
Involvement of the IL-6 Signaling Pathway in the Anti-Anhedonic Effect of the Antidepressant Agomelatine in the Chronic Mild Stress Model of Depression
 , , , , ,
, , , , ,  and
and
Abstract
:
1. Introduction
2. Results
2.1. Evaluation of Agomelatine Effect on the Impaired Sucrose Intake of CMS Rats
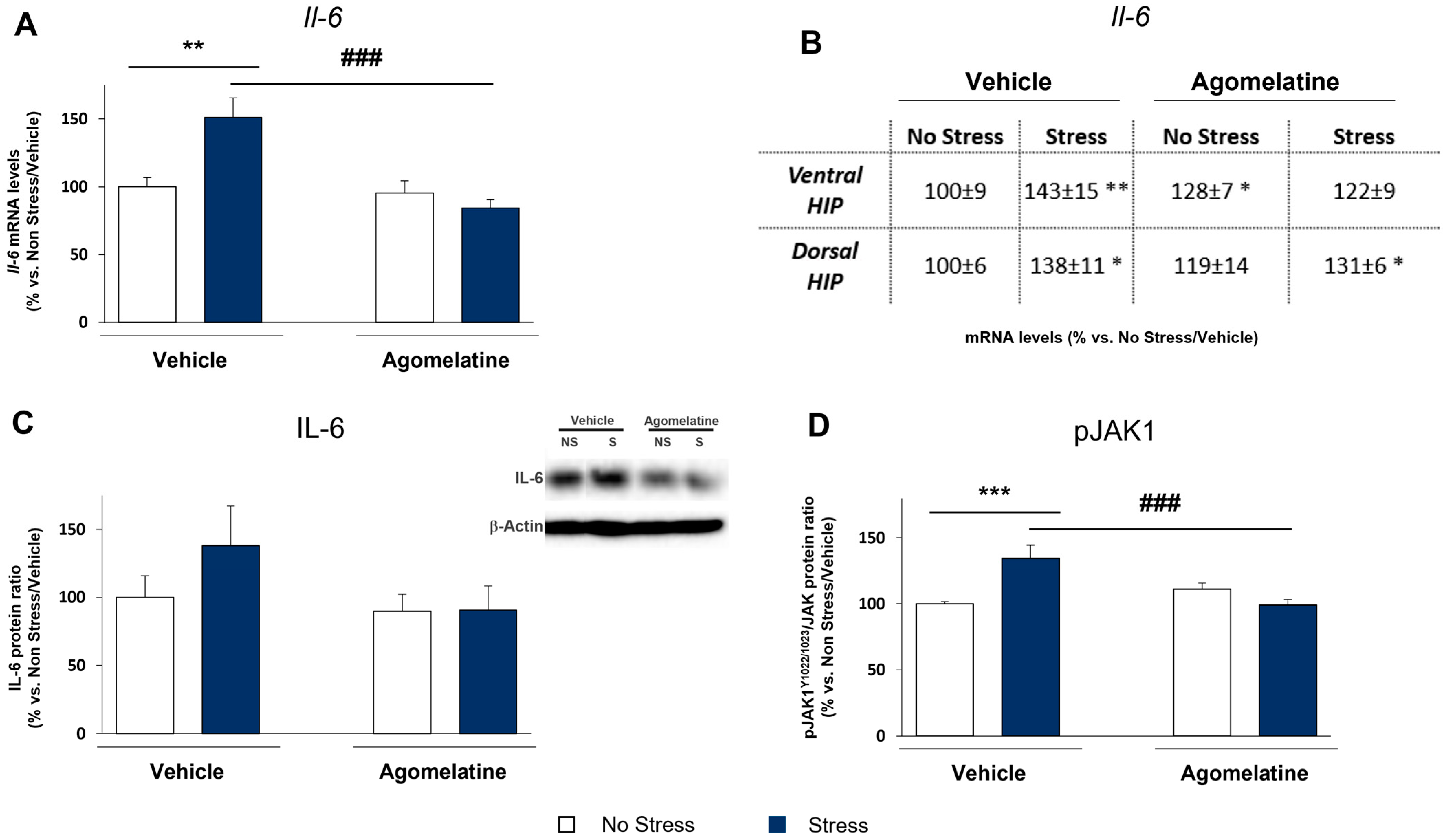
2.2. Modulation of IL-6 mRNA and Protein Levels and JAK1 Activation in Rats Exposed to CMS and Treated with Agomelatine
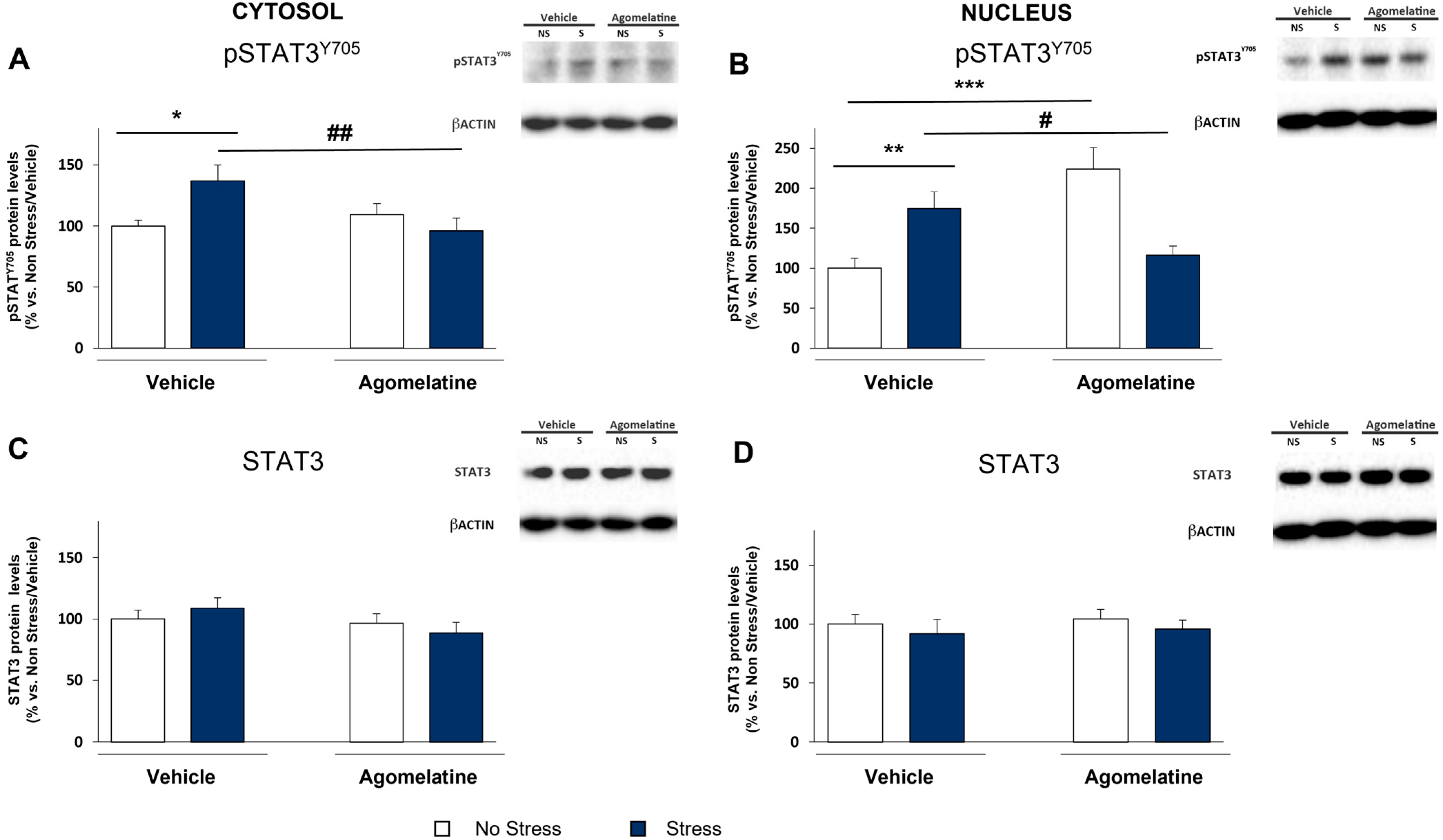
2.3. Modulation of STAT3 Activation in the Prefrontal Cortex of Rats Exposed to CMS and Treated with Agomelatine
2.4. Modulation of SOCS3 Expression in the Prefrontal Cortex of Rats Exposed to CMS and Treated with Agomelatine
2.5. MAP Kinases as Alternative Mechanisms of STAT3/SOCS3 Activation in Prefrontal Cortex of Rats Exposed to CMS and Treated with Agomelatine
3. Discussion
4. Material and Methods
4.1. Animals
4.2. Stress Procedure and Pharmacological Treatment
4.3. RNA Preparation and Gene Expression Analyses
4.4. Protein Extraction, Cellular Fraction Preparation, and Western Blot Analyses
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Konnopka, A.; König, H. Economic Burden of Anxiety Disorders: A Systematic Review and Meta-Analysis. PharmacoEconomics 2020, 38, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Kraus, C.; Kadriu, B.; Lanzenberger, R.; Zarate, C.A., Jr.; Kasper, S. Prognosis and improved outcomes in major depression: A review. Transl. Psychiatry 2019, 9, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Troubat, R.; Barone, P.; Leman, S.; DeSmidt, T.; Cressant, A.; Atanasova, B.; Brizard, B.; El Hage, W.; Surget, A.; Belzung, C.; et al. Neuroinflammation and depression: A review. Eur. J. Neurosci. 2021, 53, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Afridi, R.; Suk, K. Neuroinflammatory Basis of Depression: Learning From Experimental Models. Front. Cell. Neurosci. 2021, 15, 691067. [Google Scholar] [CrossRef] [PubMed]
- Dowlati, Y.; Herrmann, N.; Swardfager, W.; Liu, H.; Sham, L.; Reim, E.K.; Lanctôt, K.L. A Meta-Analysis of Cytokines in Major Depression. Biol. Psychiatry 2010, 67, 446–457. [Google Scholar] [CrossRef]
- Goldsmith, D.R.; Rapaport, M.H.; Miller, B.J. A meta-analysis of blood cytokine network alterations in psychiatric patients: Comparisons between schizophrenia, bipolar disorder and depression. Mol. Psychiatry 2016, 21, 1696–1709. [Google Scholar] [CrossRef]
