
A Novel Antibody-Drug Conjugate Targeting Nectin-2 Suppresses Ovarian Cancer Progression in Mouse Xenograft Models

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
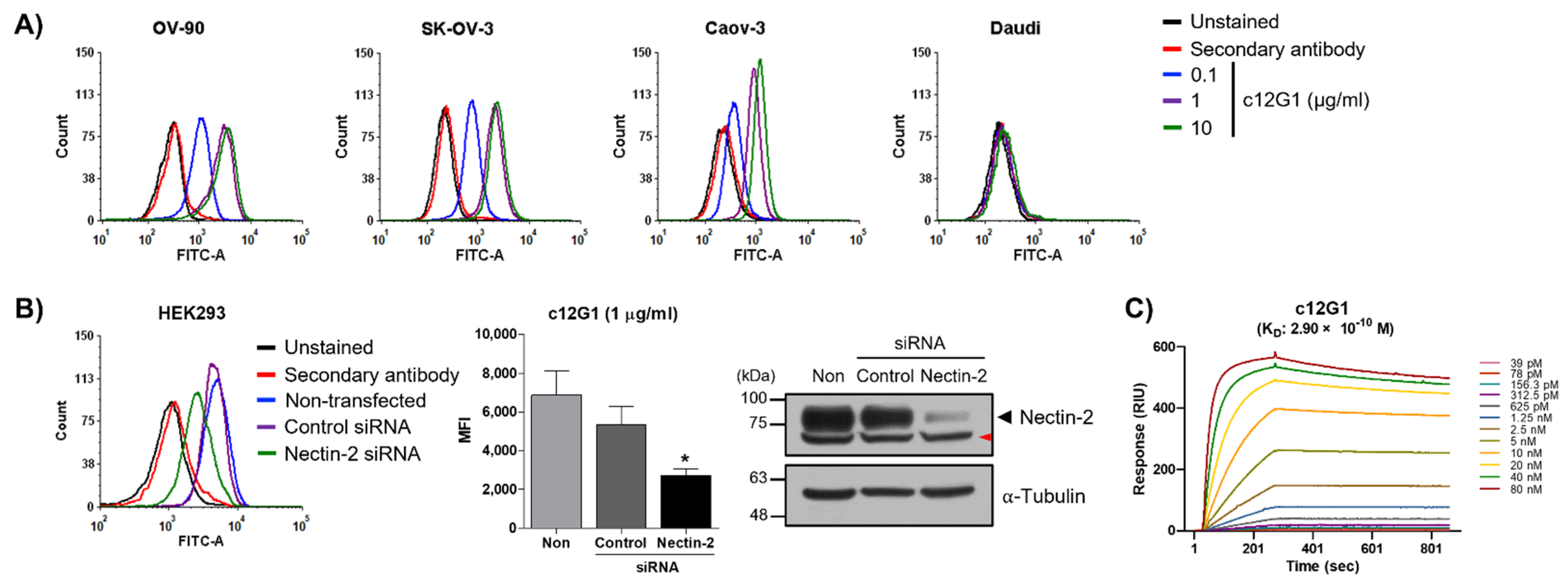
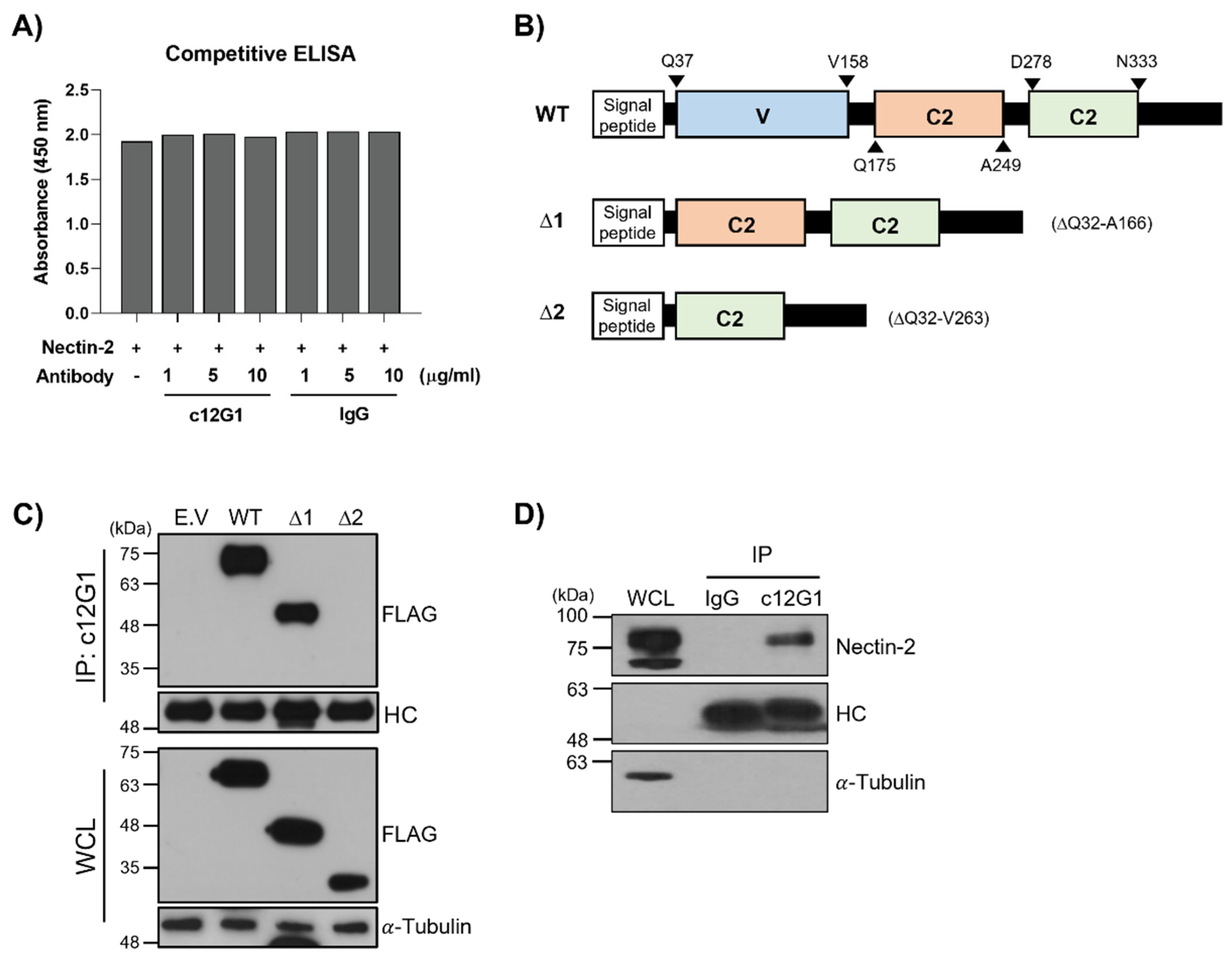
2.1. Generation and Characterization of Anti-Nectin-2 Antibody
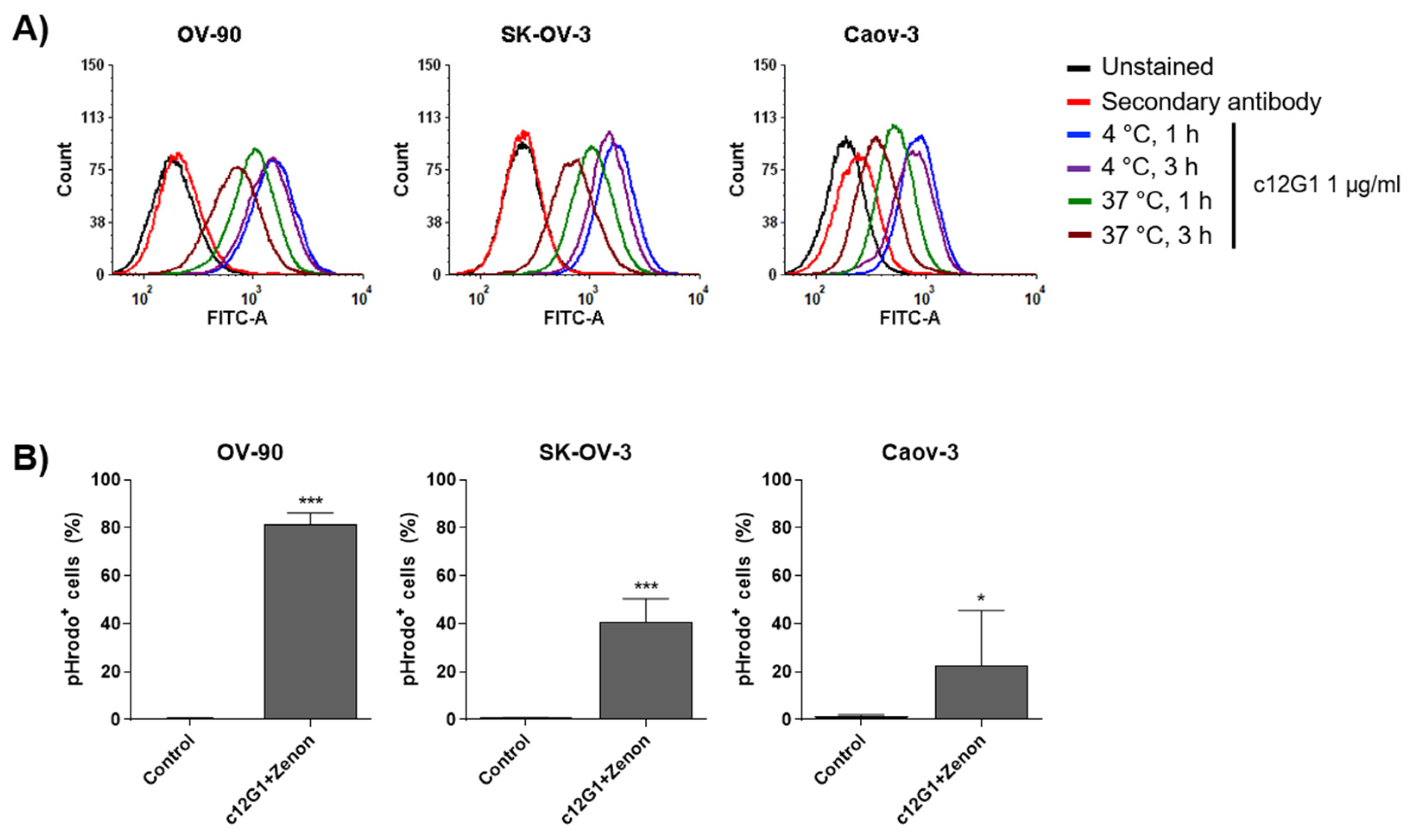
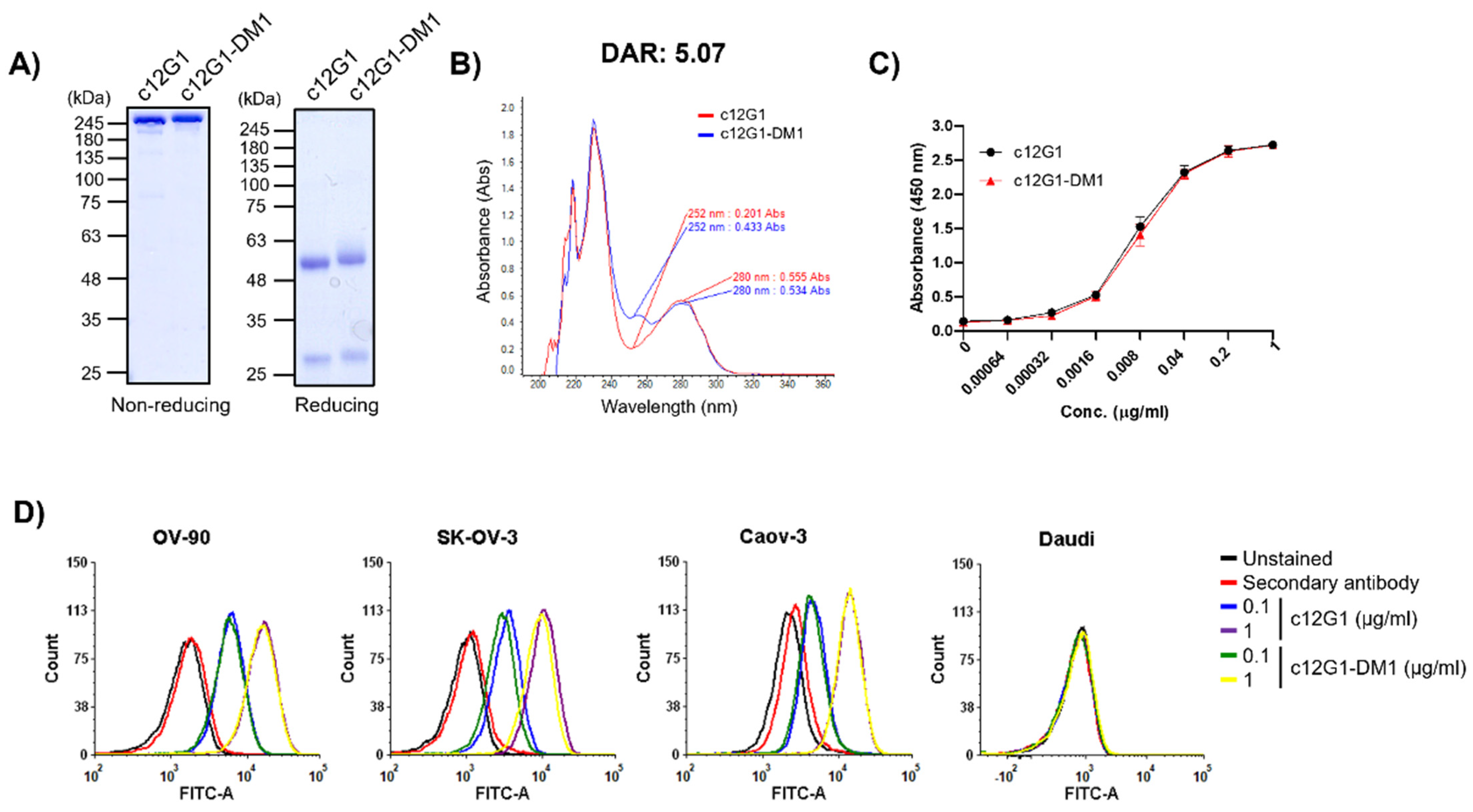
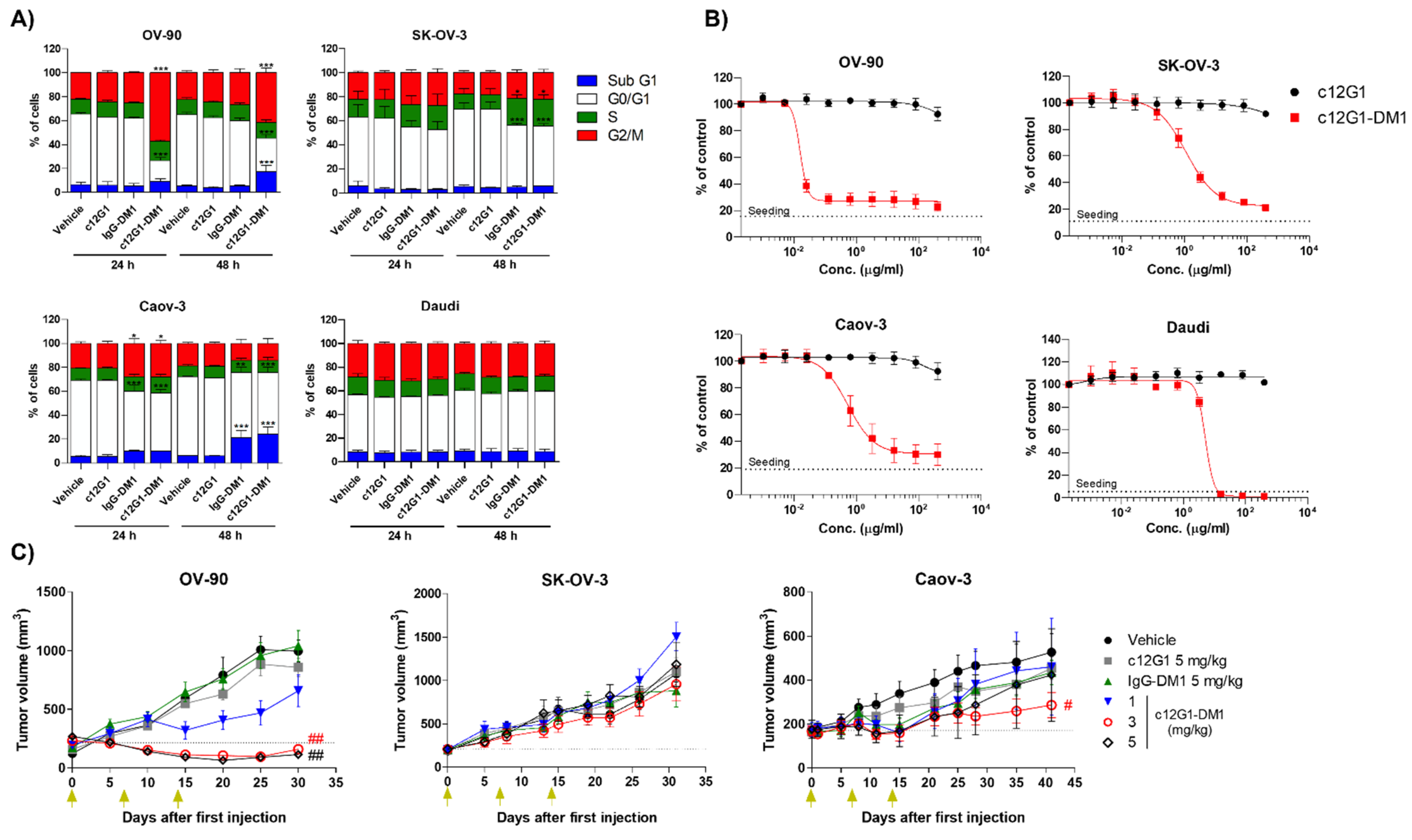
2.2. c12G1 ADC Exhibits Anti-Tumor Activity In Vitro and In Vivo
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture
4.2. Antibody Generation
4.3. qRT-PCR
4.4. Western Blot
4.5. si-RNA Study
4.6. Flow Cytometry
4.7. SPR Assay
4.8. ELISA
4.9. Effector Function Assay
4.10. Internalization Assay
4.11. Generation of ADCs
4.12. Cell Cycle and In Vitro Cytotoxicity Analyses
4.13. In Vivo Experiments
| Tumor growth inhibition rate = [1 − (RTV in the treated group)/(RTV in the control group) × 100 (%)] RTV = (tumor volume on the measured day)/(tumor volume on day 0) |
4.14. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADC | Antibody-drug conjugate |
| ADCC | Antibody-dependent cell-mediated cytotoxicity |
| CDC | Complement-dependent cytotoxicity |
| DAR | Drug-to-antibody ratio |
| EOC | Epithelial ovarian cancer |
| IC50 | Half-maximal inhibitory concentration |
| PVRIG | Poliovirus receptor-related immunoglobulin domain-containing |
| SPR | Surface plasmon resonance |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-G.; Wang, J.; Sun, J.-Y.; He, Z.-Y.; Zhang, W.-W.; Zhou, J. Real-world impact of survival by period of diagnosis in epithelial ovarian cancer between 1990 and 2014. Front. Oncol. 2019, 9, 639. [Google Scholar] [CrossRef] [Green Version]
