1 integrase. Retrovirology 2019, 16, 30. [Google Scholar] [CrossRef] [PubMed]
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef] [PubMed]
- Kapitonova, M.A.; Shadrina, O.A.; Korolev, S.P.; Gottikh, M.B. Main Approaches to Controlled Protein Degradation in the Cell. Mol. Biol. 2021, 55, 543–561. [Google Scholar] [CrossRef]
- Park, J.; Cho, J.; Song, E.J. Ubiquitin-proteasome system (UPS) as a target for anticancer treatment. Arch. Pharm. Res. 2020, 43, 1144–1161. [Google Scholar] [CrossRef]
- Ali, H.; Mano, M.; Braga, L.; Naseem, A.; Marini, B.; Vu, D.M.; Collesi, C.; Meroni, G.; Lusic, M.; Giacca, M. Cellular TRIM33 restrains HIV-1 infection by targeting viral integrase for proteasomal degradation. Nat. Commun. 2019, 10, 926. [Google Scholar] [CrossRef] [Green Version]
- Cardote, T.A.F.; Gadd, M.S.; Ciulli, A. Crystal Structure of the Cul2-Rbx1-EloBC-VHL Ubiquitin Ligase Complex. Structure 2017, 25, 901–911.e3. [Google Scholar] [CrossRef] [Green Version]
- Mousnier, A.; Kubat, N.; Massias-Simon, A.; Ségéral, E.; Rain, J.C.; Benarous, R.; Emiliani, S.; Dargemont, C. Von Hippel-Lindau binding protein 1-mediated degradation of integrase affects HIV-1 gene expression at a postintegration step. Proc. Natl. Acad. Sci. USA 2007, 104, 13615–13620. [Google Scholar] [CrossRef] [Green Version]
- Rain, J.C.; Cribier, A.; Gérard, A.; Emiliani, S.; Benarous, R. Yeast two-hybrid detection of integrase–host factor interactions. Methods 2009, 47, 291–297. [Google Scholar] [CrossRef]
- Mulder, L.C.F.; Chakrabarti, L.A.; Muesing, M.A. Interaction of HIV-1 integrase with DNA repair protein hRad18. J. Biol. Chem. 2002, 277, 27489–27493. [Google Scholar] [CrossRef] [Green Version]
- Tateishi, S.; Sakuraba, O.; Masuyama, S.; Inoue, R.; Yamaizumi, M. Dysfunction of human Rad18 results in defective postreplication repair and hypersensitivity to multiple mutagens. Proc. Natl. Acad. Sci. USA 2000, 97, 7927–7932. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.P.; Ohguro, N.; Chylack, L.T.; Shinohara, T. Lens epithelium-derived growth factor: Increased resistance to thermal and oxidative stresses. Investig. Ophthalmol. Vis. Sci. 1999, 40, 1444–1451. [Google Scholar]
- Matsui, H.; Lin, L.R.; Singh, D.P.; Shinobara, T.; Reddy, V.N. Lens epithelium-derived growth factor: Increased survival and decreased DNA breakage of human RPE cells induced by oxidative stress. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2935–2941. [Google Scholar]
- Nakamura, M.; Singh, D.P.; Kubo, E.; Chylack, L.T.; Shinohara, T. LEDGF: Survival of embryonic chick retinal photoreceptor cells. Investig. Ophthalmol. Vis. Sci. 2000, 41, 1168–1175. [Google Scholar]
- Daugaard, M.; Baude, A.; Fugger, K.; Povlsen, L.K.; Beck, H.; Sørensen, C.S.; Petersen, N.H.T.; Sorensen, P.H.B.; Lukas, C.; Bartek, J.; et al. LEDGF (p75) promotes DNA-end resection and homologous recombination. Nat. Struct. Mol. Biol. 2012, 19, 803–810. [Google Scholar] [CrossRef]
- El Ashkar, S.; Schwaller, J.; Pieters, T.; Goossens, S.; Demeulemeester, J.; Christ, F.; Van Belle, S.; Juge, S.; Boeckx, N.; Engelman, A.; et al. LEDGF/p75 is dispensable for hematopoiesis but essential for MLL-rearranged leukemogenesis. Blood 2018, 131, 95–107. [Google Scholar] [CrossRef] [Green Version]
- Canella, A.; Van Belle, S.; Brouns, T.; Nigita, G.; Carlon, M.S.; Christ, F.; Debyser, Z. LEDGF/p75-mediated chemoresistance of mixed-lineage leukemia involves cell survival pathways and super enhancer activators. Cancer Gene Ther. 2022, 29, 133–140. [Google Scholar] [CrossRef]
- Chylack, L.T.; Fu, L.; Mancini, R.; Martin-Rehrmann, M.D.; Saunders, A.J.; Konopka, G.; Tian, D.; Hedley-Whyte, E.T.; Folkerth, R.D.; Goldstein, L.E. Lens epithelium-derived growth factor (LEDGF/p75) expression in fetal and adult human brain. Exp. Eye Res. 2004, 79, 941–948. [Google Scholar] [CrossRef]
- Llano, M.; Delgado, S.; Vanegas, M.; Poeschla, E.M. Lens epithelium-derived growth factor/p75 prevents proteasomal degradation of HIV-1 integrase. J. Biol. Chem. 2004, 279, 55570–55577. [Google Scholar] [CrossRef] [Green Version]
- Studamire, B.; Goff, S.P. Host proteins interacting with the Moloney murine leukemia virus integrase: Multiple transcriptional regulators and chromatin binding factors. Retrovirology 2008, 5, 48. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Ao, Z.; Wang, B.; Jayappa, K.D.; Yao, X. Host protein Ku70 binds and protects HIV-1 integrase from proteasomal degradation and is required for HIV replication. J. Biol. Chem. 2011, 286, 17722–17735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rathaus, M.; Lerrer, B.; Cohen, H.Y. DeubiKuitylation: A novel DUB enzymatic activity for the DNA repair protein, Ku70. Cell Cycle 2009, 8, 1843–1852. [Google Scholar] [CrossRef] [PubMed]
- Narita, T.; Weinert, B.T.; Choudhary, C. Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 156–174. [Google Scholar] [CrossRef] [PubMed]
