

Novel Hydroxypyridine Compound Protects Brain Cells against Ischemic Damage In Vitro and In Vivo

 , , and
, , and
Abstract
:1. Introduction
2. Results
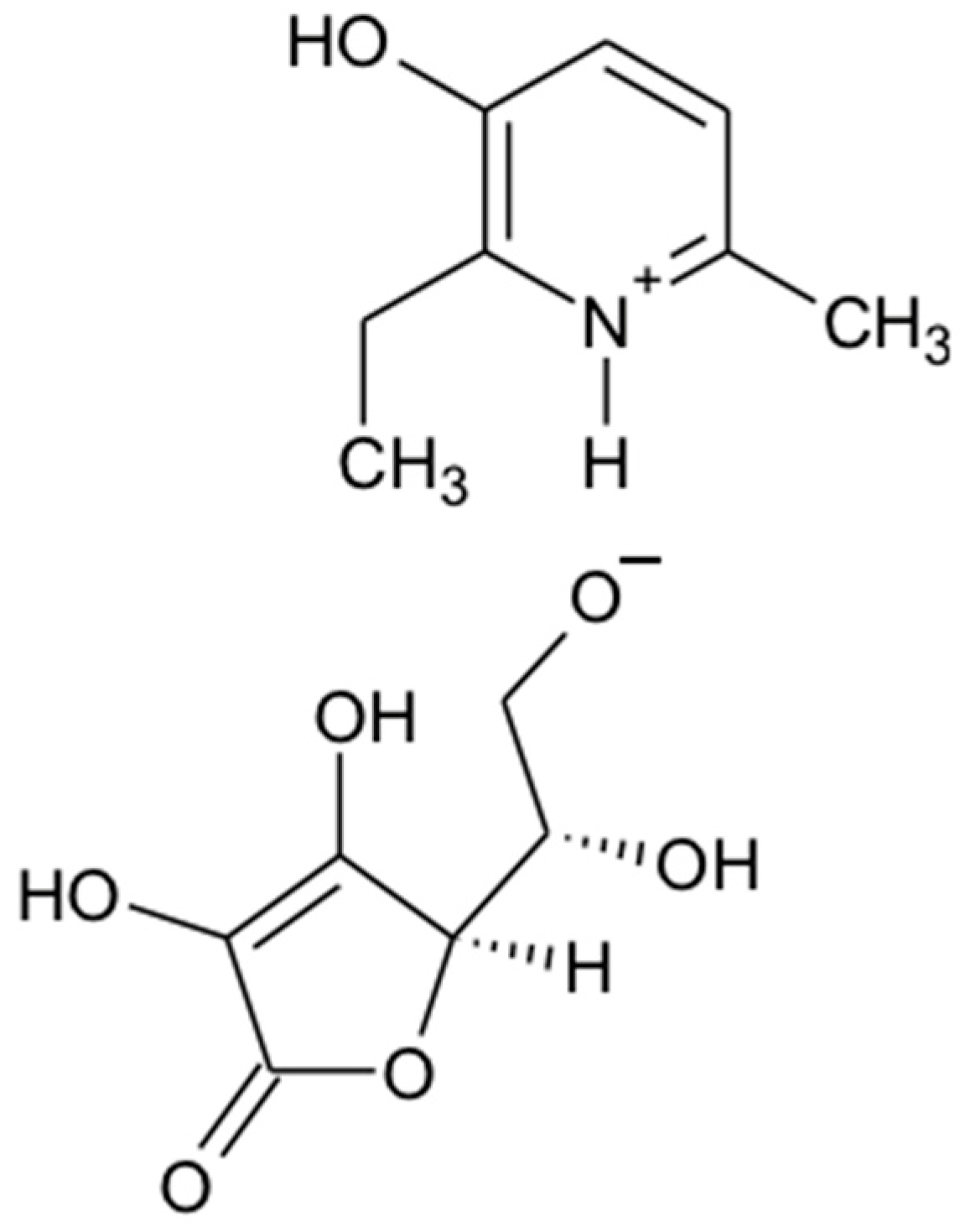
2.1. Synthesis of 3-EA
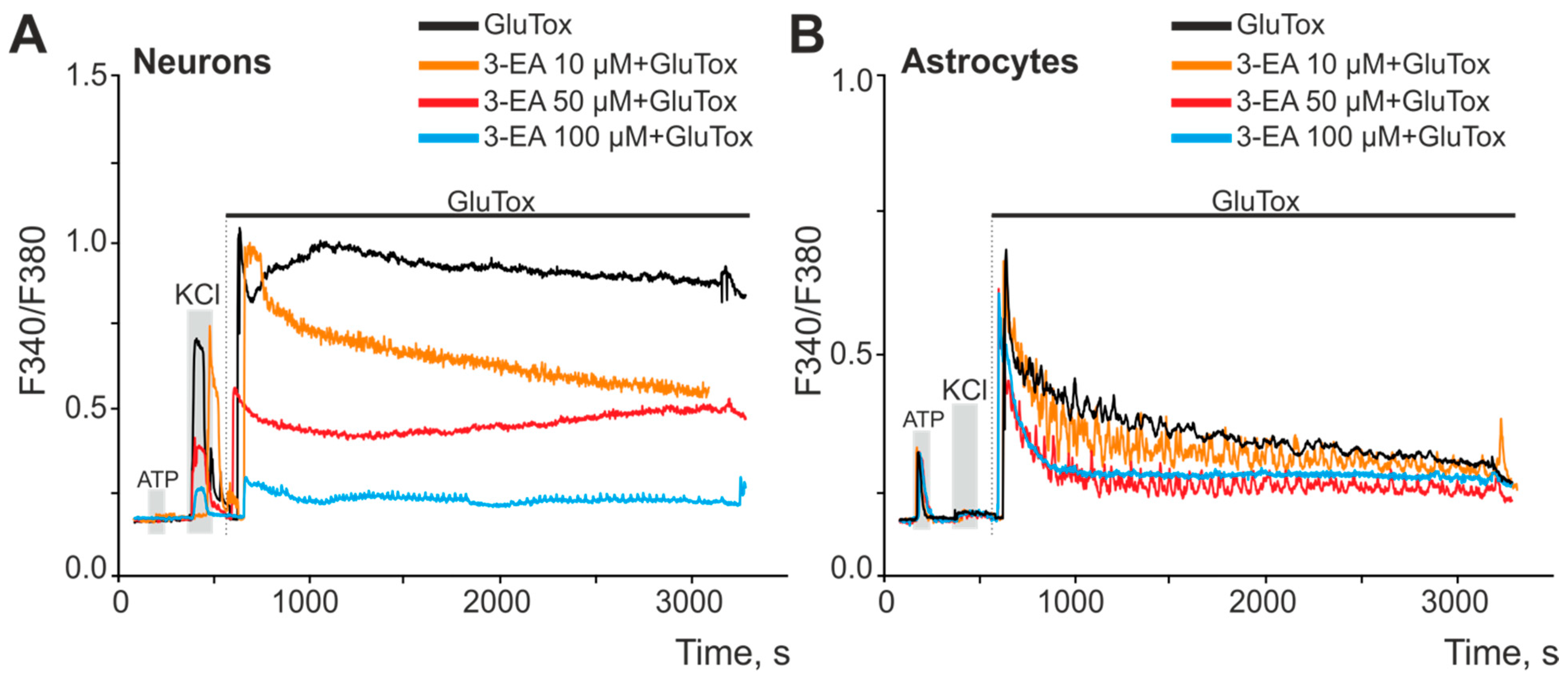
2.2. Ca2+ Dynamics in Murine Brain Cortex Cells Incubated with Different Concentrations of 3-EA
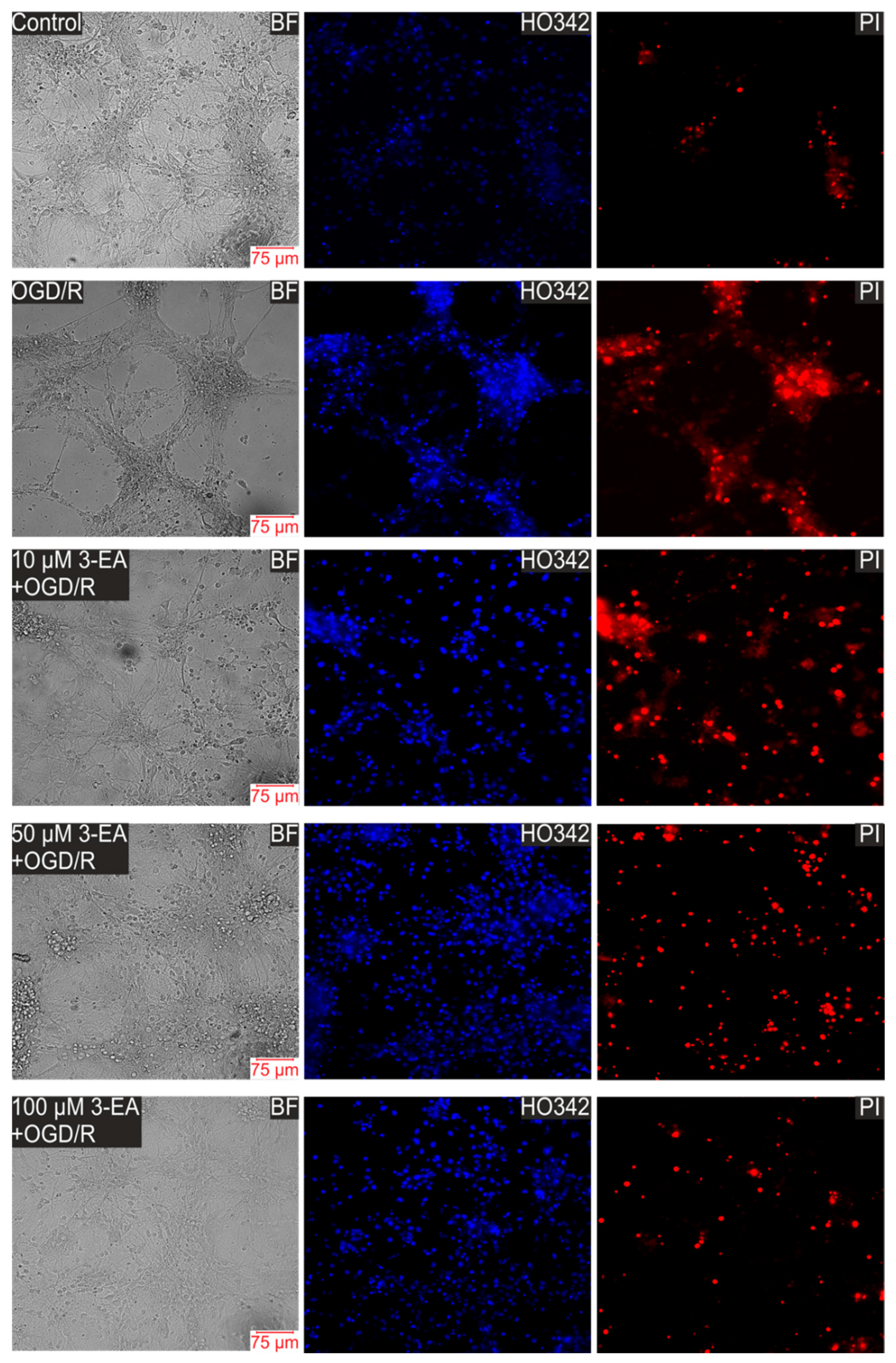
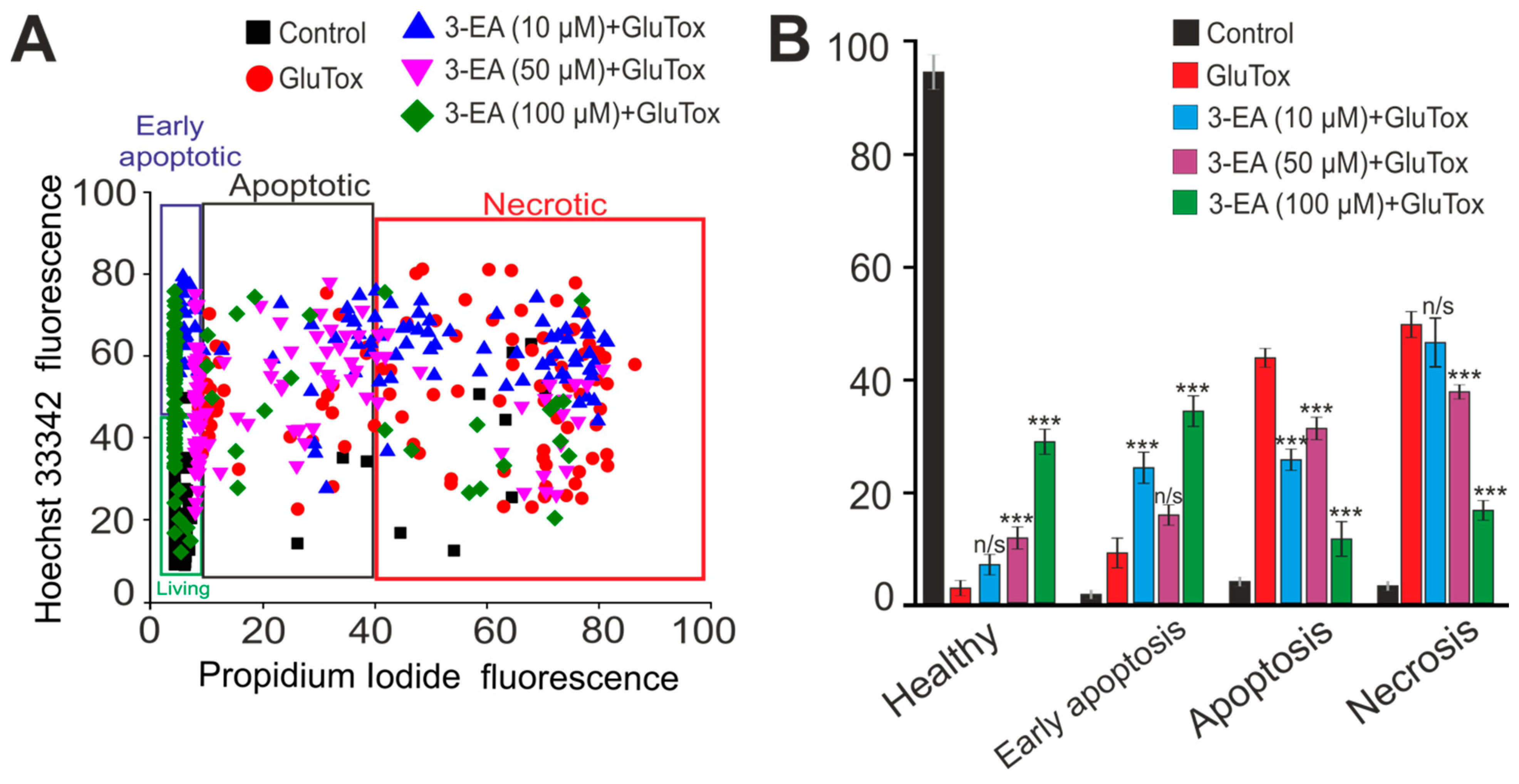
2.3. An Impact of 3-EA on Survival of Cortex Cells under GluTox Condition
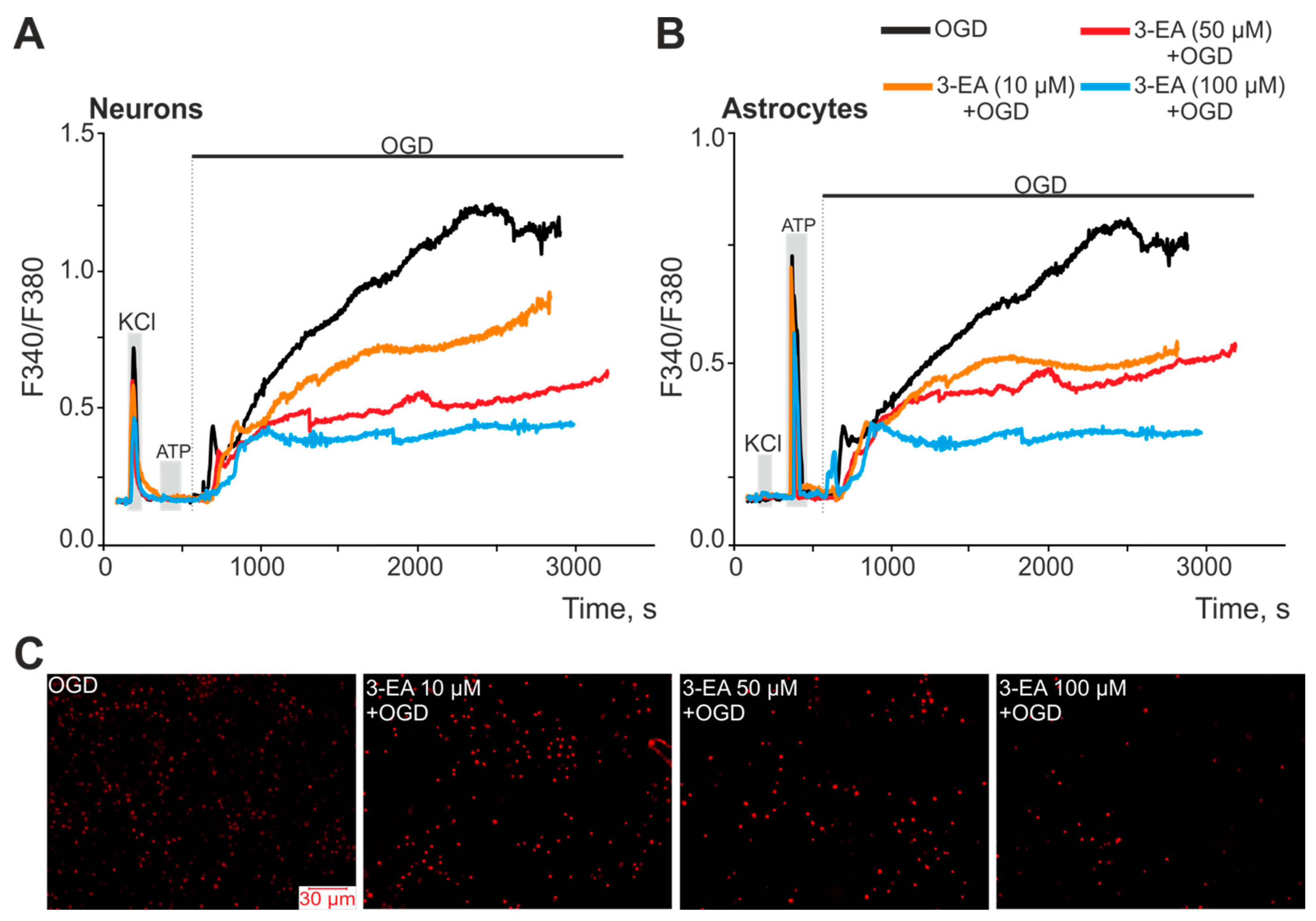
2.4. An Influence of 3-EA on Calcium Dynamics and Survival of Cells under Oxygen-Glucose Deprivation (OGD) Condition
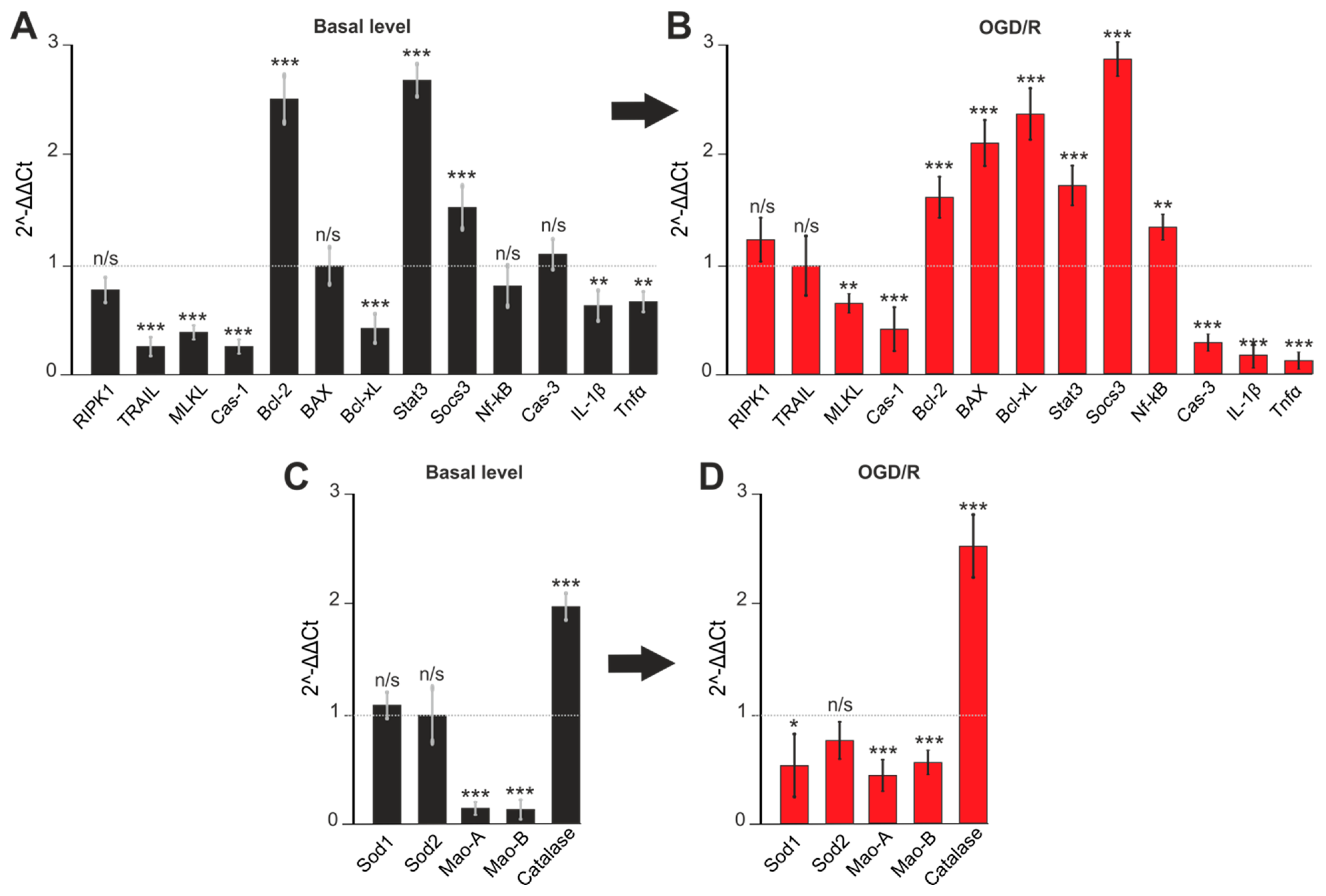
2.5. Gene Expression in Cortex Cells Treated with 100 μM 3-EA under OGD/R Condition
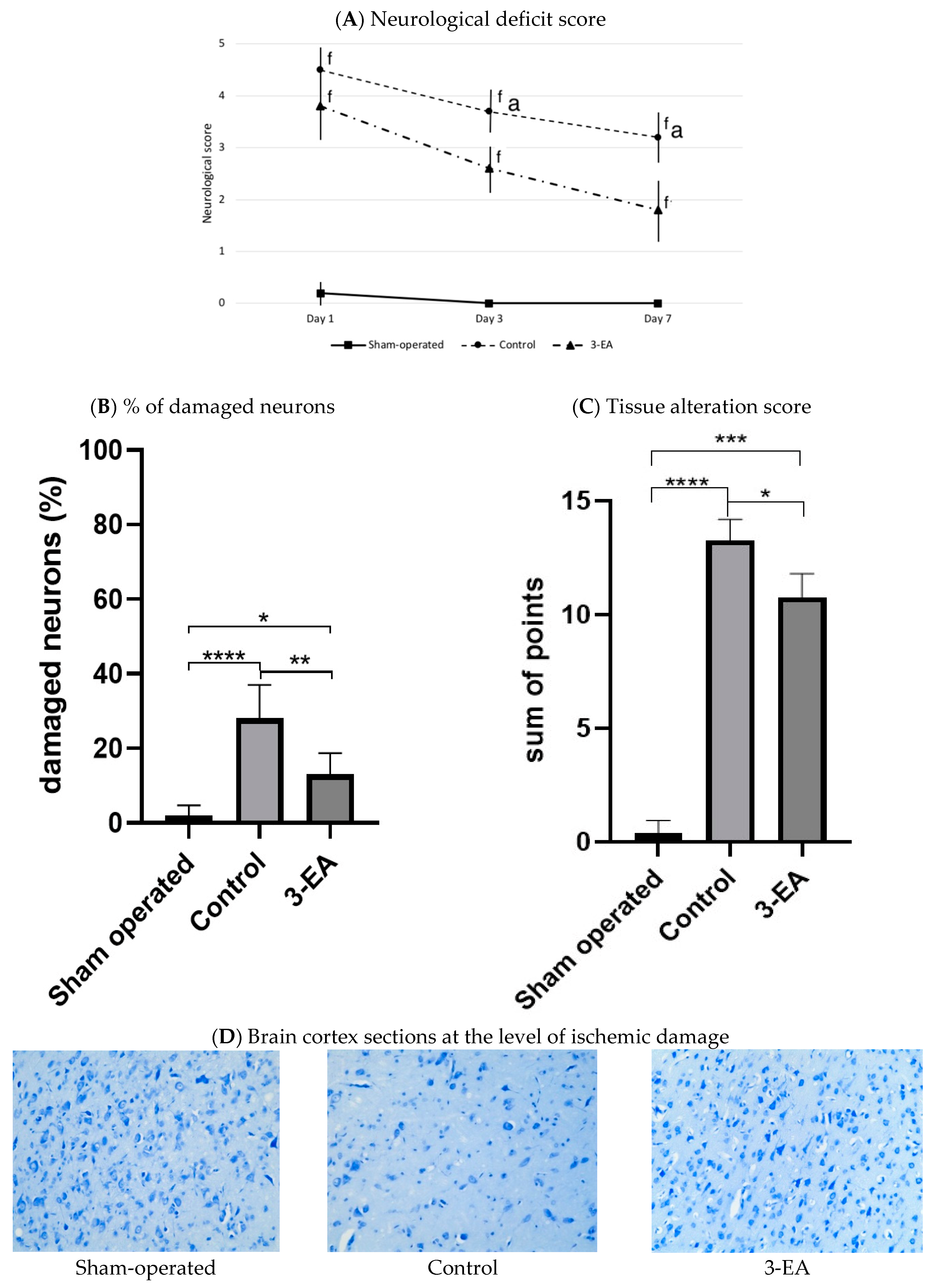
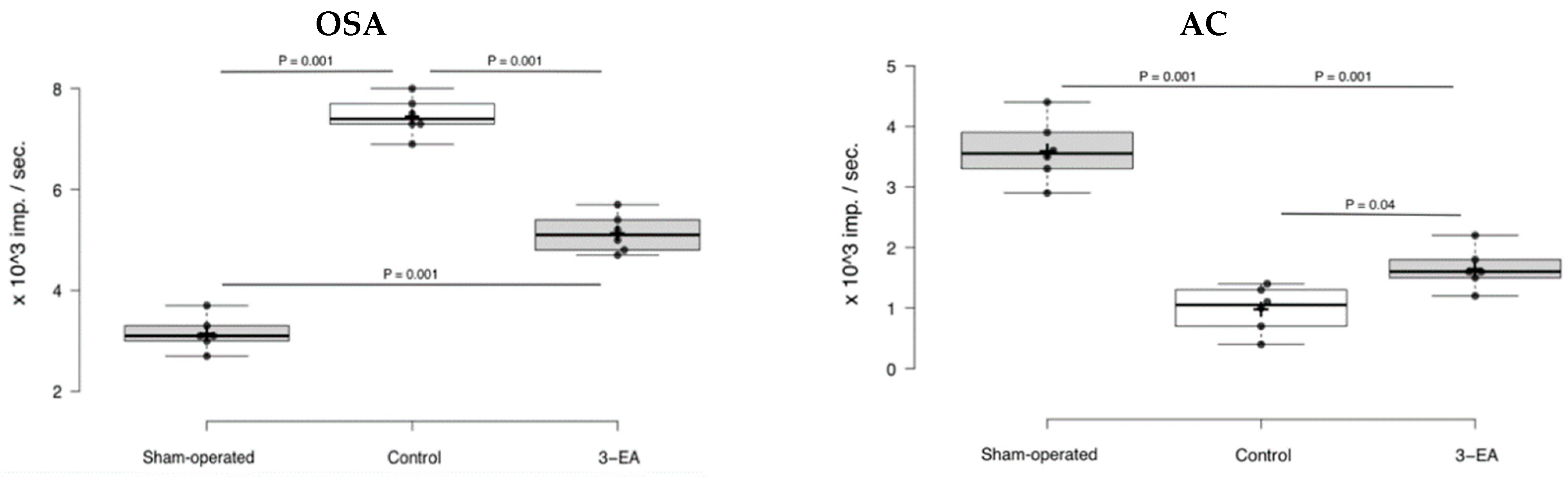
2.6. An Efficacy of 3-EA on Animal Model of Acute Brain Ischemia in Rats
3. Discussion
4. Materials and Methods
4.1. 3-EA Synthesis
4.2. Cell Culture Preparation
4.3. Modelling of Ischemia-like Conditions
4.4. Simulation of Glutamate Excitotoxicity
4.5. Measurement of Cytosolic Calcium Concentration
4.6. Assessment of Cell Viability and Apoptosis
4.7. Extraction of RNA
4.8. Real-Time Polymerase Chain Reaction (RT-qPCR)
4.9. Laboratory Animals
4.10. Experimental Stroke Modelling
4.11. Experimental Intervention and Assessments
4.12. Histological Examination
4.13. Antioxidant Capacity Assessment
4.14. Data Handling and Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix B

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gapdh | Forward 5′-aaggagccctatgagaatgtc-3′ Reverse 5′-acatcctcttctacatattcttc-3′ |
| RIPK1 | Forward 5′-ggaggatgaggcggggacc-3′ Reverse 5′-gcatgctcttcgggaggcttcaaaac-3′ |