- Lheureux, S.; Braunstein, M.; Oza, A.M. Epithelial ovarian cancer: Evolution of management in the era of precision medicine. CA A Cancer J. Clin. 2019, 69, 280–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojas, V.; Hirshfield, K.M.; Ganesan, S.; Rodriguez-Rodriguez, L. Molecular characterization of epithelial ovarian cancer: Implications for diagnosis and treatment. Int. J. Mol. Sci. 2016, 17, 2113. [Google Scholar] [CrossRef] [Green Version]
- Meinhold-Heerlein, I.; Fotopoulou, C.; Harter, P.; Kurzeder, C.; Mustea, A.; Wimberger, P.; Hauptmann, S.; Sehouli, J. The new WHO classification of ovarian, fallopian tube, and primary peritoneal cancer and its clinical implications. Arch. Gynecol. Obstet. 2016, 293, 695–700. [Google Scholar] [CrossRef] [PubMed]
- McCluggage, W.G. Morphological subtypes of ovarian carcinoma: A review with emphasis on new developments and pathogenesis. Pathology 2011, 43, 420–432. [Google Scholar] [CrossRef] [PubMed]
- Kurman, R.J.; Shih, I.-M. Molecular pathogenesis and extraovarian origin of epithelial ovarian cancer—Shifting the paradigm. Hum. Pathol. 2011, 42, 918–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurman, R. Origin and molecular pathogenesis of ovarian high-grade serous carcinoma. Ann. Oncol. 2013, 24, x16–x21. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.; Ali, S.; Wang, K.; Palmer, G.; Yelensky, R.; Lipson, D.; Miller, V.; Zajchowski, D.; Shawver, L.; Stephens, P. Comprehensive genomic profiling of epithelial ovarian cancer by next generation sequencing-based diagnostic assay reveals new routes to targeted therapies. Gynecol. Oncol. 2013, 130, 554–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Network, C.G.A.R. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609. [Google Scholar] [CrossRef]
