
Rat Group IIA Secreted Phospholipase A2 Binds to Cytochrome c Oxidase and Inhibits Its Activity: A Possible Episode in the Development of Alzheimer’s Disease
 , , , ,
, , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Recombinant rGIIA and rGIIA(D49S) Are Functional
2.2. rGIIA Binds to the Same Mitochondrial Target as the Neurotoxic Atx
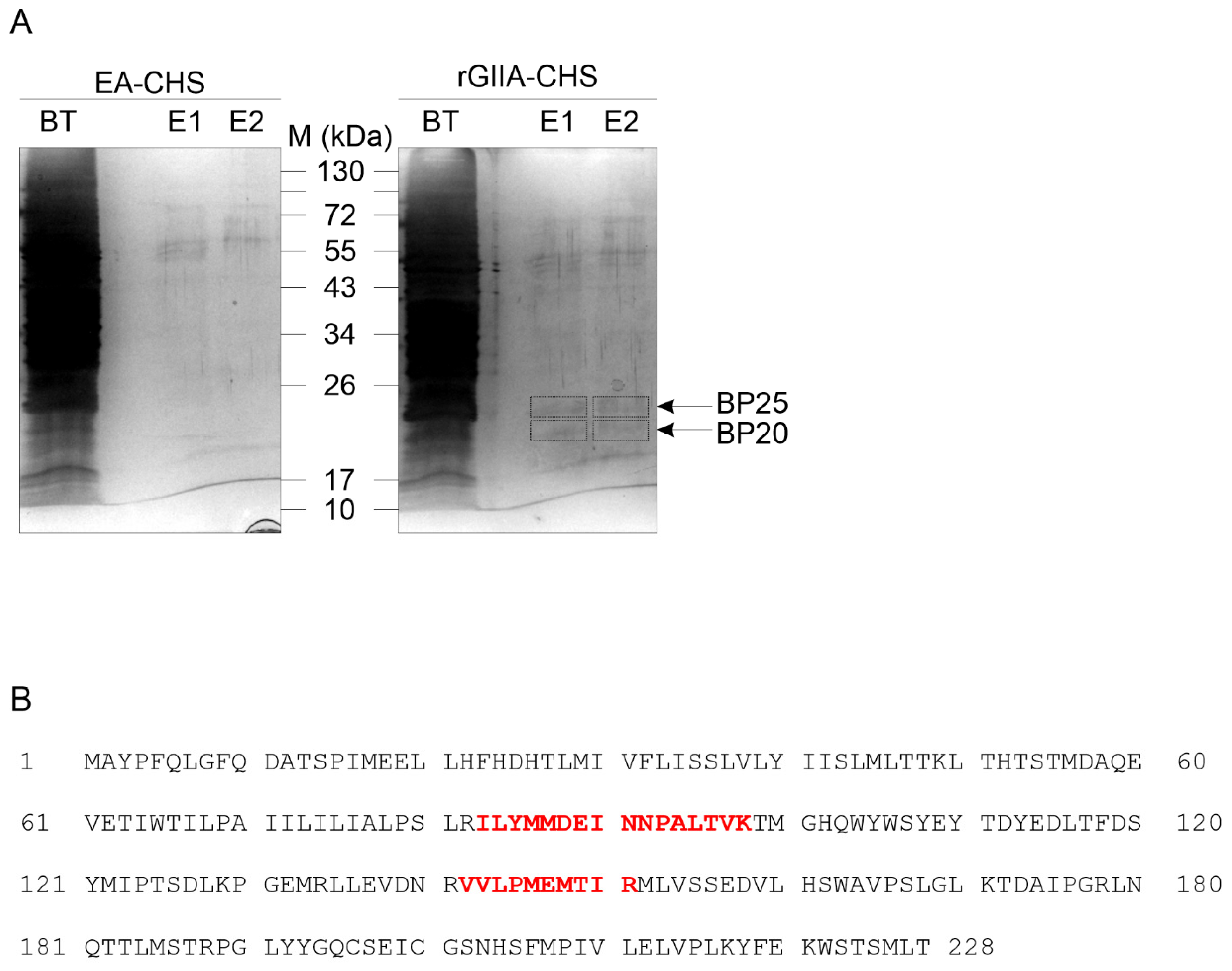
2.3. rGIIA-Binding Protein in Mitochondria Is CCOX-II
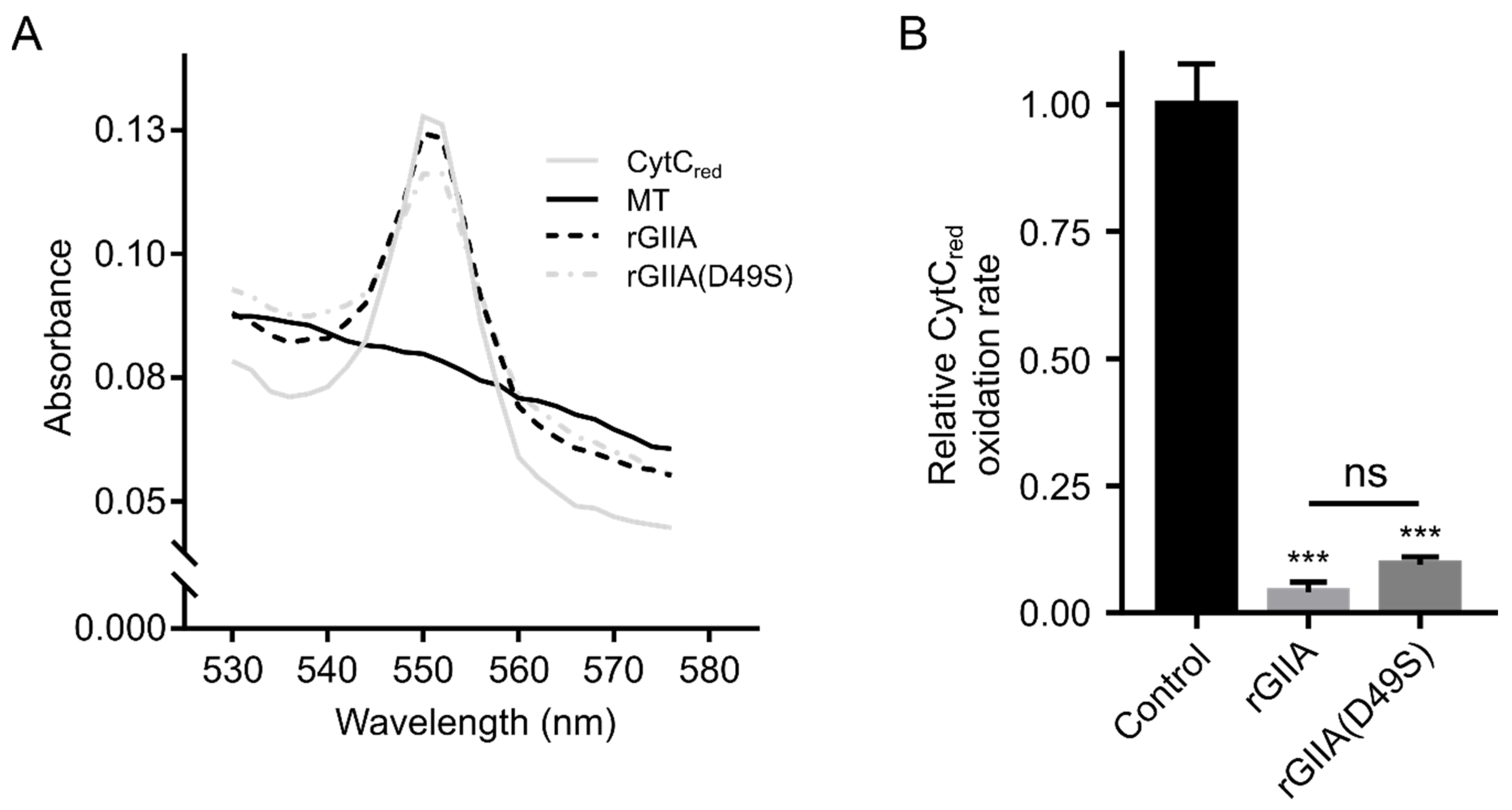
2.4. rGIIA Inhibits CCOX Activity in Isolated Mitochondria
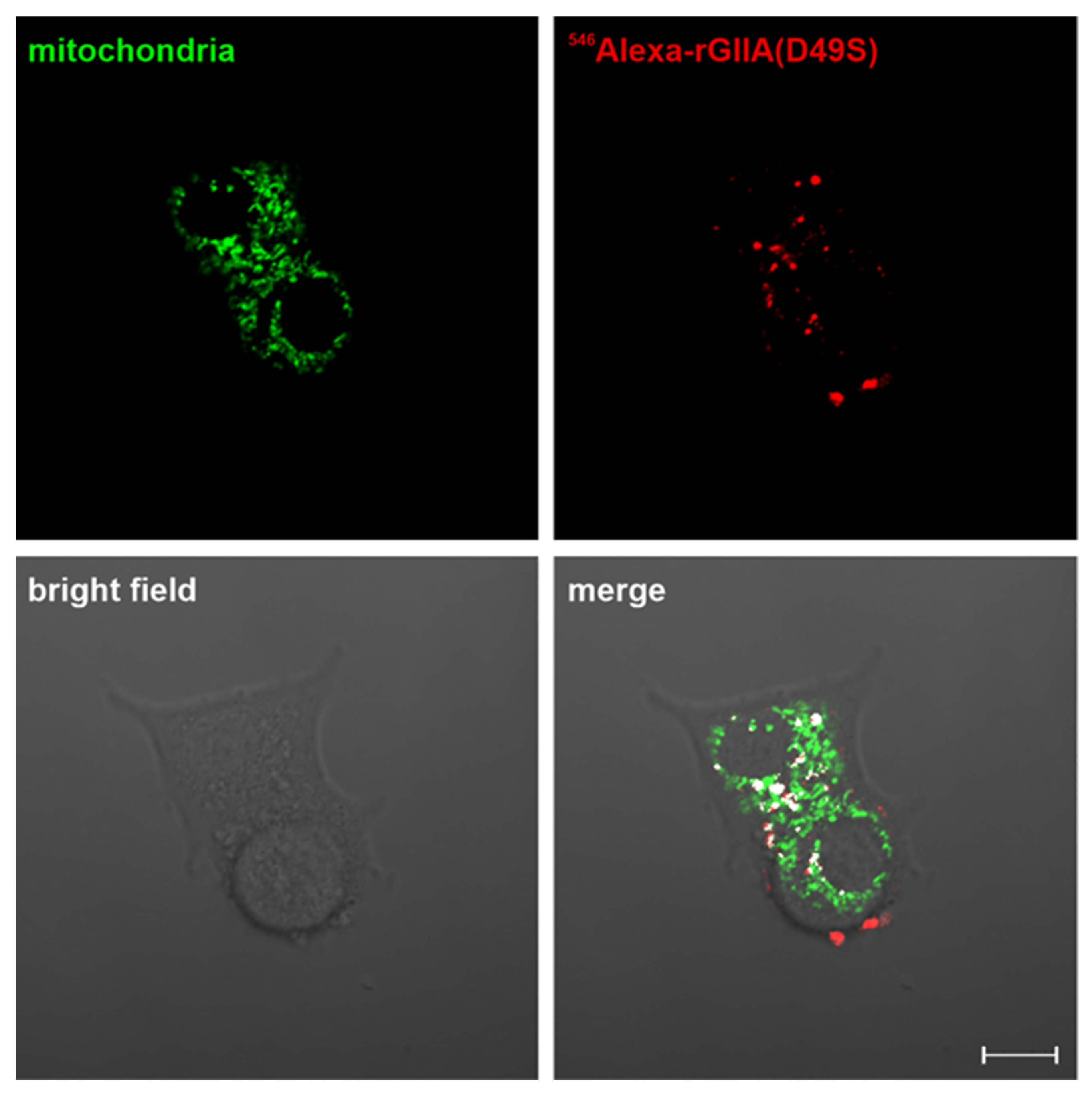
2.5. rGIIA(D49S) Internalizes into PC12 Cells and Colocalizes with Mitochondria
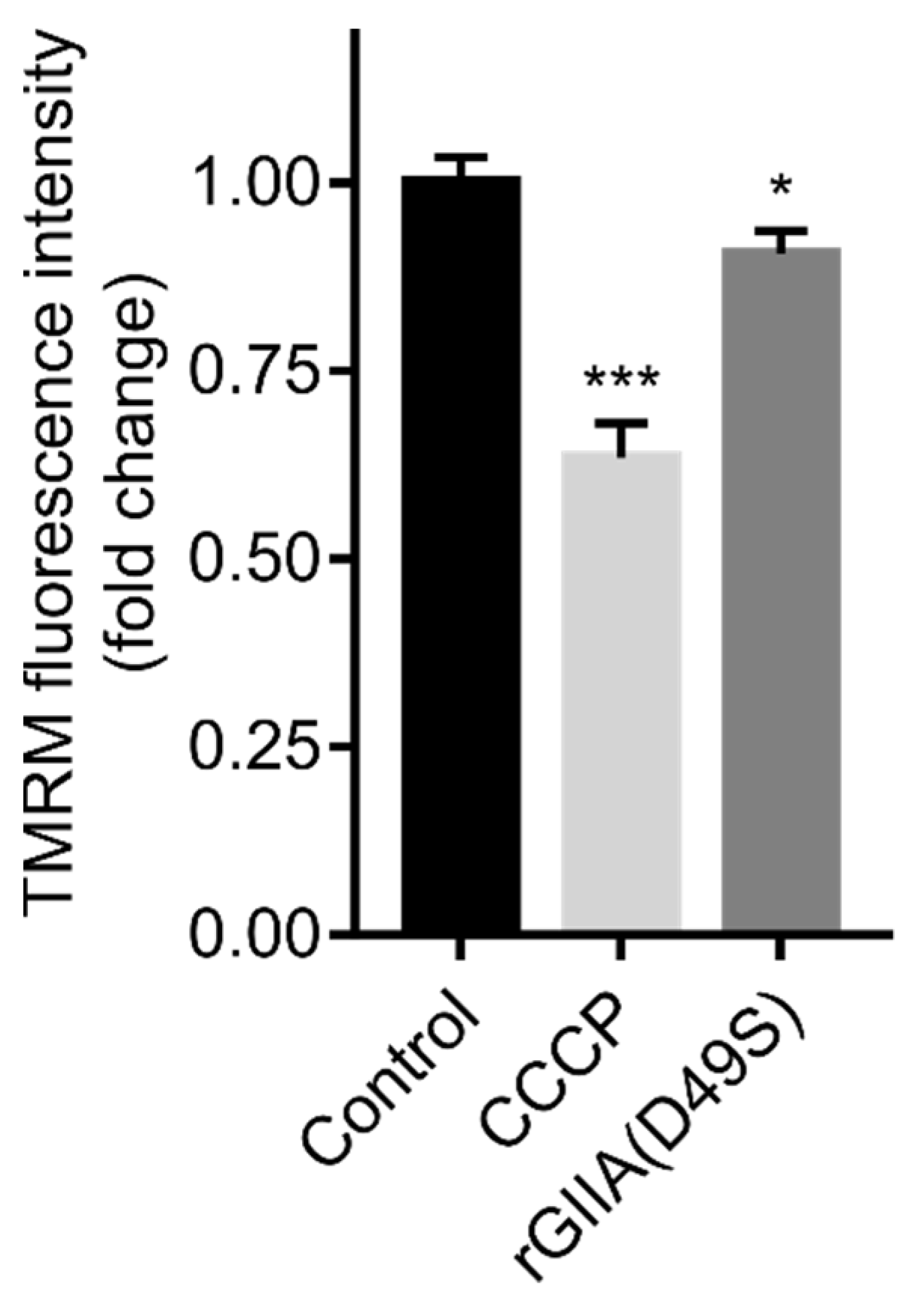
2.6. rGIIA(D49S) Showed Only Minor Effect on Mitochondrial Membrane Potential in PC12 Cells
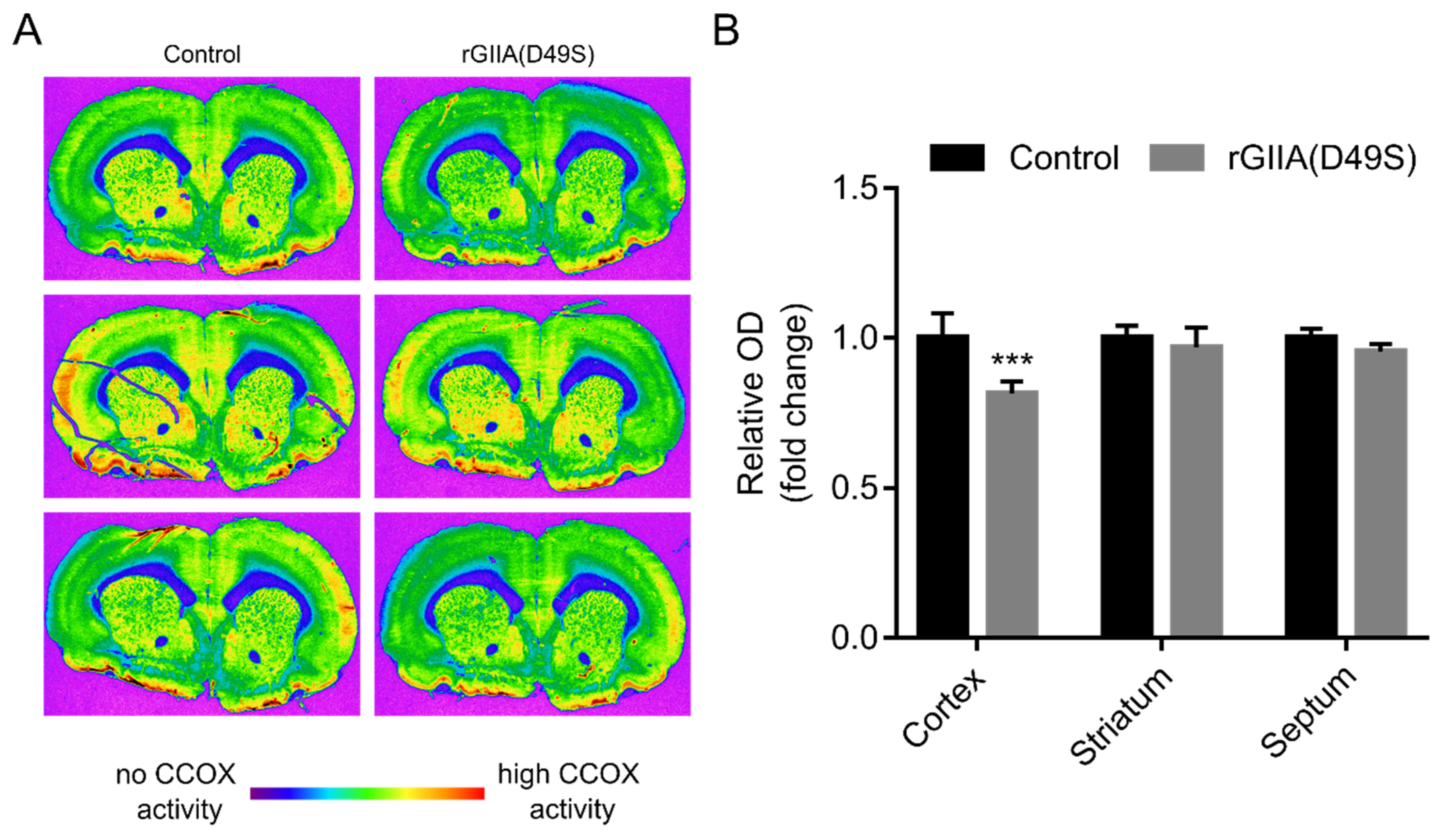
2.7. rGIIA(D49S) Inhibited CCOX Activity in Rat Brain Tissue Sections
3. Discussion
4. Materials and Methods
4.1. Production and Characterization of the Recombinant Rat GIIA Proteins
4.2. Heterologous Competition Assay
4.3. Identification of rGIIA-Binding Protein in Porcine Cerebral Cortex Mitochondria
4.4. Culturing of PC12 Cells
4.5. Isolation of Mitochondria from PC12 Cells and Measurement of CCOX Activity
4.6. Fluorescence Confocal Microscopy Colocalization Study on PC12 Cells
4.7. Measurement of TMRM Fluorescence Intensity and Cell Death
4.8. Assessment of CCOX Activity in Rat Brain Tissue Sections
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Murakami, M. Novel functions of phospholipase A2s: Overview. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 763–765. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Sato, H.; Taketomi, Y. Updating phospholipase A2 biology. Biomolecules 2020, 10, 1457. [Google Scholar] [CrossRef] [PubMed]