| TRAIL | Forward 5′-ctaaccacaacacggaacctg-3′ Reverse 5′-cagcagatggttgatggaggc-3′ |
| MLKL | Forward 5′-caaagagcactaaagcagagag-3′ Reverse 5′-ggcaatcctgacccactgg-3′ |
| Cas-1 | Forward 5′-ttattcaggcatgccgtggag-3′ Reverse 5′-tcctccaagtcacaagaccag-3′ |
| Bcl-2 | Forward 5′-ctacgagtgggatgctggagatg-3′ Reverse 5′-tcaggctggaaggagaagatgc-3′ |
| Bcl-xL | Forward 5′-tggccacagcagcagtttg-3′ Reverse 5′-tctccggtaccgcagttcaa-3′ |
| Bax | Forward 5′-taaagtgcccgagctgatcagaac-3′ Reverse 5′-cttcccagccaccctggtctt-3′ |
| Caspase3 | Forward 5′-tcagaggcgactactgccggag-3′ Reverse 5′-cgtgagcatggacacaatacacgggt-3′ |
| Stat3 | Forward 5′-ttctgggcacgaacacaaaagt-3′ Reverse 5′-gcctccattcccacatctctg-3′ |
| Socs3 | Forward 5′-aagaacctacgcatccagtgtga-3′ Reverse 5′-atgtagtggtgcaccagcttgag-3′ |
| NF-kB | Forward 5′-aagtgcaaaggaaacgccagaa-3′ Reverse 5′-actaccgaacatgcctccacca-3′ |
| IL1b | Forward 5′-aatctcgcagcagcacatcaaca-3′ Reverse 5′-tccacgggaaagacacaggtagc-3′ |
| TNFα | Forward 5′-acggcatggatctcaaagacaac-3′ Reverse 5′-tcctggtatgagatagcaaatcgg-3′ |
| Catalase | Forward 5′-gctgacacagttcgtgaccctcg-3′ Reverse 5′-acaggcaagtttttgatgccctggt-3′ |
| Mao-A | Forward 5′-ggcggcatctcaggattggct-3′ Reverse 5′-tatgccaaggggttccacacaggt-3′ |
| Mao-B | Forward 5′-gcctcagtgtggtggttctggaag-3′ Reverse 5′-cactgggaatctcttggcccatctcatc-3′ |
| Sod1 | Forward 5′-tcgagcagaaggcaagcggtg-3′ Reverse 5′-cggccaatgatggaatgctctcctgag-3′ |
| Sod2 | Forward 5′-ctcccggcacaagcacagc-3′ Reverse 5′-tcctttgggttctccaccaccctt-3′ |
| Histological Sign | Score | |||
|---|---|---|---|---|
| 0 | 1 | 2 | 3 | |
| Pericellular and perivascular edema | Absent | <25% of area | 25–50% of area | >50% of area |
| Hyperchromic shrivelled neurons | <10 | 10–20 | >20 | |
| Irregular-shaped neurons | <3 | 3–10 | >10 | |
| Hypochromic neurons | <3 | 3–10 | >10 | |
| Satellitosis | 10–14 | 15–25 | >25 | |
