
Improving Fmoc Solid Phase Synthesis of Human Beta Defensin 3
 ,
,
Abstract
:1. Introduction
2. Results
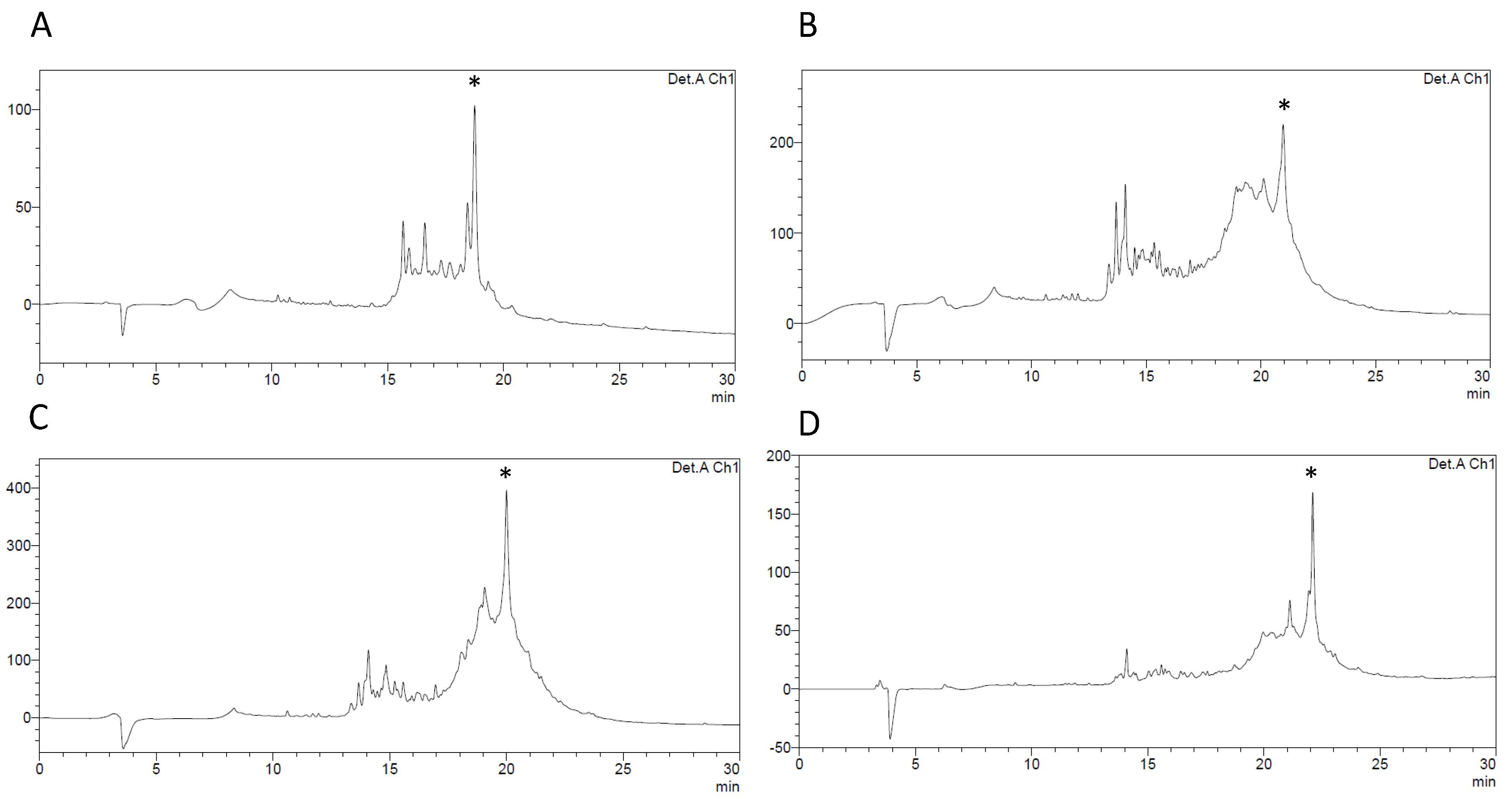
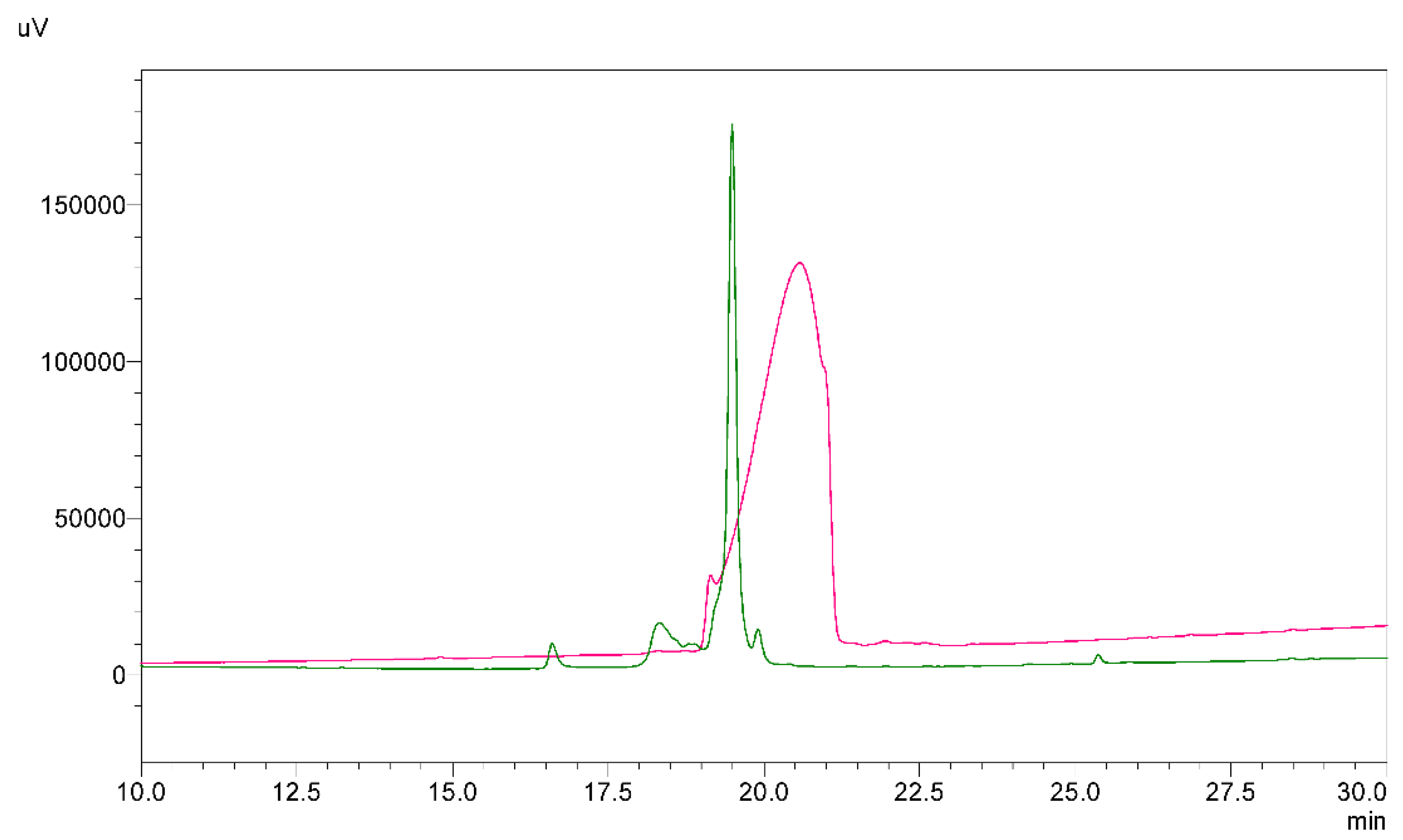
2.1. Chemical Synthesis of a Linear Precursor of HBD-3
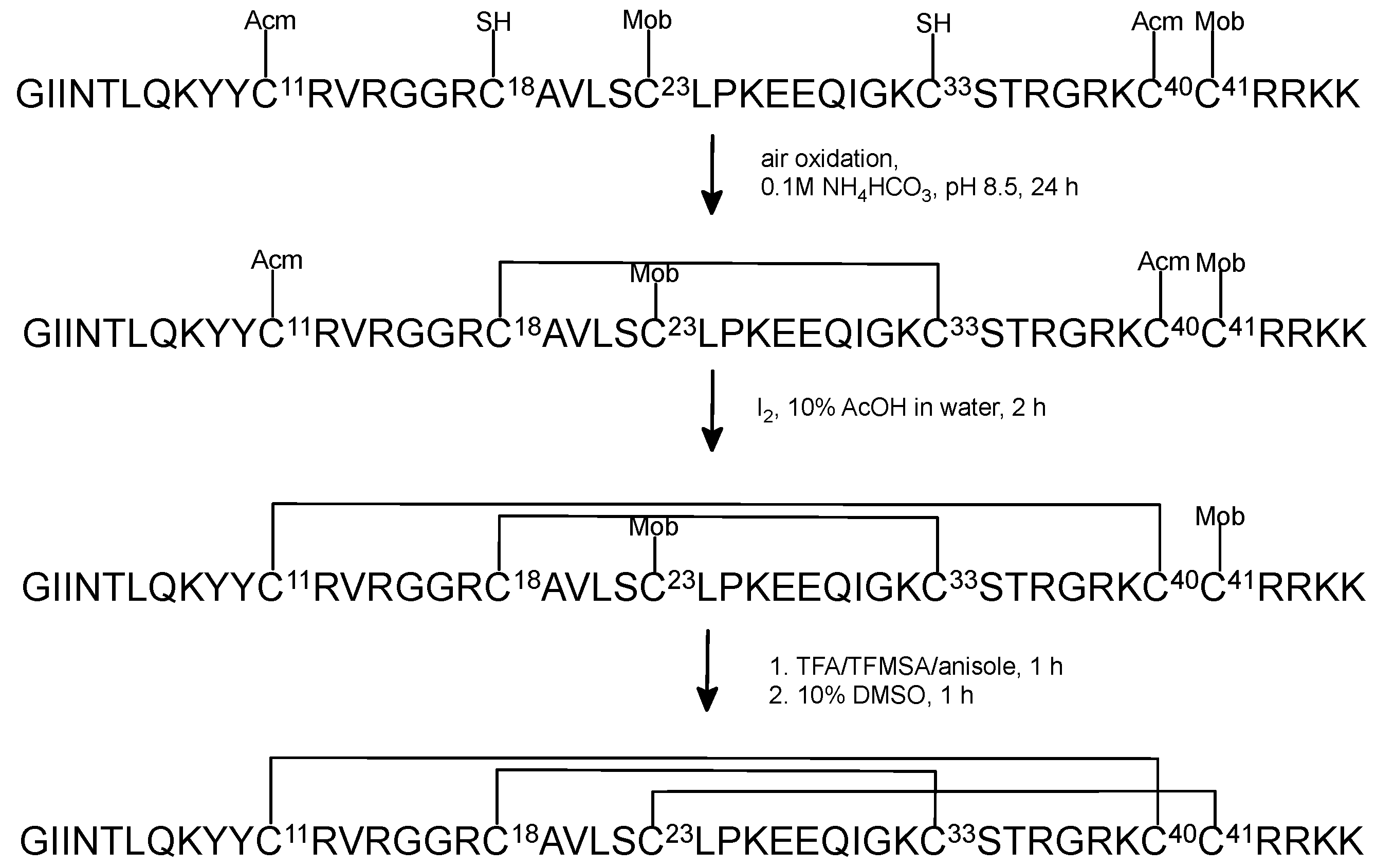
2.2. Oxidative Folding with the Orthogonal Thiol Trt/Acm/Mob Strategy
2.3. Oxidative Folding with the Orthogonal Thiol Mmt/Trt/Acm Strategy
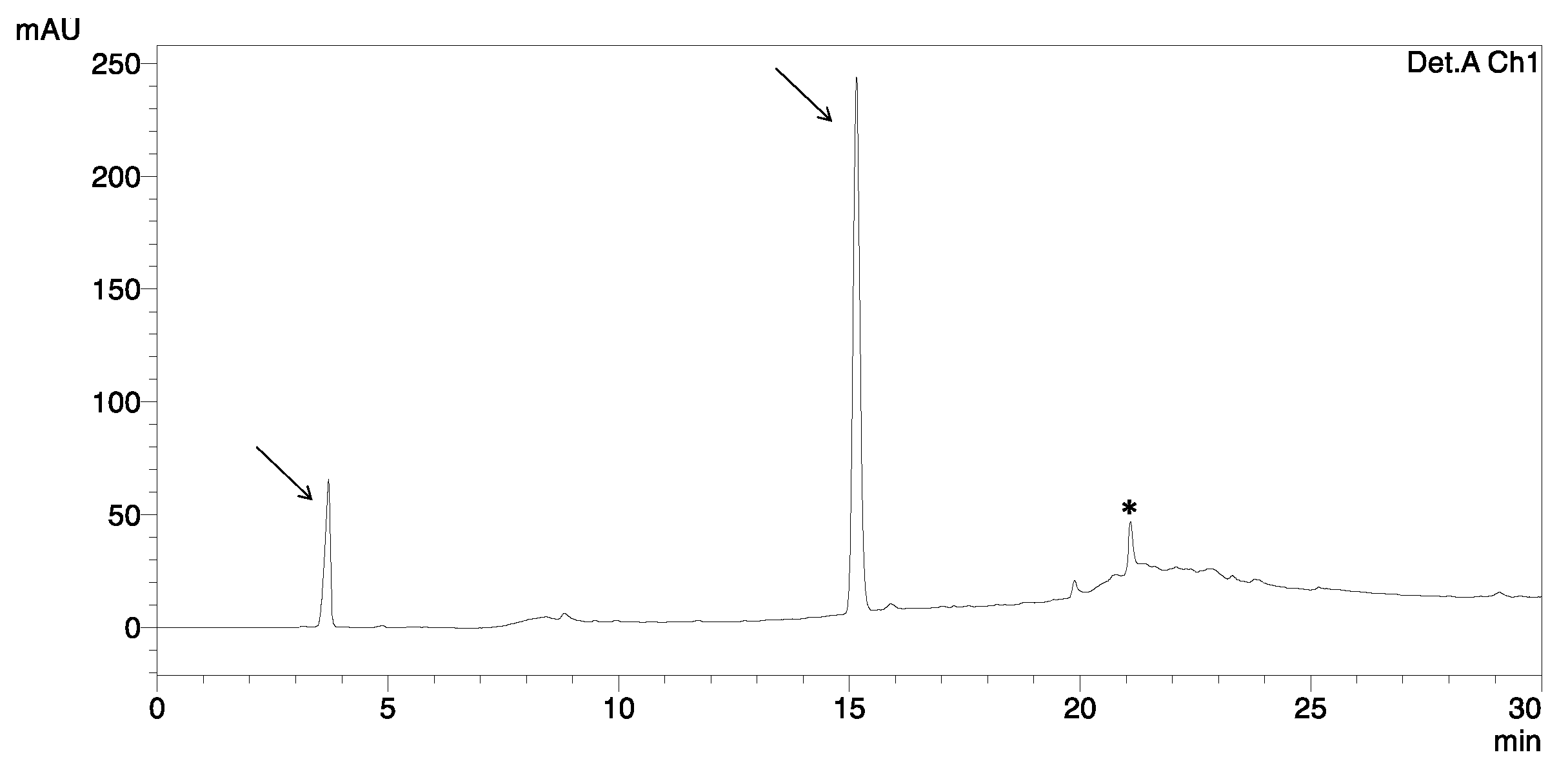
2.4. Chemical Synthesis and Oxidative Folding of [Sec18,33]HBD-3
2.5. Regioselective Oxidative Folding versus Direct Oxidative Folding of HBD-3
2.6. Antimicrobial Activity against E.coli and S. aureus
3. Discussion
4. Materials and Methods
4.1. The Chemicals
4.2. In Silico Prediction of HBD-3 Aggregation Potential
4.3. Peptide Synthesis
4.4. Analysis and Purification
4.5. Oxidative Folding
4.6. Evaluation of Antibacterial Properties
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Maisetta, G.; Batoni, G.; Esin, S.; Florio, W.; Bottai, D.; Favilli, F.; Campa, M. In vitro bactericidal activity of human beta-defensin 3 against multidrug-resistant nosocomial strains. Antimicrob. Agents. Chemother. 2006, 50, 806–809. [Google Scholar] [CrossRef] [Green Version]
- Feng, Z.; Jia, X.; Adams, M.D.; Ghosh, S.K.; Bonomo, R.A.; Weinberg, A. Epithelial innate immune response to Acinetobacter baumannii challenge. Infect Immun. 2014, 82, 4458–4465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harder, J.; Bartels, J.; Christophers, E.; Schroder, J.M. Isolation and characterization of human beta -defensin-3, a novel human inducible peptide antibiotic. J. Biol. Chem. 2001, 276, 5707–5713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batoni, G.; Maisetta, G.; Esin, S.; Campa, M. Human beta-defensin-3: A promising antimicrobial peptide. Mini Rev. Med. Chem. 2006, 6, 1063–1073. [Google Scholar] [CrossRef]