- Dore, E.; Boilard, E. Roles of secreted phospholipase A2 group IIA in inflammation and host defense. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Taketomi, Y.; Miki, Y.; Murakami, M. Old but new: Group IIA phospholipase A2 as a modulator of gut microbiota. Metabolites 2022, 12, 352. [Google Scholar] [CrossRef]
- Nardicchi, V.; Ferrini, M.; Pilolli, F.; Angeli, E.B.; Persichetti, E.; Beccari, T.; Mannucci, R.; Arcuri, C.; Donato, R.; Dorman, R.V.; et al. NGF induces the expression of group IIA secretory phospholipase A2 in PC12 cells: The newly synthesized enzyme is addressed to growing neurites. Mol. Neurobiol. 2014, 50, 15–25. [Google Scholar] [CrossRef]
- Macchioni, L.; Corazzi, L.; Nardicchi, V.; Mannucci, R.; Arcuri, C.; Porcellati, S.; Sposini, T.; Donato, R.; Goracci, G. Rat brain cortex mitochondria release group II secretory phospholipase A2 under reduced membrane potential. J. Biol. Chem. 2004, 279, 37860–37869. [Google Scholar] [CrossRef] [Green Version]
- Goracci, G.; Ferrini, M.; Nardicchi, V. Low molecular weight phospholipases A2 in mammalian brain and neural cells: Roles in functions and dysfunctions. Mol. Neurobiol. 2010, 41, 274–289. [Google Scholar] [CrossRef]
- Moses, G.S.D.; Jensen, M.D.; Lue, L.-F.; Walker, D.G.; Sun, A.Y.; Simonyi, A.; Sun, G.Y. Secretory PLA2-IIA: A new inflammatory factor for Alzheimer’s disease. J. Neuroinflammation 2006, 3, 28. [Google Scholar] [CrossRef] [Green Version]
- Villanueva, E.B.; Little, J.P.; Lambeau, G.; Klegeris, A. Secreted phospholipase A2 group IIA is a neurotoxin released by stimulated human glial cells. Mol. Cell. Neurosci. 2012, 49, 430–438. [Google Scholar] [CrossRef]