| Neuronophagy | 1–2 | 3–4 | >4 | |
| Pyknotic neurons | 1–3 | 4–15 | >15 | |
References
- GBD 2019 Stroke Collaborators. Global, regional, and national burden of stroke and its risk-factors, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet Neurol. 2021, 20, 795–820. [Google Scholar] [CrossRef]
- Katan, M.; Luft, A. Global Burden of Stroke. Semin. Neurol. 2018, 38, 208–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimberly, W.T.; Dutra, B.G.; Boers, A.M.M.; Alves, H.C.B.R.; Berkhemer, O.A.; van den Berg, L.; Sheth, K.N.; Roos, Y.B.W.E.M.; van der Lugt, A.; Beenen, L.F.M.; et al. Association of Reperfusion with Brain Edema in Patients with Acute Ischemic Stroke: A Secondary Analysis of the MR CLEAN Trial. JAMA Neurol. 2018, 75, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Orellana-Urzúa, S.; Rojas, I.; Líbano, L.; Rodrigo, R. Pathophysiology of Ischemic Stroke: Role of Oxidative Stress. Curr. Pharm. Des. 2020, 26, 4246–4260. [Google Scholar] [CrossRef]
- Ramezani, M.; Simani, L.; Abedi, S.; Pakdaman, H. Is Selenium Supplementation Beneficial in Acute Ischemic Stroke? Neurologist 2021, 27, 51–55. [Google Scholar] [CrossRef]
- Reiter, R.J.; Mayo, J.C.; Tan, D.X.; Sainz, R.M.; Alatorre-Jimenez, M.; Qin, L. Melatonin as an antioxidant: Under promises but over delivers. J. Pineal. Res. 2016, 61, 253–278. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Cao, Y.; Lu, X.; Wang, J.; Saitgareeva, A.; Kong, X.; Song, C.; Li, J.; Tian, K.; Zhang, S.; et al. Xanthohumol protects neuron from cerebral ischemia injury in experimental stroke. Mol. Biol. Rep. 2020, 47, 2417–2425. [Google Scholar] [CrossRef]
- Wang, S.; Ma, F.; Huang, L.; Zhang, Y.; Peng, Y.; Xing, C.; Feng, Y.; Wang, X.; Peng, Y. Dl-3-n-Butylphthalide (NBP): A Promising Therapeutic Agent for Ischemic Stroke. CNS Neurol. Disord. Drug Targets 2018, 17, 338–347. [Google Scholar] [CrossRef]