- Kurman, R.J.; Shih, I.-M. The dualistic model of ovarian carcinogenesis: Revisited, revised, and expanded. Am. J. Pathol. 2016, 186, 733–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.; Ahmad, S.; Brudie, L.A. Prevention of Ovarian Cancer. In Preventive Oncology for the Gynecologist; Springer: Berlin/Heidelberg, Germany, 2019; pp. 257–272. [Google Scholar]
- Pignata, S.; Cecere, S.; Du Bois, A.; Harter, P.; Heitz, F. Treatment of recurrent ovarian cancer. Ann. Oncol. 2017, 28, viii51–viii56. [Google Scholar] [CrossRef] [PubMed]
- Matulonis, U.A. Management of newly diagnosed or recurrent ovarian cancer. Clin. Adv. Hematol. Oncol. 2018, 16, 426–437. [Google Scholar] [PubMed]
- Marth, C.; Reimer, D.; Zeimet, A. Front-line therapy of advanced epithelial ovarian cancer: Standard treatment. Ann. Oncol. 2017, 28, viii36–viii39. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.; Tinker, A.V.; Friedlander, M. “Platinum resistant” ovarian cancer: What is it, who to treat and how to measure benefit? Gynecol. Oncol. 2014, 133, 624–631. [Google Scholar] [CrossRef]
- Perren, T.; Swart, A.; Pfisterer, J.; Ledermann, J.; Pujade-Lauraine, E.; Kristensen, G.; Carey, M.; Beale, P.; Cervantes, A.; Kurzeder, C.; et al. A phase 3 trial of bevacizumab in ovarian cancer. N. Engl. J. Med. 2011, 365, 2484–2496. [Google Scholar] [CrossRef] [Green Version]
- Coleman, R.L.; Brady, M.F.; Herzog, T.J.; Sabbatini, P.; Armstrong, D.K.; Walker, J.L.; Kim, B.-G.; Fujiwara, K.; Tewari, K.S.; O’Malley, D.M. Bevacizumab and paclitaxel–carboplatin chemotherapy and secondary cytoreduction in recurrent, platinum-sensitive ovarian cancer (NRG Oncology/Gynecologic Oncology Group study GOG-0213): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2017, 18, 779–791. [Google Scholar] [CrossRef] [Green Version]