- Yang, X.; Sheng, W.; Ridgley, D.M.; Haidekker, M.A.; Sun, G.Y.; Lee, J.C. Astrocytes regulate α-secretase-cleaved soluble amyloid precursor protein secretion in neuronal cells: Involvement of group IIA secretory phospholipase A2. Neuroscience 2015, 300, 508–517. [Google Scholar] [CrossRef]
- Mary, A.; Eysert, F.; Checler, F.; Chami, M. Mitophagy in Alzheimer’s disease: Molecular defects and therapeutic approaches. Mol. Psychiatry 2022, 1–15. [Google Scholar] [CrossRef]
- Šribar, J.; Oberčkal, J.; Križaj, I. Understanding the molecular mechanism underlying the presynaptic toxicity of secreted phospholipases A2: An update. Toxicon 2014, 89, 9–16. [Google Scholar] [CrossRef]
- Šribar, J.; Kovačič, L.; Oberčkal, J.; Ivanušec, A.; Petan, T.; Fox, J.W.; Križaj, I. The neurotoxic secreted phospholipase A2 from the Vipera a. ammodytes venom targets cytochrome c oxidase in neuronal mitochondria. Sci. Rep. 2019, 9, 283. [Google Scholar] [CrossRef] [Green Version]
- Ivanušec, A.; Šribar, J.; Veranič, P.; Križaj, I. The phospholipase activity of ammodytoxin, a prototype snake venom β-neurotoxin, is not obligatory for cell Internalisation and translocation to mitochondria. Toxins 2022, 14, 375. [Google Scholar] [CrossRef]
- Lee, J.C.-M.; Simonyi, A.; Sun, A.Y.; Sun, G.Y. Phospholipases A2 and neural membrane dynamics: Implications for Alzheimer’s disease. J. Neurochem. 2011, 116, 813–819. [Google Scholar] [CrossRef]
- Sun, G.Y.; Geng, X.; Teng, T.; Yang, B.; Appenteng, M.K.; Greenlief, C.M.; Lee, J.C. Dynamic role of phospholipases A2 in health and diseases in the central nervous system. Cells 2021, 10, 2963. [Google Scholar] [CrossRef]
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat. Rev. Dis. Prim. 2015, 1, 15056. [Google Scholar] [CrossRef]