- Rumaks, J.; Pupure, J.; Svirskis, S.; Isajevs, S.; Duburs, G.; Kalvinsh, I.; Klusa, V. Search for stroke-protecting agents in endothelin-1-induced ischemic stroke model in rats. Medicina 2012, 48, 525–531. [Google Scholar] [CrossRef]
- Kurganov, N.A.; Blinova, E.V.; Semeleva, E.V.; Gromova, I.A.; Blinov, D.S.; Novikov, A.V.; Mashkova, J.N.; Vasilkina, O.V. 2-aminoaethanesulfonic acid compounds possess protective property in reperfusion-induced heart jnjury. Res. Results Pharmacol. 2018, 4, 19–26. [Google Scholar] [CrossRef]
- Peresypkina, A.; Pazhinsky, A.; Pokrovskii, M.; Beskhmelnitsyna, E.; Pobeda, A.; Korokin, M. Correction of Experimental Retinal Ischemia by l-Isomer of Ethylmethylhydroxypyridine Malate. Antioxidants 2019, 8, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blinov, D.S.; Sernov, L.N.; Balashov, V.P.; Blinova, E.V.; Pivkina, L.V.; Gogina, E.D.; Van’kova, L.V.; Vertyankin, M.V.; Boiko, G.G.; Krasilina, T.V. Antiischemic activity of new domestic antioxidant 3-hydroxypyridine etoxidol derivative. Bull. Exp. Biol. Med. 2012, 152, 583–586. [Google Scholar] [CrossRef] [PubMed]
- Yasnetsov, V.V.; Tsublova, E.G.; Yasnetsov, V.V.; Skachilova, S.Y.; Karsanova, S.K.; Ivanov, Y.V. Studying some pharmacological effects of new 3-hydroxypyridine derivative. Eksp. Klin. Farmakol. 2016, 79, 3–8. [Google Scholar] [PubMed]
- Sakina, N.L.; Golubkov, A.M.; Smirnov, L.D.; Ostrovsky, M.A.; Dontsov, A.E. Studying the photoprotective activity of a new class of heteroaromatic antioxidants. Bull. Exp. Biol. Med. 2009, 147, 193–195. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Liu, H.; Xiao, Y.; Wu, L.; Shu, P. Vitamin C Intake and Ischemic Stroke. Front. Nutr. 2022, 9, 935991. [Google Scholar] [CrossRef]