- Osimo, E.F.; Pillinger, T.; Rodriguez, I.M.; Khandaker, G.M.; Pariante, C.M.; Howes, O.D. Inflammatory markers in depression: A meta-analysis of mean differences and variability in 5166 patients and 5083 controls. Brain Behav. Immun. 2020, 87, 901–909. [Google Scholar] [CrossRef]
- Bai, S.; Guo, W.; Feng, Y.; Deng, H.; Li, G.; Nie, H.; Guo, G.; Yu, H.; Ma, Y.; Wang, J.; et al. Efficacy and safety of anti-inflammatory agents for the treatment of major depressive disorder: A systematic review and meta-analysis of randomised controlled trials. J. Neurol. Neurosurg. Psychiatry 2020, 91, 21–32. [Google Scholar] [CrossRef]
- Köhler, O.; Benros, M.E.; Nordentoft, M.; Farkouh, M.E.; Iyengar, R.L.; Mors, O.; Krogh, J. Effect of anti-inflammatory treatment on depression, depressive symptoms, and adverse effects: A systematic review and meta-analysis of randomized clinical trials. JAMA Psychiatry 2014, 71, 1381–1391. [Google Scholar] [CrossRef]
- Liu, Y.; Ho, R.C.-M.; Mak, A. Interleukin (IL)-6, tumour necrosis factor alpha (TNF-α) and soluble interleukin-2 receptors (sIL-2R) are elevated in patients with major depressive disorder: A meta-analysis and meta-regression. J. Affect. Disord. 2012, 139, 230–239. [Google Scholar] [CrossRef]
- Howren, M.B.; Lamkin, D.M.; Suls, J. Associations of Depression With C-Reactive Protein, IL-1, and IL-6: A Meta-Analysis. Psychosom. Med. 2009, 71, 171–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, C.A.; Jones, S.A. Erratum: Corrigendum: IL-6 as a keystone cytokine in health and disease. Nat. Immunol. 2017, 18, 1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maes, M.; Anderson, G.; Kubera, M.; Berk, M. Targeting classical IL-6 signalling or IL-6trans-signalling in depression? Expert Opin. Ther. Targets 2014, 18, 495–512. [Google Scholar] [CrossRef]
- Reeh, H.; Rudolph, N.; Billing, U.; Christen, H.; Streif, S.; Bullinger, E.; Schliemann-Bullinger, M.; Findeisen, R.; Schaper, F.; Huber, H.J.; et al. Response to IL-6 trans- and IL-6 classic signalling is determined by the ratio of the IL-6 receptor α to gp130 expression: Fusing experimental insights and dynamic modelling. Cell Commun. Signal. 2019, 17, 46. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.; Tanaka, T.; Narazaki, M.; Kishimoto, T. Targeting Interleukin-6 Signaling in Clinic. Immunity 2019, 50, 1007–1023. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, K.; Hasegawa, T.; Takeda, M. Serum level of soluble interleukin 6 receptor is a useful biomarker for identification of treatment-resistant major depressive disorder. Neuropsychopharmacol. Rep. 2020, 40, 130–137. [Google Scholar] [CrossRef] [Green Version]