- Swerdlow, R.H. The mitochondrial hypothesis: Dysfunction, bioenergetic defects, and the metabolic link to Alzheimer’s disease. Int. Rev. Neurobiol. 2020, 154, 207–233. [Google Scholar] [CrossRef]
- Johri, A. Disentangling mitochondria in Alzheimer’s disease. Int. J. Mol. Sci. 2021, 22, 11520. [Google Scholar] [CrossRef]
- Mounier, C.M.; Bon, C.; Kini, R.M. Anticoagulant Venom and Mammalian Secreted Phospholipases A2: Protein- versus Phospholipid-Dependent Mechanism of Action. Pathophysiol. Haemost. Thromb. 2001, 31, 279–287. [Google Scholar] [CrossRef]
- Greene, L.A.; Tischler, A.S. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc. Natl. Acad. Sci. USA 1976, 73, 2424–2428. [Google Scholar] [CrossRef] [Green Version]
- Lomonte, B.; Križaj, I. Snake venom phospholipase A2 toxins. In Handbook of Venoms and Toxins of Reptiles; Mackessy, S., Ed.; CRC Press: Boca Raton, FL, USA, 2021; pp. 389–412. ISBN 9780429054204. [Google Scholar]
- Kadenbach, B. Complex IV—The regulatory center of mitochondrial oxidative phosphorylation. Mitochondrion 2021, 58, 296–302. [Google Scholar] [CrossRef]
- Gillardon, F.; Rist, W.; Kussmaul, L.; Vogel, J.; Berg, M.; Danzer, K.; Kraut, N.; Hengerer, B. Proteomic and functional alterations in brain mitochondria from Tg2576 mice occur before amyloid plaque deposition. Proteomics 2007, 7, 605–616. [Google Scholar] [CrossRef]
- Song, T.; Song, X.; Zhu, C.; Patrick, R.; Skurla, M.; Santangelo, I.; Green, M.; Harper, D.; Ren, B.; Forester, B.P.; et al. Mitochondrial dysfunction, oxidative stress, neuroinflammation, and metabolic alterations in the progression of Alzheimer’s disease: A meta-analysis of in vivo magnetic resonance spectroscopy studies. Ageing Res. Rev. 2021, 72, 101503. [Google Scholar] [CrossRef]