- Myung, S.; Ju, W.; Cho, B.; Oh, S.; Park, S.M.; Koo, B.; Park, B.J. Efficacy of vitamin and antioxidant supplements in prevention of cardiovascular disease: Systematic review and meta-analysis of randomized controlled trials. BMJ 2013, 346, f10. [Google Scholar] [CrossRef] [Green Version]
- Turovsky, E.A.; Varlamova, E.G.; Plotnikov, E.Y. Mechanisms Underlying the Protective Effect of the Peroxiredoxin-6 Are Mediated via the Protection of Astrocytes during Ischemia/Reoxygenation. Int. J. Mol. Sci. 2021, 22, 8805. [Google Scholar] [CrossRef]
- Baev, A.Y.; Vinokurov, A.Y.; Novikova, I.N.; Dremin, V.V.; Potapova, E.V.; Abramov, A.Y. Interaction of Mitochondrial Calcium and ROS in Neurodegeneration. Cells 2022, 11, 706. [Google Scholar] [CrossRef]
- Gaidin, S.G.; Turovskaya, M.V.; Gavrish, M.S.; Babaev, A.A.; Mal’tseva, V.N.; Blinova, E.V.; Turovsky, E.A. The selective BDNF overexpression in neurons protects neuroglial networks against OGD and glutamate-induced excitotoxicity. Int. J. Neurosci. 2020, 130, 363–383. [Google Scholar] [CrossRef]
- Abramov, A.Y.; Scorziello, A.; Duchen, M.R. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J. Neurosci. 2007, 27, 1129–1138. [Google Scholar] [CrossRef] [Green Version]
- Turovsky, E.A.; Varlamova, E.G.; Gudkov, S.V.; Plotnikov, E.Y. The Protective Mechanism of Deuterated Linoleic Acid Involves the Activation of the Ca2+ Signaling System of Astrocytes in Ischemia In Vitro. Int. J. Mol. Sci. 2021, 22, 13216. [Google Scholar] [CrossRef]
- Molina, C.A.; Alvarez-Sabín, J. Recanalization and reperfusion therapies for acute ischemic stroke. Cerebrovasc. Dis. 2009, 27 (Suppl. 1), 162–167. [Google Scholar] [CrossRef] [PubMed]