- Burger, R.; Brady, M.; Bookman, M.; Fleming, G.; Monk, B.; Huang, H. Gynecologic Oncologic Group Phase III trial of bevacizumab (BEV) in the primary treatment of advanced epithelial ovarian cancer (EOC), primary peritoneal cancer (PPC), and fallopian tube cancer (FTC): A Gynaecologic Oncology Group study. N. Engl. J. Med. 2011, 365, 2473–2483. [Google Scholar] [CrossRef] [Green Version]
- Aghajanian, C.; Blank, S.V.; Goff, B.A.; Judson, P.L.; Teneriello, M.G.; Husain, A.; Sovak, M.A.; Yi, J.; Nycum, L.R. OCEANS: A randomized, double-blind, placebo-controlled phase III trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent epithelial ovarian, primary peritoneal, or fallopian tube cancer. J. Clin. Oncol. 2012, 30, 2039–2045. [Google Scholar] [CrossRef] [Green Version]
- O’Malley, D.M.; Matulonis, U.A.; Birrer, M.J.; Castro, C.M.; Gilbert, L.; Vergote, I.; Martin, L.P.; Mantia-Smaldone, G.M.; Martin, A.G.; Bratos, R.; et al. Phase Ib study of mirvetuximab soravtansine, a folate receptor alpha (FRα)-targeting antibody-drug conjugate (ADC), in combination with bevacizumab in patients with platinum-resistant ovarian cancer. Gynecol. Oncol. 2020, 157, 379–385. [Google Scholar] [CrossRef]
- Oshima, T.; Sato, S.; Kato, J.; Ito, Y.; Watanabe, T.; Tsuji, I.; Hori, A.; Kurokawa, T.; Kokubo, T. Nectin-2 is a potential target for antibody therapy of breast and ovarian cancers. Mol. Cancer 2013, 12, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, W.M.; Spear, P.G. Structural features of nectin-2 (HveB) required for herpes simplex virus entry. J. Virol. 