- Cereseto, A.; Manganaro, L.; Gutierrez, M.I.; Terreni, M.; Fittipaldi, A.; Lusic, M.; Marcello, A.; Giacca, M. Acetylation of HIV-1 integrase by p300 regulates viral integration. EMBO J. 2005, 24, 3070–3081. [Google Scholar] [CrossRef]
- Topper, M.; Luo, Y.; Zhadina, M.; Mohammed, K.; Smith, L.; Muesing, M.A. Posttranslational acetylation of the human immunodeficiency virus type 1 integrase carboxyl-terminal domain is dispensable for viral replication. J. Virol. 2007, 81, 3012–3017. [Google Scholar] [CrossRef] [Green Version]
- Apolonia, L.; Waddington, S.N.; Fernandes, C.; Ward, N.J.; Bouma, G.; Blundell, M.P.; Thrasher, A.J.; Collins, M.K.; Philpott, N.J. Stable gene transfer to muscle using non-integrating lentiviral vectors. Mol. Ther. 2007, 15, 1947–1954. [Google Scholar] [CrossRef]
- Terreni, M.; Valentini, P.; Liverani, V.; Gutierrez, M.I.; Di Primio, C.; Di Fenza, A.; Tozzini, V.; Allouch, A.; Albanese, A.; Giacca, M.; et al. GCN5-dependent acetylation of HIV-1 integrase enhances viral integration. Retrovirology 2010, 7, 18. [Google Scholar] [CrossRef] [Green Version]
- Winans, S.; Goff, S.P. Mutations altering acetylated residues in the CTD of HIV-1 integrase cause defects in proviral transcription at early times after integration of viral DNA. PLoS Pathog. 2020, 16, e1009147. [Google Scholar] [CrossRef]
- Winans, S.; Yu, H.J.; de los Santos, K.; Wang, G.Z.; KewalRamani, V.N.; Goff, S.P. A point mutation in HIV-1 integrase redirects proviral integration into centromeric repeats. Nat. Commun. 2022, 13, 1474. [Google Scholar] [CrossRef]
- Larguet, F.; Caté, C.; Barbeau, B.; Rassart, E.; Edouard, E. Histone deacetylase 1 interacts with HIV-1 Integrase and modulates viral replication. Virol. J. 2019, 16, 138. [Google Scholar] [CrossRef]
- Allouch, A.; Di Primio, C.; Alpi, E.; Lusic, M.; Arosio, D.; Giacca, M.; Cereseto, A. The TRIM family protein KAP1 inhibits HIV-1 integration. Cell Host Microbe 2011, 9, 484–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorin, M.; Cano, J.; Das, S.; Mathew, S.; Wu, X.; Davies, K.P.; Shi, X.; Cheng, S.W.G.; Ott, D.; Kalpana, G.V. Recruitment of a SAP18-HDAC1 Complex into HIV-1 Virions and Its Requirement for Viral Replication. PLOS Pathog. 2009, 5, e1000463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahringer, J. NuRD and SIN3: Histone deacetylase complexes in development. Trends Genet. 2000, 16, 351–356. [Google Scholar] [CrossRef]
- Kalpana, G.V.; Marmon, S.; Wang, W.; Crabtree, G.R.; Goff, S.P. Binding and stimulation of HIV-1 integrase by a human homolog of yeast transcription factor SNF5. Science 1994, 266, 2002–2006. [Google Scholar] [CrossRef]
- Grassi, D.A.; Jönsson, M.E.; Brattås, P.L.; Jakobsson, J. TRIM28 and the control of transposable elements in the brain. Brain Res. 2019, 1705, 43–47. [Google Scholar] [CrossRef]
- Taura, M.; Song, E.; Ho, Y.C.; Iwasaki, A. Apobec3A maintains HIV-1 latency through recruitment of epigenetic silencing machinery to the long terminal repeat. Proc. Natl. Acad. Sci. USA 2019, 116, 2282–2289. [Google Scholar] [CrossRef] [Green Version]
- Ait-Ammar, A.; Bellefroid, M.; Daouad, F.; Martinelli, V.; Van Assche, J.; Wallet, C.; Rodari, A.; De Rovere, M.; Fahrenkrog, B.; Schwartz, C.; et al. Inhibition of HIV-1 gene transcription by KAP1 in myeloid lineage. Sci. Rep. 2021, 11, 2692. [Google Scholar] [CrossRef]
- Ran, X.; Ao, Z.; Trajtman, A.; Xu, W.; Kobinger, G.; Keynan, Y.; Yao, X. HIV-1 envelope glycoprotein stimulates viral transcription and increases the infectivity of the progeny virus through the manipulation of cellular machinery. Sci. Rep. 2017, 7, 9487. [Google Scholar] [CrossRef] [Green Version]
- Ran, X.; Ao, Z.; Olukitibi, T.; Yao, X. Characterization of the Role of Host Cellular Factor Histone Deacetylase 10 during HIV-1 Replication. Viruses 2019, 12, 28. [Google Scholar] [CrossRef] [Green Version]
- Hardman, G.; Perkins, S.; Brownridge, P.J.; Clarke, C.J.; Byrne, D.P.; Campbell, A.E.; Kalyuzhnyy, A.; Myall, A.; Eyers, P.A.; Jones, A.R.; et al. Strong anion exchange-mediated phosphoproteomics reveals extensive human non-canonical phosphorylation. EMBO J. 2019, 38, e100847. [Google Scholar] [CrossRef]
- Cohen, P. The origins of protein phosphorylation. Nat. Cell Biol. 2002, 4, E127–E130. [Google Scholar] [CrossRef] [PubMed]
- Manganaro, L.; Lusic, M.; Gutierrez, M.I.; Cereseto, A.; Del Sal, G.; Giacca, M. Concerted action of cellular JNK and Pin1 restricts HIV-1 genome integration to activated CD4+ T lymphocytes. Nat. Med. 2010, 16, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wu, Y.R.; Yang, H.Y.; Li, X.Z.; Jie, M.M.; Hu, C.J.; Wu, Y.Y.; Yang, S.M.; Yang, Y.B. Prolyl isomerase Pin1: A promoter of cancer and a target for therapy. Cell Death Dis. 2018, 9, 883. [Google Scholar] [CrossRef] [Green Version]