- D’Orsi, B.; Mateyka, J.; Prehn, J.H.M. Control of mitochondrial physiology and cell death by the Bcl-2 family proteins Bax and Bok. Neurochem. Int. 2017, 109, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Jurcau, A.; Simion, A. Neuroinflammation in Cerebral Ischemia and Ischemia/Reperfusion Injuries: From Pathophysiology to Therapeutic Strategies. Int. J. Mol. Sci. 2021, 23, 14. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Wang, K.; Wan, W.; Cheng, Y.; Pu, X.; Ye, X. Resveratrol provides neuroprotection by regulating the JAK2/STAT3/PI3K/AKT/mTOR pathway after stroke in rats. Genes Dis. 2018, 5, 245–255. [Google Scholar] [CrossRef]
- Yoshimura, A.; Suzuki, M.; Sakaguchi, R.; Hanada, T.; Yasukawa, H. SOCS, Inflammation, and Autoimmunity. Front. Immunol. 2012, 3, 20. [Google Scholar] [CrossRef] [Green Version]
- Yeung, A.W.K.; Georgieva, M.G.; Atanasov, A.G.; Tzvetkov, N.T. Monoamine Oxidases (MAOs) as Privileged Molecular Targets in Neuroscience: Research Literature Analysis. Front. Mol. Neurosci. 2019, 12, 143. [Google Scholar] [CrossRef] [Green Version]
- Matveychuk, D.; MacKenzie, E.M.; Kumpula, D.; Song, M.-S.; Holt, A.; Kar, S.; Todd, K.G.; Wood, P.L.; Baker, G.B. Overview of the Neuroprotective Effects of the MAO-Inhibiting Antidepressant Phenelzine. Cell. Mol. Neurobiol. 2022, 42, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Rathitharan, G.; Meyer, J.H.; Furukawa, Y.; Ang, L.C.; Boileau, I.; Guttman, M.; Hornykiewicz, O.; Kish, S.J. Brain monoamine oxidase B and A in human parkinsonian dopamine deficiency disorders. Brain 2017, 140, 2460–2474. [Google Scholar] [CrossRef] [Green Version]
- Novikova, I.N.; Manole, A.; Zherebtsov, E.A.; Stavtsev, D.D.; Vukolova, M.N.; Dunaev, A.V.; Angelova, P.R.; Abramov, A.Y. Adrenaline induces calcium signal in astrocytes and vasoconstriction via activation of monoamine oxidase. Free Radic. Biol. Med. 2020, 159, 15–22. [Google Scholar] [CrossRef]