2001, 75, 11185–11195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deuss, F.A.; Gully, B.S.; Rossjohn, J.; Berry, R. Recognition of nectin-2 by the natural killer cell receptor T cell immunoglobulin and ITIM domain (TIGIT). J. Biol. Chem. 2017, 292, 11413–11422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ooshio, T.; Irie, K.; Morimoto, K.; Fukuhara, A.; Imai, T.; Takai, Y. Involvement of LMO7 in the association of two cell-cell adhesion molecules, nectin and E-cadherin, through afadin and alpha-actinin in epithelial cells. J. Biol. Chem. 2004, 279, 31365–31373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bekes, I.; Löb, S.; Holzheu, I.; Janni, W.; Baumann, L.; Wöckel, A.; Wulff, C. Nectin-2 in ovarian cancer: How is it expressed and what might be its functional role? Cancer Sci. 2019, 110, 1872–1882. [Google Scholar] [CrossRef] [Green Version]
- Pende, D.; Bottino, C.; Castriconi, R.; Cantoni, C.; Marcenaro, S.; Rivera, P.; Spaggiari, G.M.; Dondero, A.; Carnemolla, B.; Reymond, N. PVR (CD155) and Nectin-2 (CD112) as ligands of the human DNAM-1 (CD226) activating receptor: Involvement in tumor cell lysis. Mol. Immunol. 2005, 42, 463–469. [Google Scholar] [CrossRef]
- Oshima, T.; Miyashita, H.; Ishimura, Y.; Ito, Y.; Tanaka, Y.; Hori, A.; Kokubo, T.; Kurokawa, T. Fc engineering of anti-Nectin-2 antibody improved thrombocytopenic adverse event in monkey. PLoS ONE 2018, 13, e0196422. [Google Scholar]
- Whelan, S.; Ophir, E.; Kotturi, M.F.; Levy, O.; Ganguly, S.; Leung, L.; Vaknin, I.; Kumar, S.; Dassa, L.; Hansen, K.; et al. PVRIG and PVRL2 Are Induced in Cancer and Inhibit CD8(+) T-cell Function. Cancer Immunol. Res. 2019, 7, 257–268. [Google Scholar] [CrossRef] [Green Version]