- Ferris, L.K.; Mburu, Y.K.; Mathers, A.R.; Fluharty, E.R.; Larregina, A.T.; Ferris, R.L.; Falo, L.D., Jr. Human beta-defensin 3 induces maturation of human langerhans cell-like dendritic cells: An antimicrobial peptide that functions as an endogenous adjuvant. J. Investig. Dermatol. 2013, 133, 460–468. [Google Scholar] [CrossRef] [Green Version]
- Mohan, T.; Mitra, D.; Rao, D.N. Nasal delivery of PLG microparticle encapsulated defensin peptides adjuvanted gp41 antigen confers strong and long-lasting immunoprotective response against HIV-1. Immunol. Res. 2014, 58, 139–153. [Google Scholar] [CrossRef] [PubMed]
- Hazrati, E.; Galen, B.; Lu, W.; Wang, W.; Ouyang, Y.; Keller, M.J.; Lehrer, R.I.; Herold, B.C. Human alpha- and beta-defensins block multiple steps in herpes simplex virus infection. J. Immunol. 2006, 177, 8658–8666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leikina, E.; Delanoe-Ayari, H.; Melikov, K.; Cho, M.S.; Chen, A.; Waring, A.J.; Wang, W.; Xie, Y.; Loo, J.A.; Lehrer, R.I.; et al. Carbohydrate-binding molecules inhibit viral fusion and entry by crosslinking membrane glycoproteins. Nat. Immunol. 2005, 6, 995–1001. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Dubyak, G.R.; Lederman, M.M.; Weinberg, A. Cutting edge: Human beta defensin 3—A novel antagonist of the HIV-1 coreceptor CXCR4. J. Immunol. 2006, 177, 782–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Xie, Z.; Feng, J.; Li, W.; Cao, Z.; Wu, Y. Pharmacological characterization of human beta-defensins 3 and 4 on potassium channels: Evidence of diversity in beta-defensin-potassium channel interactions. Peptides 2018, 108, 14–18. [Google Scholar] [CrossRef]
- Nix, M.A.; Kaelin, C.B.; Ta, T.; Weis, A.; Morton, G.J.; Barsh, G.S.; Millhauser, G.L. Molecular and functional analysis of human β-defensin 3 action at melanocortin receptors. Chem. Biol. 2013, 20, 784–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulsen, C.E.; Carroll, K.S. Orchestrating Redox Signaling Networks through Regulatory Cysteine Switches. ACS Chem. Biol. 2010, 5, 47–62. [Google Scholar] [CrossRef] [Green Version]