- Zhao, R.-Z.; Jiang, S.; Zhang, L.; Yu, Z.-B. Mitochondrial electron transport chain, ROS generation and uncoupling (review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Napolitano, G.; Fasciolo, G.; Venditti, P. Mitochondrial management of reactive oxygen species. Antioxidants 2021, 10, 1824. [Google Scholar] [CrossRef]
- Ikawa, M.; Okazawa, H.; Nakamoto, Y.; Yoneda, M. PET imaging for oxidative stress in neurodegenerative disorders associated with mitochondrial dysfunction. Antioxidants 2020, 9, 861. [Google Scholar] [CrossRef]
- Angelova, P.R.; Esteras, N.; Abramov, A.Y. Mitochondria and lipid peroxidation in the mechanism of neurodegeneration: Finding ways for prevention. Med. Res. Rev. 2021, 41, 770–784. [Google Scholar] [CrossRef]
- Paradies, G.; Ruggiero, F.M.; Petrosillo, G.; Quagliariello, E. Peroxidative damage to cardiac mitochondria: Cytochrome oxidase and cardiolipin alterations. FEBS Lett. 1998, 424, 155–158. [Google Scholar] [CrossRef] [Green Version]
- Bobba, A.; Amadoro, G.; Valenti, D.; Corsetti, V.; Lassandro, R.; Atlante, A. Mitochondrial respiratory chain Complexes I and IV are impaired by β-amyloid via direct interaction and through Complex I-dependent ROS production, respectively. Mitochondrion 2013, 13, 298–311. [Google Scholar] [CrossRef]
- Johnson, E.C.B.; Carter, E.K.; Dammer, E.B.; Duong, D.M.; Gerasimov, E.S.; Liu, Y.; Liu, J.; Betarbet, R.; Ping, L.; Yin, L.; et al. Large-scale deep multi-layer analysis of Alzheimer’s disease brain reveals strong proteomic disease-related changes not observed at the RNA level. Nat. Neurosci. 2022, 25, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Holper, L.; Ben-Shachar, D.; Mann, J.J. Multivariate meta-analyses of mitochondrial complex I and IV in major depressive disorder, bipolar disorder, schizophrenia, Alzheimer disease, and Parkinson disease. Neuropsychopharmacology 2019, 44, 837–849. [Google Scholar] [CrossRef] [PubMed]