- Casey, J.R.; Grinstein, S.; Orlowski, J. Sensors and regulators of intracellular pH. Nat. Rev. Mol. Cell Biol. 2010, 11, 50–61. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, Y.; Wu, Y.; Jiang, X.; Tao, Y.; Yao, Y.; Peng, Y.; Chen, X.; Fu, Y.; Yu, L. Therapeutic potential of an anti-HER2 single chain antibody–DM1 conjugates for the treatment of HER2-positive cancer. Signal Transduct. Target. Ther. 2017, 2, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.H.; Kim, J.O.; Park, J.Y.; Seo, M.D.; Park, S.G. Antibody-Drug Conjugate Targeting c-Kit for the Treatment of Small Cell Lung Cancer. Int. J. Mol. Sci. 2022, 23, 2264. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Paniccia, A.; Schulick, A.C.; Chen, W.; Koenig, M.R.; Byers, J.T.; Yao, S.; Bevers, S.; Edil, B.H. Identification of CD112R as a novel checkpoint for human T cells. J. Exp. Med. 2016, 213, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Buckel, L.; Savariar, E.N.; Crisp, J.L.; Jones, K.A.; Hicks, A.M.; Scanderbeg, D.J.; Nguyen, Q.T.; Sicklick, J.K.; Lowy, A.M.; Tsien, R.Y. Tumor Radiosensitization by Monomethyl Auristatin E: Mechanism of Action and Targeted DeliveryMMAE-Mediated Radiosensitization. Cancer Res. 2015, 75, 1376–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hale, S.J.; Perrins, R.D.; García, C.E.; Pace, A.; Peral, U.; Patel, K.R.; Robinson, A.; Williams, P.; Ding, Y.; Saito, G. DM1 loaded ultrasmall gold nanoparticles display significant efficacy and improved tolerability in murine models of hepatocellular carcinoma. Bioconjugate Chem. 2018, 30, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Legigan, T.; Clarhaut, J.; Renoux, B.; Tranoy-Opalinski, I.; Monvoisin, A.; Jayle, C.; Alsarraf, J.; Thomas, M.; Papot, S. Synthesis and biological evaluations of a monomethylauristatin E glucuronide prodrug for selective cancer chemotherapy. Eur. J. Med. Chem. 2013, 67, 75–80. [Google Scholar] [CrossRef]