- Saleh, S.; Lu, H.K.; Evans, V.; Harisson, D.; Zhou, J.; Jaworowski, A.; Sallmann, G.; Cheong, K.Y.; Mota, T.M.; Tennakoon, S.; et al. HIV integration and the establishment of latency in CCL19-treated resting CD4(+) T cells require activation of NF-κB. Retrovirology 2016, 13, 49. [Google Scholar] [CrossRef]
- Jaspart, A.; Calmels, C.; Cosnefroy, O.; Bellecave, P.; Pinson, P.; Claverol, S.; Guyonnet-Dupérat, V.; Dartigues, B.; Benleulmi, M.S.; Mauro, E.; et al. GCN2 phosphorylates HIV-1 integrase and decreases HIV-1 replication by limiting viral integration. Sci. Rep. 2017, 7, 2283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masson, G.R. Towards a model of GCN2 activation. Biochem. Soc. Trans. 2019, 47, 1481–1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosnefroy, O.; Jaspart, A.; Calmels, C.; Parissi, V.; Fleury, H.; Ventura, M.; Reigadas, S.; Andréola, M.L. Activation of GCN2 upon HIV-1 infection and inhibition of translation. Cell. Mol. Life Sci. 2013, 70, 2411–2421. [Google Scholar] [CrossRef]
- Vertegaal, A.C.O. Signalling mechanisms and cellular functions of SUMO. Nat. Rev. Mol. Cell Biol. 2022, 23, 715–731. [Google Scholar] [CrossRef]
- Varejão, N.; Lascorz, J.; Codina-Fabra, J.; Bellí, G.; Borràs-Gas, H.; Torres-Rosell, J.; Reverter, D. Structural basis for the E3 ligase activity enhancement of yeast Nse2 by SUMO-interacting motifs. Nat. Commun. 2021, 12, 7013. [Google Scholar] [CrossRef]
- Li, Z.; Wu, S.; Wang, J.; Li, W.; Lin, Y.; Ji, C.; Xue, J.; Chen, J. Evaluation of the interactions of HIV-1 integrase with small ubiquitin-like modifiers and their conjugation enzyme Ubc9. Int. J. Mol. Med. 2012, 30, 1053–1060. [Google Scholar] [CrossRef] [Green Version]
- Zamborlini, A.; Coiffic, A.; Beauclair, G.; Delelis, O.; Paris, J.; Koh, Y.; Magne, F.; Giron, M.L.; Tobaly-Tapiero, J.; Deprez, E.; et al. Impairment of human immunodeficiency virus type-1 integrase SUMOylation correlates with an early replication defect. J. Biol. Chem. 2011, 286, 21013–21022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.J.; Chiang, C.M. Sumoylation in gene regulation, human disease, and therapeutic action. F1000Prime Rep. 2013, 5, 45. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Jayappa, K.D.; Ao, Z.; Qiu, X.; Su, R.C.; Yao, X. Noncovalent SUMO-interaction motifs in HIV integrase play important roles in SUMOylation, cofactor binding, and virus replication. Virol. J. 2019, 16, 42. [Google Scholar] [CrossRef] [PubMed]
- Jaber, T.; Bohl, C.R.; Lewis, G.L.; Wood, C.; West, J.T.; Weldon, R.A. Human Ubc9 contributes to production of fully infectious human immunodeficiency virus type 1 virions. J. Virol. 2009, 83, 10448–10459. [Google Scholar] [CrossRef] [PubMed]
- Gurer, C.; Berthoux, L.; Luban, J. Covalent modification of human immunodeficiency virus type 1 p6 by SUMO-1. J. Virol. 2005, 79, 910–917. [Google Scholar] [CrossRef] [Green Version]
- Mete, B.; Pekbilir, E.; Bilge, B.N.; Georgiadou, P.; Çelik, E.; Sutlu, T.; Tabak, F.; Sahin, U. Human immunodeficiency virus type 1 impairs sumoylation. Life Sci. Alliance 2022, 5, e202101103. [Google Scholar] [CrossRef]
- Strambio-De-Castillia, C.; Niepel, M.; Rout, M.P. The nuclear pore complex: Bridging nuclear transport and gene regulation. Nat. Rev. Mol. Cell Biol. 2010, 11, 490–501. [Google Scholar] [CrossRef]
- Piller, S.C.; Caly, L.; Jans, D.A. Nuclear Import of the Pre-Integration Complex (PIC): The Achilles Heel of HIV? Curr. Drug Targets 2005, 4, 409–429. [Google Scholar] [CrossRef]
- Depienne, C.; Roques, P.; Créminon, C.; Fritsch, L.; Casseron, R.; Dormont, D.; Dargemont, C.; Benichou, S. Cellular Distribution and Karyophilic Properties of Matrix, Integrase, and Vpr Proteins from the Human and Simian Immunodeficiency Viruses. Exp. Cell Res. 2000, 260, 387–395. [Google Scholar] [CrossRef]
- Depienne, C.; Mousnier, A.; Leh, H.; Le Rouzic, E.; Dormont, D.; Benichou, S.; Dargemont, C. Characterization of the Nuclear Import Pathway for HIV-1 Integrase. J. Biol. Chem. 2001, 276, 18102–18107. [Google Scholar] [CrossRef] [Green Version]
- Woodward, C.L.; Prakobwanakit, S.; Mosessian, S.; Chow, S.A. Integrase Interacts with Nucleoporin NUP153 To Mediate the Nuclear Import of Human Immunodeficiency Virus Type 1. J. Virol. 2009, 83, 6522–6533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoneda, Y.; Imamoto-Sonobe, N.; Yamaizumi, M.; Uchida, T. Reversible inhibition of protein import into the nucleus by wheat germ agglutinin injected into cultured cells. Exp. Cell Res. 1987, 173, 586–595. [Google Scholar] [CrossRef]
- Petit, C.; Schwartz, O.; Mammano, F. The Karyophilic Properties of Human Immunodeficiency Virus Type 1 Integrase Are Not Required for Nuclear Import of Proviral DNA. J. Virol. 2000, 74, 7119–7126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsurutani, N.; Kubo, M.; Maeda, Y.; Ohashi, T.; Yamamoto, N.; Kannagi, M.; Masuda, T. Identification of Critical Amino Acid Residues in Human Immunodeficiency Virus Type 1 IN Required for Efficient Proviral DNA Formation at Steps prior to Integration in Dividing and Nondividing Cells. J. Virol. 2000, 74, 4795–4806. [Google Scholar] [CrossRef] [PubMed]