- Leonard, S.E.; Garcia, F.J.; Goodsell, D.S.; Carroll, K.S. Redox-based probes for protein tyrosine phosphatases. Angew. Chem. Int. Ed. Engl. 2011, 50, 4423–4427. [Google Scholar] [CrossRef]
- Berg, J.M.; Godwin, H.A. Lessons from zinc-binding peptides. Annu. Rev. Biophys. Biomol. Struct. 1997, 26, 357–371. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.R.; Campbell, R.E.; Gross, L.A.; Martin, B.R.; Walkup, G.K.; Yao, Y.; Llopis, J.; Tsien, R.Y. New biarsenical ligands and tetracysteine motifs for protein labeling in vitro and in vivo: Synthesis and biological applications. J. Am. Chem. Soc. 2002, 124, 6063–6076. [Google Scholar] [CrossRef]
- Chalker, J.M.; Bernardes, G.J.L.; Lin, Y.A.; Davis, B.G. Chemical Modification of Proteins at Cysteine: Opportunities in Chemistry and Biology. Chem. -Asian J. 2009, 4, 630–640. [Google Scholar] [CrossRef] [PubMed]
- Dawson, P.E.; Muir, T.W.; Clarklewis, I.; Kent, S.B.H. Synthesis of Proteins by Native Chemical Ligation. Science 1994, 266, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Steiner, A.M.; Bulaj, G. Optimization of oxidative folding methods for cysteine-rich peptides: A study of conotoxins containing three disulfide bridges. J. Pept. Sci. 2011, 17, 1–7. [Google Scholar] [CrossRef]
- Daly, N.L.; Love, S.; Alewood, P.F.; Craik, D.J. Chemical synthesis and folding pathways of large cyclic polypeptides: Studies of the cystine knot polypeptide kalata B1. Biochemistry 1999, 38, 10606–10614. [Google Scholar] [CrossRef]
- Kubo, S.; Chino, N.; Kimura, T.; Sakakibara, S. Oxidative folding of omega-conotoxin MVIIC: Effects of temperature and salt. Biopolymers 1996, 38, 733–744. [Google Scholar] [CrossRef]