- Victor, B.C.; Anbalagan, A.; Mohamed, M.M.; Sloane, B.F.; Cavallo-Medved, D. Inhibition of cathepsin B activity attenuates extracellular matrix degradation and inflammatory breast cancer invasion. Breast Cancer Res. 2011, 13, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Poreba, M.; Groborz, K.; Vizovisek, M.; Maruggi, M.; Turk, D.; Turk, B.; Powis, G.; Drag, M.; Salvesen, G.S. Fluorescent probes towards selective cathepsin B detection and visualization in cancer cells and patient samples. Chem. Sci. 2019, 10, 8461–8477. [Google Scholar] [CrossRef] [Green Version]
- Mijanović, O.; Branković, A.; Panin, A.N.; Savchuk, S.; Timashev, P.; Ulasov, I.; Lesniak, M.S. Cathepsin B: A sellsword of cancer progression. Cancer Lett. 2019, 449, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Barok, M.; Joensuu, H.; Isola, J. Trastuzumab emtansine: Mechanisms of action and drug resistance. Breast Cancer Res. 2014, 16, 209. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Liu, D.-W.; Zheng, M.-J.; Deng, L.; Wang, H.-M.; Jin, S.; Liu, J.-J.; Hao, Y.-Y.; Zhu, L.-C.; Lin, B. Human epididymis protein 4 promotes P-glycoprotein-mediated chemoresistance in ovarian cancer cells through interactions with Annexin II. Mol. Med. Rep. 2021, 24, 496. [Google Scholar] [CrossRef]
- Li, M.; Qiao, D.; Pu, J.; Wang, W.; Zhu, W.; Liu, H. Elevated Nectin-2 expression is involved in esophageal squamous cell carcinoma by promoting cell migration and invasion. Oncol. Lett. 2018, 15, 4731–4736. [Google Scholar] [CrossRef] [PubMed]