- Youdim, M.B.; Bakhle, Y. Monoamine oxidase: Isoforms and inhibitors in Parkinson’s disease and depressive illness. Br. J. Pharmacol. 2006, 147, S287–S296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armogida, M.; Spalloni, A.; Amantea, D.; Nutini, M.; Petrelli, F.; Longone, P.; Bagetta, G.; Nisticò, R.; Mercuri, N.B. The protective role of catalase against cerebral ischemia in vitro and in vivo. Int. J. Immunopathol. Pharmacol. 2011, 24, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Zhao, H.; Yenari, M.A.; Sapolsky, R.M.; Steinberg, G.K. Catalase over-expression protects striatal neurons from transient focal cerebral ischemia. Neuroreport 2004, 15, 413–416. [Google Scholar] [CrossRef] [PubMed]
- Varlamova, E.G.; Turovsky, E.A.; Babenko, V.A.; Plotnikov, E.Y. The Mechanisms Underlying the Protective Action of Selenium Nanoparticles against Ischemia/Reoxygenation Are Mediated by the Activation of the Ca2+ Signaling System of Astrocytes and Reactive Astrogliosis. Int. J. Mol. Sci. 2021, 22, 12825. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Chiang, T.; Messing, R.O.; Chou, W.H. Mouse model of middle cerebral artery occlusion. J. Vis. Exp. 2011, 48, e2761. [Google Scholar] [CrossRef] [Green Version]
- Bederson, J.B.; Pitts, L.H.; Tsuji, M.; Nishimura, M.C.; Davis, R.L.; Bartkowski, H. Rat middle cerebral artery occlusion: Evaluation of the model and development of a neurologic examination. Stroke 1986, 17, 472–476. [Google Scholar] [CrossRef] [Green Version]
- Dzhatdoeva, A.A.; Polimova, A.M.; Proskurina, E.V.; Vladimirov, Y.A. Tissue chemiluminescence as a method of evaluation of superoxide radical producing ability of mitochondria. Bull. Russ. State Med. Univ. 2016, 1, 49–55. [Google Scholar] [CrossRef]