- Zhang, J.W.; Diamond, S.; Arvedson, T.; Sasu, B.J.; Miranda, L.P. Oxidative Folding of Hepcidin at Acidic pH. Biopolymers 2010, 94, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Reinwarth, M.; Glotzbach, B.; Tomaszowski, M.; Fabritz, S.; Avrutina, O.; Kolmar, H. Oxidative folding of peptides with cystine-knot architectures: Kinetic studies and optimization of folding conditions. Chembiochem 2013, 14, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Laps, S.; Atamleh, F.; Kamnesky, G.; Sun, H.; Brik, A. General synthetic strategy for regioselective ultrafast formation of disulfide bonds in peptides and proteins. Nat. Commun. 2021, 12, 870. [Google Scholar] [CrossRef] [PubMed]
- Shen, F.; Zhang, Z.P.; Li, J.B.; Lin, Y.; Liu, L. Hydrazine-sensitive thiol protecting group for peptide and protein chemistry. Org. Lett. 2011, 13, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Tombling, B.J.; Wang, C.K.; Craik, D.J. EGF-like and Other Disulfide-rich Microdomains as Therapeutic Scaffolds. Angew. Chem. Int. Ed. Engl. 2020, 59, 11218–11232. [Google Scholar] [CrossRef] [PubMed]
- Kotzur, N.; Briand, B.; Beyermann, M.; Hagen, V. Wavelength-selective photoactivatable protecting groups for thiols. J. Am. Chem. Soc. 2009, 131, 16927–16931. [Google Scholar] [CrossRef]
- Spears, R.J.; McMahon, C.; Chudasama, V. Cysteine protecting groups: Applications in peptide and protein science. Chem. Soc. Rev. 2021, 50, 11098–11155. [Google Scholar] [CrossRef]
- Klüver, E.; Schulz-Maronde, S.; Scheid, S.; Meyer, B.; Forssmann, W.G.; Adermann, K. Structure-activity relation of human beta-defensin 3: Influence of disulfide bonds and cysteine substitution on antimicrobial activity and cytotoxicity. Biochemistry 2005, 44, 9804–9816. [Google Scholar] [CrossRef]