- Limón, A.; Devroe, E.; Lu, R.; Ghory, H.Z.; Silver, P.A.; Engelman, A. Nuclear Localization of Human Immunodeficiency Virus Type 1 Preintegration Complexes (PICs): V165A and R166A Are Pleiotropic Integrase Mutants Primarily Defective for Integration, Not PIC Nuclear Import. J. Virol. 2002, 76, 10598–10607. [Google Scholar] [CrossRef] [Green Version]
- Dvorin, J.D.; Bell, P.; Maul, G.G.; Yamashita, M.; Emerman, M.; Malim, M.H. Reassessment of the roles of integrase and the central DNA flap in human immunodeficiency virus type 1 nuclear import. J. Virol. 2002, 76, 12087–12096. [Google Scholar] [CrossRef] [Green Version]
- Pemberton, L.F.; Paschal, B.M. Mechanisms of Receptor-Mediated Nuclear Import and Nuclear Export. Traffic 2005, 6, 187–198. [Google Scholar] [CrossRef]
- Pumroy, R.A.; Cingolani, G. Diversification of importin-α isoforms in cellular trafficking and disease states. Biochem. J. 2015, 466, 13–28. [Google Scholar] [CrossRef] [Green Version]
- Hearps, A.C.; Jans, D.A. HIV-1 integrase is capable of targeting DNA to the nucleus via an importin alpha/beta-dependent mechanism. Biochem. J. 2006, 398, 475–484. [Google Scholar] [CrossRef]
- Levin, A.; Hayouka, Z.; Friedler, A.; Loyter, A. Transportin 3 and importin α are required for effective nuclear import of HIV-1 integrase in virus-infected cells. Nucleus 2010, 1, 422–431. [Google Scholar] [CrossRef] [Green Version]
- Ao, Z.; Huang, G.; Yao, H.; Xu, Z.; Labine, M.; Cochrane, A.W.; Yao, X. Interaction of human immunodeficiency virus type 1 integrase with cellular nuclear import receptor importin 7 and its impact on viral replication. J. Biol. Chem. 2007, 282, 13456–13467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jäkel, S.; Albig, W.; Kutay, U.; Bischoff, F.R.; Schwamborn, K.; Doenecke, D.; Görlich, D. The importin β/importin 7 heterodimer is a functional nuclear import receptor for histone H1. EMBO J. 1999, 18, 2411–2423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaitseva, L.; Cherepanov, P.; Leyens, L.; Wilson, S.J.; Rasaiyaah, J.; Fassati, A. HIV-1 exploits importin 7 to maximize nuclear import of its DNA genome. Retrovirology 2009, 6, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fassati, A.; Görlich, D.; Harrison, I.; Zaytseva, L.; Mingot, J.M. Nuclear import of HIV-1 intracellular reverse transcription complexes is mediated by importin 7. EMBO J. 2003, 22, 3675–3685. [Google Scholar] [CrossRef]
- Tabasi, M.; Nombela, I.; Janssens, J.; Lahousse, A.P.; Christ, F.; Debyser, Z. Role of Transportin-SR2 in HIV-1 Nuclear Import. Viruses 2021, 13, 829. [Google Scholar] [CrossRef]
- Christ, F.; Thys, W.; De Rijck, J.; Gijsbers, R.; Albanese, A.; Arosio, D.; Emiliani, S.; Rain, J.C.; Benarous, R.; Cereseto, A.; et al. Transportin-SR2 Imports HIV into the Nucleus. Curr. Biol. 2008, 18, 1192–1202. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, L.; Matreyek, K.A.; Oztop, I.; Lee, K.; Tipper, C.H.; Li, X.; Dar, M.J.; KewalRamani, V.N.; Engelman, A. The Requirement for Cellular Transportin 3 (TNPO3 or TRN-SR2) during Infection Maps to Human Immunodeficiency Virus Type 1 Capsid and Not Integrase. J. Virol. 2010, 84, 397–406. [Google Scholar] [CrossRef] [Green Version]
- De Houwer, S.; Demeulemeester, J.; Thys, W.; Taltynov, O.; Zmajkovicova, K.; Christ, F.; Debyser, Z. Identification of residues in the C-terminal domain of HIV-1 integrase that mediate binding to the transportin-SR2 protein. J. Biol. Chem. 2012, 287, 34059–34068. [Google Scholar] [CrossRef] [Green Version]
- Larue, R.; Gupta, K.; Wuensch, C.; Shkriabai, N.; Kessl, J.J.; Danhart, E.; Feng, L.; Taltynov, O.; Christ, F.; Van Duyne, G.D.; et al. Interaction of the HIV-1 intasome with transportin 3 protein (TNPO3 or TRN-SR2). J. Biol. Chem. 2012, 287, 34044–34058. [Google Scholar] [CrossRef] [Green Version]
- De Houwer, S.; Demeulemeester, J.; Thys, W.; Rocha, S.; Dirix, L.; Gijsbers, R.; Christ, F.; Debyser, Z. The HIV-1 integrase mutant R263A/K264A is 2-fold defective for TRN-SR2 binding and viral nuclear import. J. Biol. Chem. 2014, 289, 25351–25361. [Google Scholar] [CrossRef] [Green Version]
- Janssens, J.; Blokken, J.; Lampi, Y.; De Wit, F.; Zurnic Bonisch, I.; Nombela, I.; Van de Velde, P.; Van Remoortel, B.; Gijsbers, R.; Christ, F.; et al. CRISPR/Cas9-Induced Mutagenesis Corroborates the Role of Transportin-SR2 in HIV-1 Nuclear Import. Microbiol. Spectr. 2021, 9, e01336-21. [Google Scholar] [CrossRef] [PubMed]