- Della Pepa, C.; Tonini, G.; Pisano, C.; Di Napoli, M.; Cecere, S.C.; Tambaro, R.; Facchini, G.; Pignata, S. Ovarian cancer standard of care: Are there real alternatives? Chin. J. Cancer 2015, 34, 17–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shor, B.; Kahler, J.; Dougher, M.; Xu, J.; Mack, M.; Rosfjord, E.; Wang, F.; Melamud, E.; Sapra, P. Enhanced Antitumor Activity of an Anti-5T4 Antibody-Drug Conjugate in Combination with PI3K/mTOR inhibitors or Taxanes. Clin. Cancer Res. 2016, 22, 383–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, Y.L.; Sapra, P.; Bolton, J.; Chua, J.X.; Durrant, L.G.; Stern, P.L. Correction to: Combination Treatment with an Antibody-Drug Conjugate (A1mcMMAF) Targeting the Oncofetal Glycoprotein 5T4 and Carboplatin Improves Survival in a Xenograft Model of Ovarian Cancer. Target Oncol. 2019, 14, 769. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sim, Y.H.; Um, Y.J.; Park, J.-Y.; Seo, M.-D.; Park, S.G. A Novel Antibody-Drug Conjugate Targeting Nectin-2 Suppresses Ovarian Cancer Progression in Mouse Xenograft Models. Int. J. Mol. Sci. 2022, 23, 12358. https://doi.org/10.3390/ijms232012358
Sim YH, Um YJ, Park J-Y, Seo M-D, Park SG. A Novel Antibody-Drug Conjugate Targeting Nectin-2 Suppresses Ovarian Cancer Progression in Mouse Xenograft Models. International Journal of Molecular Sciences. 2022; 23(20):12358. https://doi.org/10.3390/ijms232012358
Chicago/Turabian StyleSim, Yun Hee, Yun Jung Um, Jeong-Yang Park, Min-Duk Seo, and Sang Gyu Park. 2022. "A Novel Antibody-Drug Conjugate Targeting Nectin-2 Suppresses Ovarian Cancer Progression in Mouse Xenograft Models" International Journal of Molecular Sciences 23, no. 20: 12358. https://doi.org/10.3390/ijms232012358