- Schibli, D.J.; Hunter, H.N.; Aseyev, V.; Starner, T.D.; Wiencek, J.M.; McCray, P.B.; Tack, B.F.; Vogel, H.J. The solution structures of the human beta-defensins lead to a better understanding of the potent bactericidal activity of HBD3 against Staphylococcus aureus. J. Biol. Chem. 2002, 277, 8279–8289. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L. Interaction of Human β Defensin Type 3 (hBD-3) with Different PIP2-Containing Membranes, a Molecular Dynamics Simulation Study. J. Chem. Inf. Model. 2021, 61, 4670–4686. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Hoover, D.M.; Yang, D.; Boulegue, C.; Santamaria, F.; Oppenheim, J.J.; Lubkowski, J.; Lu, W. Engineering disulfide bridges to dissect antimicrobial and chemotactic activities of human beta-defensin 3. Proc. Natl. Acad. Sci. USA 2003, 100, 8880–8885. [Google Scholar] [CrossRef] [Green Version]
- Conchillo-Sole, O.; de Groot, N.S.; Aviles, F.X.; Vendrell, J.; Daura, X.; Ventura, S. AGGRESCAN: A server for the prediction and evaluation of "hot spots" of aggregation in polypeptides. BMC Bioinform. 2007, 8, 65. [Google Scholar] [CrossRef] [Green Version]
- Walewska, A.; Zhang, M.M.; Skalicky, J.J.; Yoshikami, D.; Olivera, B.M.; Bulaj, G. Integrated Oxidative Folding of Cysteine/Selenocysteine Containing Peptides: Improving Chemical Synthesis of Conotoxins. Angew. Chem. -Int. Ed. 2009, 48, 2221–2224. [Google Scholar] [CrossRef] [PubMed]
- Pegoraro, S.; Fiori, S.; Rudolph-Bohner, S.; Watanabe, T.X.; Moroder, L. Isomorphous replacement of cystine with selenocystine in endothelin: Oxidative refolding, biological and conformational properties of [Sec3,Sec11,Nle7]-endothelin-1. J. Mol. Biol. 1998, 284, 779–792. [Google Scholar] [CrossRef] [PubMed]
- Pegoraro, S.; Fiori, S.; Cramer, J.; Rudolph-Bohner, S.; Moroder, L. The disulfide-coupled folding pathway of apamin as derived from diselenide-quenched analogs and intermediates. Protein Sci. 1999, 8, 1605–1613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gowd, K.H.; Yarotskyy, V.; Elmslie, K.S.; Skalicky, J.J.; Olivera, B.M.; Bulaj, G. Site-specific effects of diselenide bridges on the oxidative folding of a cystine knot peptide, omega-selenoconotoxin GVIA. Biochemistry 2010, 49, 2741–2752. [Google Scholar] [CrossRef] [Green Version]