- De Iaco, A.; Luban, J. Inhibition of HIV-1 infection by TNPO3 depletion is determined by capsid and detectable after viral cDNA enters the nucleus. Retrovirology 2011, 8, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.J.; Lopez, L.A.; Exline, C.M.; Haworth, K.G.; Cannon, P.M. Lack of adaptation to human tetherin in HIV-1 Group O and P. Retrovirology 2011, 8, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brass, A.L.; Dykxhoorn, D.M.; Benita, Y.; Yan, N.; Engelman, A.; Xavier, R.J.; Lieberman, J.; Elledge, S.J. Identification of host proteins required for HIV infection through a functional genomic screen. Science 2008, 319, 921–926. [Google Scholar] [CrossRef]
- Shen, Q.; Wu, C.; Freniere, C.; Tripler, T.N.; Xiong, Y. Nuclear Import of HIV-1. Viruses 2021, 13, 2242. [Google Scholar] [CrossRef]
- Dharan, A.; Campbell, E.M. Teaching old dogmas new tricks: Recent insights into the nuclear import of HIV-1. Curr. Opin. Virol. 2022, 53, 101203. [Google Scholar] [CrossRef]
- Burdick, R.C.; Li, C.; Munshi, M.H.; Rawson, J.M.O.; Nagashima, K.; Hu, W.S.; Pathak, V.K. HIV-1 uncoats in the nucleus near sites of integration. Proc. Natl. Acad. Sci. USA 2020, 117, 5486–5493. [Google Scholar] [CrossRef]
- Li, C.; Burdick, R.C.; Nagashima, K.; Hu, W.S.; Pathak, V.K. HIV-1 cores retain their integrity until minutes before uncoating in the nucleus. Proc. Natl. Acad. Sci. USA 2021, 118, e2019467118. [Google Scholar] [CrossRef]
- Dharan, A.; Bachmann, N.; Talley, S.; Zwikelmaier, V.; Campbell, E.M. Nuclear pore blockade reveals that HIV-1 completes reverse transcription and uncoating in the nucleus. Nat. Microbiol. 2020, 5, 1088–1095. [Google Scholar] [CrossRef]
- Selyutina, A.; Persaud, M.; Lee, K.; KewalRamani, V.; Diaz-Griffero, F. Nuclear Import of the HIV-1 Core Precedes Reverse Transcription and Uncoating. Cell Rep. 2020, 32, 108201. [Google Scholar] [CrossRef]
- Zila, V.; Margiotta, E.; Turoňová, B.; Müller, T.G.; Zimmerli, C.E.; Mattei, S.; Allegretti, M.; Börner, K.; Rada, J.; Müller, B.; et al. Cone-shaped HIV-1 capsids are transported through intact nuclear pores. Cell 2021, 184, 1032–1046.e18. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.G.; Zila, V.; Peters, K.; Schifferdecker, S.; Stanic, M.; Lucic, B.; Laketa, V.; Lusic, M.; Müller, B.; Kräusslich, H.G. HIV-1 uncoating by release of viral cDNA from capsid-like structures in the nucleus of infected cells. Elife 2021, 10, e64776. [Google Scholar] [CrossRef]
- Mamede, J.I.; Cianci, G.C.; Anderson, M.R.; Hope, T.J. Early cytoplasmic uncoating is associated with infectivity of HIV-1. Proc. Natl. Acad. Sci. USA 2017, 114, E7169–E7178. [Google Scholar] [CrossRef] [Green Version]
- Francis, A.C.; Marin, M.; Shi, J.; Aiken, C.; Melikyan, G.B. Time-Resolved Imaging of Single HIV-1 Uncoating In Vitro and in Living Cells. PLOS Pathog. 2016, 12, e1005709. [Google Scholar] [CrossRef] [PubMed]
- Francis, A.C.; Melikyan, G.B. Single HIV-1 Imaging Reveals Progression of Infection through CA-Dependent Steps of Docking at the Nuclear Pore, Uncoating, and Nuclear Transport. Cell Host Microbe 2018, 23, 536–548.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schröder, A.R.W.; Shinn, P.; Chen, H.; Berry, C.; Ecker, J.R.; Bushman, F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell 2002, 110, 521–529. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.K.; Plumb, M.R.; Ferris, A.L.; Iben, J.R.; Wu, X.; Fadel, H.J.; Luke, B.T.; Esnault, C.; Poeschla, E.M.; Hughes, S.H.; et al. LEDGF/p75 interacts with mRNA splicing factors and targets HIV-1 integration to highly spliced genes. Genes Dev. 2015, 29, 2287–2297. [Google Scholar] [CrossRef] [Green Version]
- Lucic, B.; Chen, H.C.; Kuzman, M.; Zorita, E.; Wegner, J.; Minneker, V.; Wang, W.; Fronza, R.; Laufs, S.; Schmidt, M.; et al. Spatially clustered loci with multiple enhancers are frequent targets of HIV-1 integration. Nat. Commun. 2019, 10, 4059. [Google Scholar] [CrossRef] [Green Version]
- Francis, A.C.; Marin, M.; Singh, P.K.; Achuthan, V.; Prellberg, M.J.; Palermino-Rowland, K.; Lan, S.; Tedbury, P.R.; Sarafianos, S.G.; Engelman, A.N.; et al. HIV-1 replication complexes accumulate in nuclear speckles and integrate into speckle-associated genomic domains. Nat. Commun. 2020, 11, 3505. [Google Scholar] [CrossRef]
- Galganski, L.; Urbanek, M.O.; Krzyzosiak, W.J. Nuclear speckles: Molecular organization, biological function and role in disease. Nucleic Acids Res. 2017, 45, 10350–10368. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.K.; Bedwell, G.J.; Engelman, A.N. Spatial and Genomic Correlates of HIV-1 Integration Site Targeting. Cells 2022, 11, 655. [Google Scholar] [CrossRef] [PubMed]
- Janssens, J.; Bruggemans, A.; Christ, F.; Debyser, Z. Towards a Functional Cure of HIV-1: Insight Into the Chromatin Landscape of the Provirus. Front. Microbiol. 2021, 12, 391. [Google Scholar]
- Debyser, Z.; Vansant, G.; Bruggemans, A.; Janssens, J.; Christ, F. Insight in HIV Integration Site Selection Provides a Block-and-Lock Strategy for a Functional Cure of HIV Infection. Viruses 2018, 11, 12. [Google Scholar]