- Guardiola-Lemaitre, B.; De Bodinat, C.; Delagrange, P.; Millan, M.J.; Munoz, C.; Mocaër, E. Agomelatine: Mechanism of action and pharmacological profile in relation to antidepressant properties. J. Cereb. Blood Flow Metab. 2014, 171, 3604–3619. [Google Scholar] [CrossRef]
- Molteni, R.; Macchi, F.; Zecchillo, C.; Dell’Agli, M.; Colombo, E.; Calabrese, F.; Guidotti, G.; Racagni, G.; Riva, M.A. Modulation of the inflammatory response in rats chronically treated with the antidepressant agomelatine. Eur. Neuropsychopharmacol. 2013, 23, 1645–1655. [Google Scholar] [CrossRef]
- Rossetti, A.C.; Paladini, M.S.; Racagni, G.; Riva, M.A.; Cattaneo, A.; Molteni, R. Genome-wide analysis of LPS-induced inflammatory response in the rat ventral hippocampus: Modulatory activity of the antidepressant agomelatine. World J. Biol. Psychiatry 2017, 19, 390–401. [Google Scholar] [CrossRef]
- Rossetti, A.C.; Papp, M.; Gruca, P.; Paladini, M.S.; Racagni, G.; Riva, M.A.; Molteni, R. Stress-induced anhedonia is associated with the activation of the inflammatory system in the rat brain: Restorative effect of pharmacological intervention. Pharmacol. Res. 2016, 103, 78–93. [Google Scholar] [CrossRef]
- Willner, P. The chronic mild stress (CMS) model of depression: History, evaluation and usage. Neurobiol. Stress 2016, 6, 78–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, E.; Sansing, L.H.; Arnsten, A.F.T.; Datta, D. Chronic Stress Weakens Connectivity in the Prefrontal Cortex: Architectural and Molecular Changes. Chronic Stress 2014, 5, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Qi, Q.; Yang, Z.-M. Regulation and function of signal transducer and activator of transcription 3. World J. Biol. Chem. 2014, 5, 231–239. [Google Scholar] [CrossRef]
- McEwen, B.S.; Morrison, J.H. The Brain on Stress: Vulnerability and Plasticity of the Prefrontal Cortex over the Life Course. Neuron 2013, 79, 16–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Himmerich, H.; Fischer, J.; Bauer, K.; Kirkby, K.C.; Sack, U.; Krügel, U. Stress-induced cytokine changes in rats. Eur. Cytokine Netw. 2013, 24, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Cao, F.; Liu, Q.; Li, X.; Xu, G.; Liu, G.; Zhang, Y.; Yang, X.; Yi, S.; Xu, F.; et al. Behavioral, inflammatory and neurochemical disturbances in LPS and UCMS-induced mouse models of depression. Behav. Brain Res. 2019, 364, 494–502. [Google Scholar] [CrossRef] [PubMed]
- López-López, A.L.; Jaime, H.B.; Villanueva, M.D.C.E.; Padilla, M.B.; Palacios, G.V.; Aguilar, F.J.A. Chronic unpredictable mild stress generates oxidative stress and systemic inflammation in rats. Physiol. Behav. 2016, 161, 15–23. [Google Scholar] [CrossRef]