- Blinova, E.; Pakhomov, D.; Shimanovsky, D.; Kilmyashkina, M.; Mazov, Y.; Demura, T.; Drozdov, V.; Blinov, D.; Deryabina, O.; Samishina, E.; et al. Cerium-Containing N-Acetyl-6-Aminohexanoic Acid Formulation Accelerates Wound Reparation in Diabetic Animals. Biomolecules 2021, 11, 834. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blinova, E.; Turovsky, E.; Eliseikina, E.; Igrunkova, A.; Semeleva, E.; Golodnev, G.; Termulaeva, R.; Vasilkina, O.; Skachilova, S.; Mazov, Y.; et al. Novel Hydroxypyridine Compound Protects Brain Cells against Ischemic Damage In Vitro and In Vivo. Int. J. Mol. Sci. 2022, 23, 12953. https://doi.org/10.3390/ijms232112953
Blinova E, Turovsky E, Eliseikina E, Igrunkova A, Semeleva E, Golodnev G, Termulaeva R, Vasilkina O, Skachilova S, Mazov Y, et al. Novel Hydroxypyridine Compound Protects Brain Cells against Ischemic Damage In Vitro and In Vivo. International Journal of Molecular Sciences. 2022; 23(21):12953. https://doi.org/10.3390/ijms232112953
Chicago/Turabian StyleBlinova, Ekaterina, Egor Turovsky, Elena Eliseikina, Alexandra Igrunkova, Elena Semeleva, Grigorii Golodnev, Rita Termulaeva, Olga Vasilkina, Sofia Skachilova, Yan Mazov, and et al. 2022. "Novel Hydroxypyridine Compound Protects Brain Cells against Ischemic Damage In Vitro and In Vivo" International Journal of Molecular Sciences 23, no. 21: 12953. https://doi.org/10.3390/ijms232112953
APA StyleBlinova, E., Turovsky, E., Eliseikina, E., Igrunkova, A., Semeleva, E., Golodnev, G., Termulaeva, R., Vasilkina, O., Skachilova, S., Mazov, Y., Zhandarov, K., Simakina, E., Belanov, K., Zalogin, S., & Blinov, D. (2022). Novel Hydroxypyridine Compound Protects Brain Cells against Ischemic Damage In Vitro and In Vivo. International Journal of Molecular Sciences, 23(21), 12953. https://doi.org/10.3390/ijms232112953