- Craigie, R.; Bushman, F.D. Host Factors in Retroviral Integration and the Selection of Integration Target Sites. Microbiol. Spectr. 2014, 2, 1035–1050. [Google Scholar]
- Cherepanov, P.; Devroe, E.; Silver, P.A.; Engelman, A. Identification of an evolutionarily conserved domain in human lens epithelium-derived growth factor/transcriptional co-activator p75 (LEDGF/p75) that binds HIV-1 integrase. J. Biol. Chem. 2004, 279, 48883–48892. [Google Scholar] [CrossRef] [Green Version]
- Cherepanov, P.; Maertens, G.; Proost, P.; Devreese, B.; Van Beeumen, J.; Engelborghs, Y.; De Clercq, E.; Debyser, Z. HIV-1 integrase forms stable tetramers and associates with LEDGF/p75 protein in human cells. J. Biol. Chem. 2003, 278, 372–381. [Google Scholar] [CrossRef] [Green Version]
- Ciuffi, A.; Llano, M.; Poeschla, E.; Hoffmann, C.; Leipzig, J.; Shinn, P.; Ecker, J.R.; Bushman, F. A role for LEDGF/p75 in targeting HIV DNA integration. Nat. Med. 2005, 11, 1287–1289. [Google Scholar]
- Pradeepa, M.M.; Sutherland, H.G.; Ule, J.; Grimes, G.R.; Bickmore, W.A. Psip1/Ledgf p52 binds methylated histone H3K36 and splicing factors and contributes to the regulation of alternative splicing. PLoS Genet. 2012, 8, e1002717. [Google Scholar]
- Vandekerckhove, L.; Christ, F.; Van Maele, B.; De Rijck, J.; Gijsbers, R.; Van den Haute, C.; Witvrouw, M.; Debyser, Z. Transient and stable knockdown of the integrase cofactor LEDGF/p75 reveals its role in the replication cycle of human immunodeficiency virus. J. Virol. 2006, 80, 1886–1896. [Google Scholar] [CrossRef]
- De Rijck, J.; Bartholomeeusen, K.; Ceulemans, H.; Debyser, Z.; Gijsbers, R. High-resolution profiling of the LEDGF/p75 chromatin interaction in the ENCODE region. Nucleic Acids Res. 2010, 38, 6135–6147. [Google Scholar]
- Marshall, H.M.; Ronen, K.; Berry, C.; Llano, M.; Sutherland, H.; Saenz, D.; Bickmore, W.; Poeschla, E.; Bushman, F.D. Role of PSIP1/LEDGF/p75 in lentiviral infectivity and integration targeting. PLoS ONE 2007, 2, e1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llano, M.; Vanegas, M.; Fregoso, O.; Saenz, D.; Chung, S.; Peretz, M.; Poeschla, E.M. LEDGF/p75 determines cellular trafficking of diverse lentiviral but not murine oncoretroviral integrase proteins and is a component of functional lentiviral preintegration complexes. J. Virol. 2004, 78, 9524–9537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lampi, Y.; Van Looveren, D.; Vranckx, L.S.; Thiry, I.; Bornschein, S.; Debyser, Z.; Gijsbers, R. Targeted editing of the PSIP1 gene encoding LEDGF/p75 protects cells against HIV infection. Sci. Rep. 2019, 9, 2389. [Google Scholar] [CrossRef] [Green Version]
- Busschots, K.; Voet, A.; De Maeyer, M.; Rain, J.C.; Emiliani, S.; Benarous, R.; Desender, L.; Debyser, Z.; Christ, F. Identification of the LEDGF/p75 binding site in HIV-1 integrase. J. Mol. Biol. 2007, 365, 1480–1492. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Ao, Z.; Jayappa, K.D.; Yao, X. Characterization of the HIV-1 integrase chromatin- and LEDGF/p75-binding abilities by mutagenic analysis within the catalytic core domain of integrase. Virol. J. 2010, 7, 68. [Google Scholar] [CrossRef] [Green Version]
- Passos, D.O.; Li, M.; Yang, R.; Rebensburg, S.V.; Ghirlando, R.; Jeon, Y.; Shkriabai, N.; Kvaratskhelia, M.; Craigie, R.; Lyumkis, D. Cryo-EM structures and atomic model of the HIV-1 strand transfer complex intasome. Science 2017, 355, 89–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faure, A.; Calmels, C.; Desjobert, C.; Castroviejo, M.; Caumont-Sarcos, A.; Tarrago-Litvak, L.; Litvak, S.; Parissi, V. HIV-1 integrase crosslinked oligomers are active in vitro. Nucleic Acids Res. 2005, 33, 977–986. [Google Scholar] [CrossRef]
- Hare, S.; Di Nunzio, F.; Labeja, A.; Wang, J.; Engelman, A.; Cherepanov, P. Structural basis for functional tetramerization of lentiviral integrase. PLoS Pathog. 2009, 5, e1000515. [Google Scholar] [CrossRef] [Green Version]
- Maertens, G.N.; Engelman, A.N.; Cherepanov, P. Structure and function of retroviral integrase. Nat. Rev. Microbiol. 2022, 20, 20–34. [Google Scholar] [CrossRef]
- Rüegsegger, U.; Blank, D.; Keller, W. Human pre-mRNA cleavage factor Im is related to spliceosomal SR proteins and can be reconstituted in vitro from recombinant subunits. Mol. Cell 1998, 1, 243–253. [Google Scholar] [CrossRef]