- Morais, F.M.; Ribeiro, A.M.; Moreira, F.A.; Silva, P.V.G. Systematic review and meta-analysis on the role of mitochondrial cytochrome c oxidase in Alzheimer’s disease. Acta Neuropsychiatr. 2021, 33, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Pungerčar, J.; Križaj, I.; Liang, N.S.; Gubenšek, F. An aromatic, but not a basic, residue is involved in the toxicity of group-II phospholipase A2 neurotoxins. Biochem. J. 1999, 341, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Oberčkal, J.; Kovačič, L.; Šribar, J.; Leonardi, A.; Dolinar, K.; Pucer Janež, A.; Križaj, I. On the role of protein disulfide isomerase in the retrograde cell transport of secreted phospholipases A2. PLoS ONE 2015, 10, e0120692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Šribar, J.; Sherman, N.E.; Prijatelj, P.; Faure, G.; Gubenšek, F.; Fox, J.W.; Aitken, A.; Pungerčar, J.; Križaj, I. The neurotoxic phospholipase A2 associates, through a non-phosphorylated binding motif, with 14-3-3 protein gamma and epsilon isoforms. Biochem. Biophys. Res. Commun. 2003, 302, 691–696. [Google Scholar] [CrossRef]
- Vučemilo, N.; Čopič, A.; Gubenšek, F.; Križaj, I. Identification of a new high-affinity binding protein for neurotoxic phospholipases A2. Biochem. Biophys. Res. Commun. 1998, 251, 209–212. [Google Scholar] [CrossRef]
- Čopič, A.; Vučemilo, N.; Gubenšek, F.; Križaj, I. Identification and purification of a novel receptor for secretory phospholipase A2 in porcine cerebral cortex. J. Biol. Chem. 1999, 274, 26315–26320. [Google Scholar] [CrossRef] [Green Version]