- Muttenthaler, M.; Nevin, S.T.; Grishin, A.A.; Ngo, S.T.; Choy, P.T.; Daly, N.L.; Hu, S.H.; Armishaw, C.J.; Wang, C.I.; Lewis, R.J.; et al. Solving the alpha-conotoxin folding problem: Efficient selenium-directed on-resin generation of more potent and stable nicotinic acetylcholine receptor antagonists. J. Am. Chem. Soc. 2010, 132, 3514–3522. [Google Scholar] [CrossRef]
- Armishaw, C.J.; Daly, N.L.; Nevin, S.T.; Adams, D.J.; Craik, D.J.; Alewood, P.F. Alpha-selenoconotoxins, a new class of potent alpha7 neuronal nicotinic receptor antagonists. J. Biol. Chem. 2006, 281, 14136–14143. [Google Scholar] [CrossRef] [Green Version]
- Feher, K.; Timari, I.; Rakosi, K.; Szolomajer, J.; Illyes, T.Z.; Bartok, A.; Varga, Z.; Panyi, G.; Toth, G.K.; Kover, K.E. Probing pattern and dynamics of disulfide bridges using synthesis and NMR of an ion channel blocker peptide toxin with multiple diselenide bonds. Chem. Sci. 2016, 7, 2666–2673. [Google Scholar] [CrossRef] [Green Version]
- Postma, T.M.; Albericio, F. N-Chlorosuccinimide, an efficient reagent for on-resin disulfide formation in solid-phase peptide synthesis. Org. Lett. 2013, 15, 616–619. [Google Scholar] [CrossRef]
- Kellenberger, C.; Hietter, H.; Luu, B. Regioselective formation of the three disulfide bonds of a 35-residue insect peptide. Pept. Res. 1995, 8, 321–327. [Google Scholar] [PubMed]
- Li, T.; Guo, F.; Wang, Q.; Fang, H.; Li, Z.; Wang, D.; Wang, H. N-terminus three residues deletion mutant of human beta-defensin 3 with remarkably enhanced salt-resistance. PLoS ONE 2015, 10, e0117913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greber, K.E.; Dawgul, M. Antimicrobial Peptides Under Clinical Trials. Curr. Top Med. Chem. 2017, 17, 620–628. [Google Scholar] [CrossRef] [PubMed]