- Chin, C.R.; Perreira, J.M.; Savidis, G.; Portmann, J.M.; Aker, A.M.; Feeley, E.M.; Smith, M.C.; Brass, A.L. Direct Visualization of HIV-1 Replication Intermediates Shows that Capsid and CPSF6 Modulate HIV-1 Intra-nuclear Invasion and Integration. Cell Rep. 2015, 13, 1717–1731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sowd, G.A.; Serrao, E.; Wang, H.; Wang, W.; Fadel, H.J.; Poeschla, E.M.; Engelman, A.N. A critical role for alternative polyadenylation factor CPSF6 in targeting HIV-1 integration to transcriptionally active chromatin. Proc. Natl. Acad. Sci. USA 2016, 113, E1054–E1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Achuthan, V.; Perreira, J.M.; Sowd, G.A.; Puray-Chavez, M.; McDougall, W.M.; Paulucci-Holthauzen, A.; Wu, X.; Fadel, H.J.; Poeschla, E.M.; Multani, A.S.; et al. Capsid-CPSF6 Interaction Licenses Nuclear HIV-1 Trafficking to Sites of Viral DNA Integration. Cell Host Microbe 2018, 24, 392–404.e8. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Jurado, K.A.; Wu, X.; Shun, M.C.; Li, X.; Ferris, A.L.; Smith, S.J.; Patel, P.A.; Fuchs, J.R.; Cherepanov, P.; et al. HRP2 determines the efficiency and specificity of HIV-1 integration in LEDGF/p75 knockout cells but does not contribute to the antiviral activity of a potent LEDGF/p75-binding site integrase inhibitor. Nucleic Acids Res. 2012, 40, 11518–11530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrijvers, R.; Vets, S.; De Rijck, J.; Malani, N.; Bushman, F.D.; Debyser, Z.; Gijsbers, R. HRP-2 determines HIV-1 integration site selection in LEDGF/p75 depleted cells. Retrovirology 2012, 9, 84. [Google Scholar] [CrossRef] [Green Version]
- Schrijvers, R.; De Rijck, J.; Demeulemeester, J.; Adachi, N.; Vets, S.; Ronen, K.; Christ, F.; Bushman, F.D.; Debyser, Z.; Gijsbers, R. LEDGF/p75-independent HIV-1 replication demonstrates a role for HRP-2 and remains sensitive to inhibition by LEDGINs. PLoS Pathog. 2012, 8, e1002558. [Google Scholar] [CrossRef] [Green Version]
- Roberts, C.W.M.; Orkin, S.H. The SWI/SNF complex--chromatin and cancer. Nat. Rev. Cancer 2004, 4, 133–142. [Google Scholar] [CrossRef]
- Lesbats, P.; Botbol, Y.; Chevereau, G.; Vaillant, C.; Calmels, C.; Arneodo, A.; Andreola, M.L.; Lavigne, M.; Parissi, V. Functional coupling between HIV-1 integrase and the SWI/SNF chromatin remodeling complex for efficient in vitro integration into stable nucleosomes. PLoS Pathog. 2011, 7, e1001280. [Google Scholar] [CrossRef] [Green Version]
- Yung, E.; Sorin, M.; Pal, A.; Craig, E.; Morozov, A.; Delattre, O.; Kappes, J.; Ott, D.; Kalpana, G.V. Inhibition of HIV-1 virion production by a transdominant mutant of integrase interactor 1. Nat. Med. 2001, 7, 920–926. [Google Scholar] [CrossRef]
- Sorin, M.; Yung, E.; Wu, X.; Kalpana, G.V. HIV-1 replication in cell lines harboring INI1/hSNF5 mutations. Retrovirology 2006, 3, 56. [Google Scholar] [CrossRef]
- Maroun, M.; Delelis, O.; Coadou, G.; Bader, T.; Ségéral, E.; Mbemba, G.; Petit, C.; Sonigo, P.; Rain, J.C.; Mouscadet, J.F.; et al. Inhibition of early steps of HIV-1 replication by SNF5/Ini1. J. Biol. Chem. 2006, 281, 22736–22743. [Google Scholar] [CrossRef] [Green Version]
- Angelov, D.; Charra, M.; Seve, M.; Côté, J.; Khochbin, S.; Dimitrov, S. Differential remodeling of the HIV-1 nucleosome upon transcription activators and SWI/SNF complex binding. J. Mol. Biol. 2000, 302, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Maillot, B.; Lévy, N.; Eiler, S.; Crucifix, C.; Granger, F.; Richert, L.; Didier, P.; Godet, J.; Pradeau-Aubreton, K.; Emiliani, S.; et al. Structural and functional role of INI1 and LEDGF in the HIV-1 preintegration complex. PLoS ONE 2013, 8, e60734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desfarges, S.; San Filippo, J.; Fournier, M.; Calmels, C.; Caumont-Sarcos, A.; Litvak, S.; Sung, P.; Parissi, V. Chromosomal integration of LTR-flanked DNA in yeast expressing HIV-1 integrase: Down regulation by RAD51. Nucleic Acids Res. 2006, 34, 6215–6224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosnefroy, O.; Tocco, A.; Lesbats, P.; Thierry, S.; Calmels, C.; Wiktorowicz, T.; Reigadas, S.; Kwon, Y.; De Cian, A.; Desfarges, S.; et al. Stimulation of the Human RAD51 Nucleofilament Restricts HIV-1 Integration In Vitro and in Infected Cells. J. Virol. 2012, 86, 513–526. [Google Scholar] [CrossRef] [Green Version]
- Thierry, S.; Benleulmi, M.S.; Sinzelle, L.; Thierry, E.; Calmels, C.; Chaignepain, S.; Waffo-Teguo, P.; Merillon, J.M.; Budke, B.; Pasquet, J.M.; et al. Dual and Opposite Effects of hRAD51 Chemical Modulation on HIV-1 Integration. Chem. Biol. 2015, 22, 712–723. [Google Scholar] [CrossRef] [Green Version]
- Anisenko, A.N.; Gottikh, M.B. Role of Cellular DNA Repair Systems in HIV-1 Replication. Mol. Biol. 2019, 53, 355–366. [Google Scholar] [CrossRef]
- Daniel, R.; Katz, R.A.; Skalka, A.M. A role for DNA-PK in retroviral DNA integration. Science 1999, 284, 644–647. [Google Scholar] [CrossRef]
- Daniel, R.; Greger, J.G.; Katz, R.A.; Taganov, K.D.; Wu, X.; Kappes, J.C.; Skalka, A.M. Evidence that stable retroviral transduction and cell survival following DNA integration depend on components of the nonhomologous end joining repair pathway. J. Virol. 2004, 78, 8573–8581. [Google Scholar] [CrossRef] [Green Version]