- Šribar, J.; Čopič, A.; Paris, A.; Sherman, N.E.; Gubenšek, F.; Fox, J.W.; Križaj, I. A high affinity acceptor for phospholipase A2 with neurotoxic activity is a calmodulin. J. Biol. Chem. 2001, 276, 12493–12496. [Google Scholar] [CrossRef] [Green Version]
- Merril, C.R.; Goldman, D.; Sedman, S.A.; Ebert, M.H. Ultrasensitive stain for proteins in polyacrylamide gels shows regional variation in cerebrospinal fluid proteins. Science 1981, 211, 1437–1438. [Google Scholar] [CrossRef]
- Strojan, K.; Leonardi, A.; Bregar, V.B.; Križaj, I.; Svete, J.; Pavlin, M. Dispersion of nanoparticles in different media importantly determines the composition of their protein corona. PLoS ONE 2017, 12, e0169552. [Google Scholar] [CrossRef] [Green Version]
- Premrov Bajuk, B.; Zrimšek, P.; Kotnik, T.; Leonardi, A.; Križaj, I.; Jakovac Strajn, B. Insect protein-based diet as potential risk of allergy in dogs. Animals 2021, 11, 1942. [Google Scholar] [CrossRef]
- Leavesley, H.B.; Li, L.; Prabhakaran, K.; Borowitz, J.L.; Isom, G.E. Interaction of cyanide and nitric oxide with cytochrome c oxidase: Implications for acute cyanide toxicity. Toxicol. Sci. 2008, 101, 101–111. [Google Scholar] [CrossRef] [Green Version]
- Manders, E.M.M.; Verbeek, F.J.; Aten, J.A. Measurement of co-localization of objects in dual-colour confocal images. J. Microsc. 1993, 169, 375–382. [Google Scholar] [CrossRef]
- Perry, S.W.; Norman, J.P.; Barbieri, J.; Brown, E.B.; Gelbard, H.A. Mitochondrial membrane potential probes and the proton gradient: A practical usage guide. Biotechniques 2011, 50, 98–115. [Google Scholar] [CrossRef]