- Qing, H.; Desrouleaux, R.; Israni-Winger, K.; Mineur, Y.S.; Fogelman, N.; Zhang, C.; Rashed, S.; Palm, N.W.; Sinha, R.; Picciotto, M.R.; et al. Origin and Function of Stress-Induced IL-6 in Murine Models. Cell 2020, 182, 372–387. [Google Scholar] [CrossRef]
- Chourbaji, S.; Urani, A.; Inta, I.; Sanchis-Segura, C.; Brandwein, C.; Zink, M.; Schwaninger, M.; Gass, P. IL-6 knockout mice exhibit resistance to stress-induced development of depression-like behaviors. Neurobiol. Dis. 2006, 23, 587–594. [Google Scholar] [CrossRef]
- Engler, H.; Brendt, P.; Wischermann, J.; Wegner, A.; Röhling, R.; Schoemberg, T.; Meyer, U.; Gold, R.; Peters, J.; Benson, S.; et al. Selective increase of cerebrospinal fluid IL-6 during experimental systemic inflammation in humans: Association with depressive symptoms. Mol. Psychiatry 2017, 22, 1448–1454. [Google Scholar] [CrossRef]
- Du, R.-H.; Tan, J.; Sun, X.-Y.; Lu, M.; Ding, J.-H.; Hu, G. Fluoxetine Inhibits NLRP3 Inflammasome Activation: Implication in Depression. Int. J. Neuropsychopharmacol. 2016, 150–151, 138–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwata, M.; Ishida, H.; Kaneko, K.; Shirayama, Y. Learned helplessness activates hippocampal microglia in rats: A potential target for the antidepressant imipramine. Pharmacol. Biochem. Behav. 2016, 150-151, 138–146. [Google Scholar] [CrossRef]
- Ramirez, K.; Sheridan, J.F. Antidepressant imipramine diminishes stress-induced inflammation in the periphery and central nervous system and related anxiety- and depressive-like behaviors. Brain Behav. Immun. 2016, 57, 293–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babon, J.J.; Varghese, L.N.; Nicola, N. Inhibition of IL-6 family cytokines by SOCS3. Semin. Immunol. 2014, 26, 13–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahony, R.; Ahmed, S.; Diskin, C.; Stevenson, N.J. SOCS3 revisited: A broad regulator of disease, now ready for therapeutic use? Neurobiol. Dis. 2016, 73, 3323–3336. [Google Scholar] [CrossRef] [PubMed]
- Money, K.M.; Olah, Z.; Korade, Z.; Garbett, K.A.; Shelton, R.C.; Mirnics, K. An altered peripheral IL6 response in major depressive disorder. Neurobiol. Dis. 2016, 89, 46–54. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Zhang, W.-W.; Liu, P.; Yu, W.; Liu, T.; Yu, J. Dysregulation of SOCS-Mediated Negative Feedback of Cytokine Signaling in Carcinogenesis and Its Significance in Cancer Treatment. Front. Immunol. 2017, 8, 70. [Google Scholar] [CrossRef] [Green Version]