- Manic, G.; Maurin-Marlin, A.; Laurent, F.; Vitale, I.; Thierry, S.; Delelis, O.; Dessen, P.; Vincendeau, M.; Leib-Mösch, C.; Hazan, U.; et al. Impact of the Ku Complex on HIV-1 Expression and Latency. PLoS ONE 2013, 8, e69691. [Google Scholar] [CrossRef]
- Waninger, S.; Kuhen, K.; Hu, X.; Chatterton, J.E.; Wong-Staal, F.; Tang, H. Identification of Cellular Cofactors for Human Immunodeficiency Virus Replication via a Ribozyme-Based Genomics Approach. J. Virol. 2004, 78, 12829–12837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meek, K.; Dang, V.; Lees-Miller, S.P. DNA-PK: The means to justify the ends? Adv. Immunol. 2008, 99, 33–58. [Google Scholar] [PubMed]
- Ilgova, E.; Galkin, S.; Khrenova, M.; Serebryakova, M.; Gottikh, M.; Anisenko, A. Complex of HIV-1 Integrase with Cellular Ku Protein: Interaction Interface and Search for Inhibitors. Int. J. Mol. Sci. 2022, 23, 2908. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Phan, A.T.; Mao, D.; Wang, X.; Gao, G.; Goff, S.P.; Zhu, Y. HIV-1 exploits the Fanconi anemia pathway for viral DNA integration. Cell Rep. 2022, 39, 110840. [Google Scholar] [CrossRef]
- Anisenko, A.N.; Knyazhanskaya, E.S.; Isaguliants, M.G.; Gottikh, M.B. A qPCR assay for measuring the post-integrational DNA repair in HIV-1 replication. J. Virol. Methods 2018, 262, 12–19. [Google Scholar] [CrossRef]
- Lingappa, J.R.; Lingappa, V.R.; Reed, J.C. Addressing antiretroviral drug resistance with host-targeting drugs—First steps towards developing a host-targeting hiv-1 assembly inhibitor. Viruses 2021, 13, 451. [Google Scholar] [CrossRef]
- Dorr, P.; Westby, M.; Dobbs, S.; Griffin, P.; Irvine, B.; Macartney, M.; Mori, J.; Rickett, G.; Smith-Burchnell, C.; Napier, C.; et al. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob. Agents Chemother. 2005, 49, 4721–4732. [Google Scholar] [CrossRef] [Green Version]
- Link, J.O.; Rhee, M.S.; Tse, W.C.; Zheng, J.; Somoza, J.R.; Rowe, W.; Begley, R.; Chiu, A.; Mulato, A.; Hansen, D.; et al. Clinical targeting of HIV capsid protein with a long-acting small molecule. Nature 2020, 584, 614–618. [Google Scholar] [CrossRef]
- DeLuca, L.; Ferro, S.; Morreale, F.; DeGrazia, S.; Chimirri, A. Inhibitors of the Interactions between HIV-1 IN and the Cofactor LEDGF/p75. ChemMedChem 2011, 6, 1184–1191. [Google Scholar] [CrossRef]
- Mesplède, T.; Wainberg, M.A. Will LEDGIN molecules be able to play a role in a cure for HIV infection? EBioMedicine 2016, 8, 14. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cellular Protein Name | Affected Stage of HIV-1 Replication | Influence on HIV-1 Replication |
|---|---|---|
| TRIM33 | Reverse transcription | Negative |
| Integration | Negative | |
| VBP1 | Viral transcription immediately after integration | Positive |
| VHL | ||
| p300 | Integration | Positive |
| GCN5 | Data inconsistent: reverse transcription, integration, viral transcription immediately after integration | Positive |
| HDAC1 | Data inconsistent: reverse transcription or integration | Data inconsistent |
| KAP1 (TRIM28) | Integration | Negative |
| HDAC10 | Integration | Negative |
| JNK and Pin1 | Integration | Positive |
| GCN2 | Integration | Negative |
| UBC9 | Data inconsistent: integration or/and morphogenesis | Data inconsistent |
| SUMO3 | Unclear | Unclear |
| Importin α (Imp α3) | Nuclear import | Positive |
| Importin 7 | Nuclear import | Positive |
| Transportin-SR2 | Nuclear import | Positive effect on PIC nuclear import, however this effect may be explained by Transportin-SR2 interaction with capsid protein rather than integrase |
| NUP153 | Nuclear import | Positive |
| LEDGF/p75 | Integration site selection | Positive |
| HRP2 | Integration site selection | Positive when LEDGF/p75 expression inhibited No effect under normal LEDGF/p75 level |
| INI1 | Unknown (Integration site selection?) | Data inconsistent |
| hRAD51 | Integration | Complex: activation of hRad51 before infection reduces integration, the late activation of hRad51 activity stimulates integration |
| hRAD18 | Unknown | Unknown |
| Ku70 | Post-integration DNA repair | Positive |
| FANCI | Integration or post-integration DNA repair | Positive |
| FANCD2 | Integration or post-integration DNA repair | Positive |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rozina, A.; Anisenko, A.; Kikhai, T.; Silkina, M.; Gottikh, M. Complex Relationships between HIV-1 Integrase and Its Cellular Partners. Int. J. Mol. Sci. 2022, 23, 12341. https://doi.org/10.3390/ijms232012341
Rozina A, Anisenko A, Kikhai T, Silkina M, Gottikh M. Complex Relationships between HIV-1 Integrase and Its Cellular Partners. International Journal of Molecular Sciences. 2022; 23(20):12341. https://doi.org/10.3390/ijms232012341
Chicago/Turabian StyleRozina, Anna, Andrey Anisenko, Tatiana Kikhai, Maria Silkina, and Marina Gottikh. 2022. "Complex Relationships between HIV-1 Integrase and Its Cellular Partners" International Journal of Molecular Sciences 23, no. 20: 12341. https://doi.org/10.3390/ijms232012341