- Mookherjee, N.; Anderson, M.A.; Haagsman, H.P.; Davidson, D.J. Antimicrobial host defence peptides: Functions and clinical potential. Nat. Rev. Drug. Discov. 2020, 19, 311–332. [Google Scholar] [CrossRef]
- Dijksteel, G.S.; Ulrich, M.M.W.; Middelkoop, E.; Boekema, B.K.H.L. Review: Lessons Learned From Clinical Trials Using Antimicrobial Peptides (AMPs). Front. Microbiol. 2021, 12, 616979. [Google Scholar] [CrossRef]
- Ghosh, S.K.; McCormick, T.S.; Weinberg, A. Human Beta Defensins and Cancer: Contradictions and Common Ground. Front. Oncol. 2019, 9, 341. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Zhang, B.; Liao, C.; Zhang, W.; Wang, W.; Chang, Y.; Shao, Y. Human beta-defensin 3 contributes to the carcinogenesis of cervical cancer via activation of NF-κB signaling. Oncotarget 2016, 7, 75902–75913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semlali, A.; Al Amri, A.; Azzi, A.; Al Shahrani, O.; Arafah, M.; Kohailan, M.; Aljebreen, A.M.; Alharbi, O.; Almadi, M.A.; Azzam, N.A.; et al. Expression and new exon mutations of the human Beta defensins and their association on colon cancer development. PLoS ONE 2015, 10, e0126868. [Google Scholar] [CrossRef]
- Feng, Z.; Dubyak, G.R.; Jia, X.; Lubkowski, J.T.; Weinberg, A. Human β-defensin-3 structure motifs that are important in CXCR4 antagonism. FEBS J. 2013, 280, 3365–3375. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, M.; Umehara, Y.; Yue, H.; Trujillo-Paez, J.V.; Peng, G.; Nguyen, H.L.T.; Ikutama, R.; Okumura, K.; Ogawa, H.; Ikeda, S.; et al. The Antimicrobial Peptide Human β-Defensin-3 Accelerates Wound Healing by Promoting Angiogenesis, Cell Migration, and Proliferation Through the FGFR/JAK2/STAT3 Signaling Pathway. Front. Immunol. 2021, 12, 712781. [Google Scholar] [CrossRef]
- Harris, K.M.; Flemer, S.; Hondal, R.J. Studies on deprotection of cysteine and selenocysteine side-chain protecting groups. J. Pept. Sci. 2007, 13, 81–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llenado, R.A.; Weeks, C.S.; Cocco, M.J.; Ouellette, A.J. Electropositive charge in alpha-defensin bactericidal activity: Functional effects of Lys-for-Arg substitutions vary with the peptide primary structure. Infect. Immun. 2009, 77, 5035–5043. [Google Scholar] [CrossRef]