- Al-Samhari, M.M.; Al-Rasheed, N.M.; Al-Rejaie, S.; Al-Rasheed, N.M.; Hasan, I.; Mahmoud, A.M.; Dzimiri, N. Possible involvement of the JAK/STAT signaling pathway in N-acetylcysteine-mediated antidepressant-like effects. Exp. Biol. Med. 2015, 241, 509–518. [Google Scholar] [CrossRef] [Green Version]
- Alboni, S.; Poggini, S.; Garofalo, S.; Milior, G.; El Hajj, H.; Lecours, C.; Girard, I.; Gagnon, S.; Boisjoly-Villeneuve, S.; Brunello, N.; et al. Fluoxetine treatment affects the inflammatory response and microglial function according to the quality of the living environment. Brain Behav. Immun. 2016, 58, 261–271. [Google Scholar] [CrossRef]
- Pan, Y.; Hong, Y.; Zhang, Q.-Y.; Kong, L.-D. Impaired hypothalamic insulin signaling in CUMS rats: Restored by icariin and fluoxetine through inhibiting CRF system. Psychoneuroendocrinology 2013, 38, 122–134. [Google Scholar] [CrossRef]
- Qin, H.; Roberts, K.L.; Niyongere, S.A.; Cong, Y.; Elson, C.O.; Benveniste, E.N. Molecular Mechanism of Lipopolysaccharide-Induced SOCS-3 Gene Expression in Macrophages and Microglia. J. Immunol. 2007, 179, 5966–5976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiejak, J.; Dunlop, J.; Gao, S.; Borland, G.; Yarwood, S.J. Extracellular Signal-Regulated Kinase Mitogen-Activated Protein Kinase-Dependent SOCS-3 Gene Induction Requires c-Jun, Signal Transducer and Activator of Transcription 3, and Specificity Protein 3 Transcription Factors. Mol. Pharmacol. 2012, 81, 657–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehlting, C.; Böhmer, O.; Hahnel, M.J.; Thomas, M.; Zanger, U.M.; Gaestel, M.; Knoefel, W.T.; Am Esch, J.S.; Häussinger, D.; Bode, J.G. Oncostatin M regulates SOCS3 mRNA stability via the MEK-ERK1/2-pathway independent of p38(MAPK)/MK2. Cell. Signal. 2015, 27, 555–567. [Google Scholar] [CrossRef]
- Cecon, E.; Oishi, A.; Jockers, R. Melatonin receptors: Molecular pharmacology and signalling in the context of system bias. J. Cereb. Blood Flow Metab. 2018, 175, 3263–3280. [Google Scholar] [CrossRef] [Green Version]
- Jonas, E.A.; Porter, G.A.; Alavian, K.N. Bcl-xL in neuroprotection and plasticity. Front. Physiol. 2014, 5, 355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.; Chen, S.; Liu, J.; Amin, N.; Jin, W.; Fang, M. Agomelatine Softens Depressive-Like Behavior through the Regulation of Autophagy and Apoptosis. BioMed Res. Int. 2021, 2021, 6664591. [Google Scholar] [CrossRef] [PubMed]