- Milatović, D.; Živin, M.; Gupta, R.C.; Dettbarn, W.D. Alterations in cytochrome c oxidase activity and energy metabolites in response to kainic acid-induced status epilepticus. Brain Res. 2001, 912, 67–78. [Google Scholar] [CrossRef]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates, 6th ed.; Academic Press: San Diego, CA, USA, 2007; 456p. [Google Scholar]
- Wong-Riley, M. Changes in the visual system of monocularly sutured or enucleated cats demonstrable with cytochrome oxidase histochemistry. Brain Res. 1979, 171, 11–28. [Google Scholar] [CrossRef]
- Divac, I.; Mojsilovic-Petrovic, J.; López-Figueroa, M.O.; Petrovic-Minic, B.; Møller, M. Improved contrast in histochemical detection of cytochrome oxidase: Metallic ions protocol. J. Neurosci. Methods 1995, 56, 105–113. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ivanušec, A.; Šribar, J.; Leonardi, A.; Zorović, M.; Živin, M.; Križaj, I. Rat Group IIA Secreted Phospholipase A2 Binds to Cytochrome c Oxidase and Inhibits Its Activity: A Possible Episode in the Development of Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 12368. https://doi.org/10.3390/ijms232012368
Ivanušec A, Šribar J, Leonardi A, Zorović M, Živin M, Križaj I. Rat Group IIA Secreted Phospholipase A2 Binds to Cytochrome c Oxidase and Inhibits Its Activity: A Possible Episode in the Development of Alzheimer’s Disease. International Journal of Molecular Sciences. 2022; 23(20):12368. https://doi.org/10.3390/ijms232012368
Chicago/Turabian StyleIvanušec, Adrijan, Jernej Šribar, Adrijana Leonardi, Maja Zorović, Marko Živin, and Igor Križaj. 2022. "Rat Group IIA Secreted Phospholipase A2 Binds to Cytochrome c Oxidase and Inhibits Its Activity: A Possible Episode in the Development of Alzheimer’s Disease" International Journal of Molecular Sciences 23, no. 20: 12368. https://doi.org/10.3390/ijms232012368