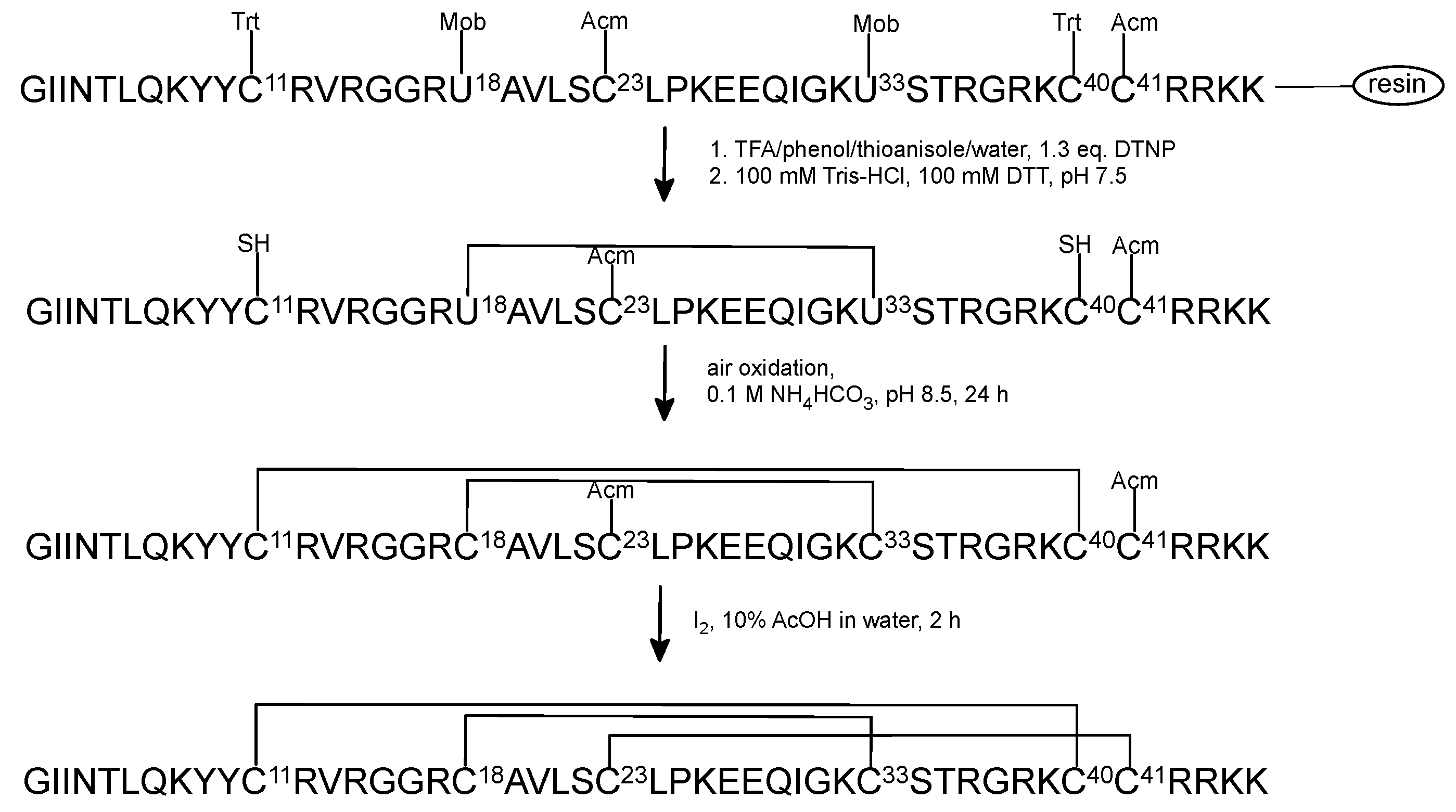


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Yield [%] | Wang Resin | TentaGel S PHB Resin | HMPB-ChemMatrix Resin | ChemMatrix Resin and Pseudoproline Blocks | [Sec18,33]HBD-3 | ** Mmt/Trt/Acm |
|---|---|---|---|---|---|---|
| purified linear peptide * | 10.1% | 22.2% | 25.5% | 38.4% | 10.6% | 5.4% |
| native peptide | 0.82% | 0.73% | 1.3% | 3.6% | 13.5% | 0.44% |
| MBC [µg/mL] | ||
|---|---|---|
| Escherichia coli PCM 2057 | Staphylococcus aureus PCM 2054 | |
| HBD-3 #1 | 12.5 | 12.5 |
| HBD-3 #2 | 25 | 25 |
| [Sec18,33]HBD-3 | 25 | 12.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walewska, A.; Kosikowska-Adamus, P.; Tomczykowska, M.; Jaroszewski, B.; Prahl, A.; Bulaj, G. Improving Fmoc Solid Phase Synthesis of Human Beta Defensin 3. Int. J. Mol. Sci. 2022, 23, 12562. https://doi.org/10.3390/ijms232012562
Walewska A, Kosikowska-Adamus P, Tomczykowska M, Jaroszewski B, Prahl A, Bulaj G. Improving Fmoc Solid Phase Synthesis of Human Beta Defensin 3. International Journal of Molecular Sciences. 2022; 23(20):12562. https://doi.org/10.3390/ijms232012562
Chicago/Turabian StyleWalewska, Aleksandra, Paulina Kosikowska-Adamus, Marta Tomczykowska, Bartosz Jaroszewski, Adam Prahl, and Grzegorz Bulaj. 2022. "Improving Fmoc Solid Phase Synthesis of Human Beta Defensin 3" International Journal of Molecular Sciences 23, no. 20: 12562. https://doi.org/10.3390/ijms232012562
APA StyleWalewska, A., Kosikowska-Adamus, P., Tomczykowska, M., Jaroszewski, B., Prahl, A., & Bulaj, G. (2022). Improving Fmoc Solid Phase Synthesis of Human Beta Defensin 3. International Journal of Molecular Sciences, 23(20), 12562. https://doi.org/10.3390/ijms232012562