- Chumboatong, W.; Thummayot, S.; Govitrapong, P.; Tocharus, C.; Jittiwat, J.; Tocharus, J. Neuroprotection of agomelatine against cerebral ischemia/reperfusion injury through an antiapoptotic pathway in rat. Neurochem. Int. 2017, 102, 114–122. [Google Scholar] [CrossRef]
- Engel, D.; Zomkowski, A.D.; Lieberknecht, V.; Rodrigues, A.L.; Gabilan, N.H. Chronic administration of duloxetine and mirtazapine downregulates proapoptotic proteins and upregulates neurotrophin gene expression in the hippocampus and cerebral cortex of mice. J. Psychiatr. Res. 2013, 47, 802–808. [Google Scholar] [CrossRef]
- Kosten, T.A.; Galloway, M.P.; Duman, R.S.; Russell, D.S.; D’Sa, C. Repeated Unpredictable Stress and Antidepressants Differentially Regulate Expression of the Bcl-2 Family of Apoptotic Genes in Rat Cortical, Hippocampal, and Limbic Brain Structures. Neuropsychopharmacology 2008, 33, 1545–1558. [Google Scholar] [CrossRef]
- Kubera, M.; Obuchowicz, E.; Goehler, L.; Brzeszcz, J.; Maes, M. In animal models, psychosocial stress-induced (neuro)inflammation, apoptosis and reduced neurogenesis are associated to the onset of depression. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2011, 35, 744–759. [Google Scholar] [CrossRef]
- Zhang, C.-L.; Song, F.; Zhang, J.; Song, Q. Hypoxia-induced Bcl-2 expression in endothelial cells via p38 MAPK pathway. Biochem. Biophys. Res. Commun. 2010, 394, 976–980. [Google Scholar] [CrossRef] [PubMed]
- Tai, T.W.; Su, F.C.; Chen, C.Y.; Jou, I.M.; Lin, C.F. Activation of p38 MAPK-regulated Bcl-xL signaling increases survival against zoledronic acid-induced apoptosis in osteoclast precursors. Bone 2014, 67, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Papp, M. Models of Affective Illness: Chronic Mild Stress in the Rat. Curr. Protoc. Pharmacol. 2012, 57, 1–11. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer | Probe |
|---|---|---|---|
| Socs3 | AGAGCGGATTCTACTGGAGT | TCGACGCTCAGTGTGAAGAA | TTTCTTATCCGCGACAGCTC |
| Bcl-xl | GAACTCTTTCGGGATGGGGTAA | ACTTGCAATCCGACTCACCA | AGCGTAGACAAGGAGATGCA |
| β-Actin | CACTTTCTACAATGAGCTGCG | CTGGATGGCTACGTACATGG | TCTGGGTCATCTTTTCACGGTTGGC |
| Il-6 | Purchased from Applied Biosystem (Italy) cod. Rn99999011_m1 | ||
| Gapdh | Purchased from Applied Biosystem (Italy) cod. Rn99999916_s1 | ||
| Target Protein | Primary Antibody | Secondary Antibody |
|---|---|---|
| IL-6 (21 kDa) | 1:500, BSA 5% in TBS-t Santa Cruz Biotech ID: AB_2127595 | HRP conjugated anti-rabbit IgG 1:500 Cell Signaling |
| pSTAT3 Tyr705 (86 kDa) | 1:500, BSA 5% in TBS-t Cell Signaling ID: AB_331586 | HRP conjugated anti-rabbit IgG 1:1000 Cell Signaling |
| pSTAT3 Ser727 (86 kDa) | 1:500, BSA 5% in TBS-t Cell Signaling ID: AB_331589 | HRP conjugated anti-rabbit IgG 1:1000 Cell Signaling |
| STAT3 (86 kDa) | 1:2000, BSA 5% in TBS-t Cell Signaling ID: AB_331269 | HRP conjugated anti-rabbit IgG 1:4000 Cell Signaling |
| SOCS3 (23 kDa) | 1:1000, BSA 5% in TBS-t Cell Signaling ID: AB_2286460 | HRP conjugated anti-rabbit IgG 1:1000 Cell Signaling |
| pJAK1 Tyr1022/1023 (120 kDa) | 1:500, BSA 5% in TBS-t Cell Signaling ID: AB_2265057 | HRP conjugated anti-rabbit IgG 1:1000 Cell Signaling |
| JAK1 (120 kDa) | 1:500, BSA 5% in TBS-t Cell Signaling ID: AB_2128499 | HRP conjugated anti-rabbit IgG 1:500 Cell Signaling |
| pp38 Thr180/Tyr182 (43 kDa) | 1:500, BSA 5% in TBS-t Cell Signaling ID: AB_331641 | HRP conjugated anti-rabbit IgG 1:500 Cell Signaling |
| p38 (43 kDa) | 1:1000, BSA 5% in TBS-t Cell Signaling ID: AB_330713 | HRP conjugated anti-rabbit IgG 1:2000 Cell Signaling |
| pERK1/2 Thr202/Tyr204 (42/44 kDa) | 1:1000, 3% nonfat dry milk in TBS-t Cell Signaling ID: AB_2315112 | HRP conjugated anti-rabbit IgG 1:2000 Cell Signaling |
| ERK1/2 (42/44 kDa) | 1:5000 1% nonfat dry milk in TBS-t Santa Cruz Biotech ID: AB_2140110 | HRP conjugated anti-rabbit IgG 1:5000 Cell Signaling |
| β-Actin (43 kDa) | 1:10,000 3% nonfat dry milk in TBS-t Sigma-Aldrich ID: AB_476744 | HRP conjugated anti-mouse IgG 1:20,000 Sigma-Aldrich |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossetti, A.C.; Paladini, M.S.; Brüning, C.A.; Spero, V.; Cattaneo, M.G.; Racagni, G.; Papp, M.; Riva, M.A.; Molteni, R. Involvement of the IL-6 Signaling Pathway in the Anti-Anhedonic Effect of the Antidepressant Agomelatine in the Chronic Mild Stress Model of Depression. Int. J. Mol. Sci. 2022, 23, 12453. https://doi.org/10.3390/ijms232012453
Rossetti AC, Paladini MS, Brüning CA, Spero V, Cattaneo MG, Racagni G, Papp M, Riva MA, Molteni R. Involvement of the IL-6 Signaling Pathway in the Anti-Anhedonic Effect of the Antidepressant Agomelatine in the Chronic Mild Stress Model of Depression. International Journal of Molecular Sciences. 2022; 23(20):12453. https://doi.org/10.3390/ijms232012453
Chicago/Turabian StyleRossetti, Andrea C., Maria Serena Paladini, Cesar Augusto Brüning, Vittoria Spero, Maria Grazia Cattaneo, Giorgio Racagni, Mariusz Papp, Marco A. Riva, and Raffaella Molteni. 2022. "Involvement of the IL-6 Signaling Pathway in the Anti-Anhedonic Effect of the Antidepressant Agomelatine in the Chronic Mild Stress Model of Depression" International Journal of Molecular Sciences 23, no. 20: 12453. https://doi.org/10.3390/ijms232012453