

Stellate Ganglia and Cardiac Sympathetic Overactivation in Heart Failure

{kind=link}
{kind=link}
Abstract
:1. Introduction
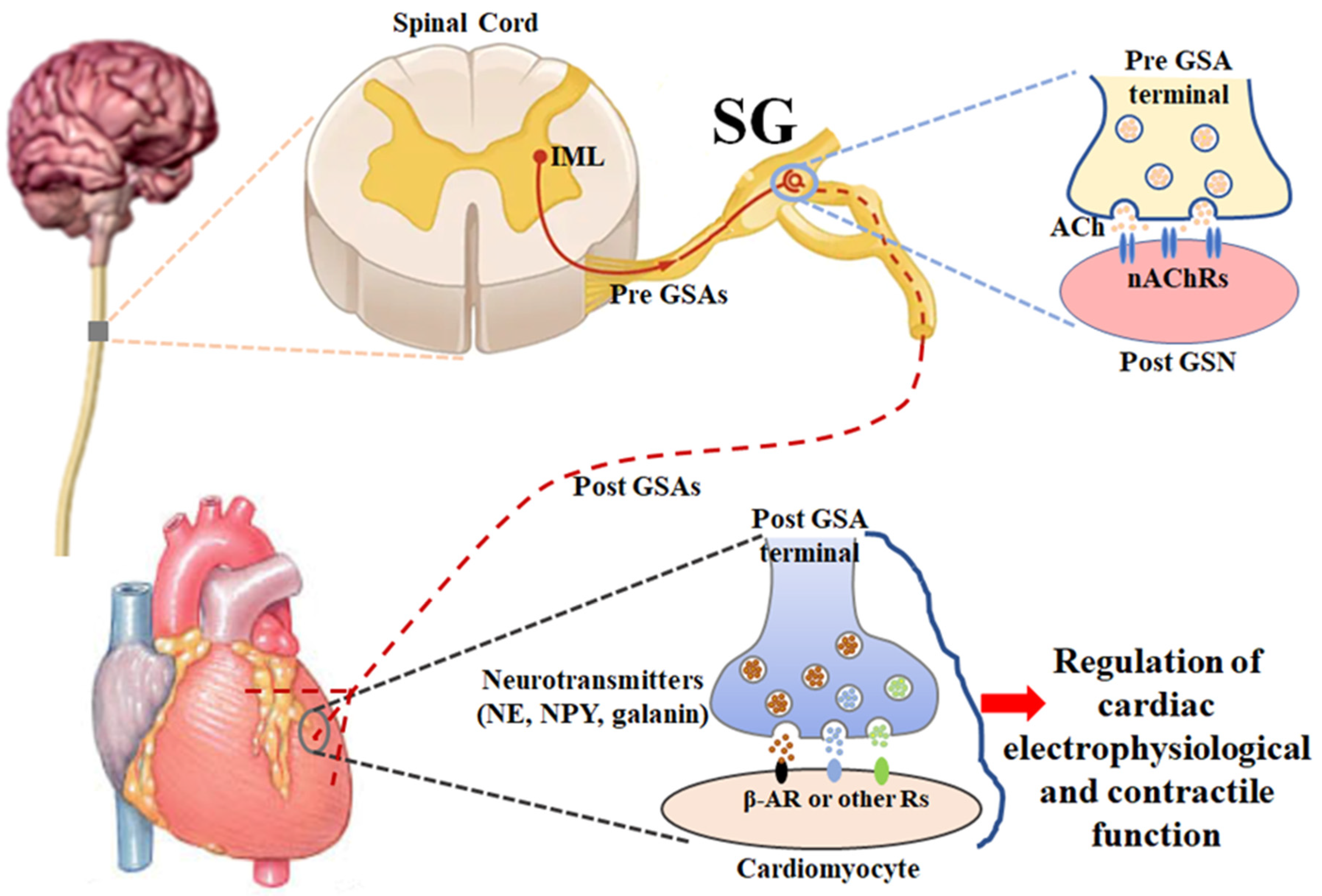
2. Anatomy and Physiology of Stellate Ganglia (Figure 1)
3. Remodeling of Cardiac Postganglionic Sympathetic Neurons and Its Role in Cardiac Sympathetic Overactivation, Malignant Arrhythmias, and Cardiac Sudden Death in HF
3.1. Structural Remodeling in Cardiac Postganglionic Sympathetic Neurons Located in Stellate Ganglia
3.2. Functional Remodeling in Cardiac Postganglionic Sympathetic Neurons Located in Stellate Ganglia
3.3. Structural Remodeling in Cardiac Postganglionic Sympathetic Nerve Terminals
3.4. Functional Remodeling in Cardiac Postganglionic Sympathetic Nerve Terminals
4. Mechanisms Underlying the Remodeling of Cardiac Postganglionic Sympathetic Neurons in HF
4.1. Mechanisms Associated with the Remodeling of Cardiac Postganglionic Sympathetic Cell Somata
4.2. Mechanisms Associated with the Remodeling in Cardiac Postganglionic Sympathetic Never Terminals
5. Conclusions
Funding
Conflicts of Interest
References
- Sweitzer, N.K. Looking ahead: Circulation: Heart failure in 2022. Circ. Heart Fail. 2022, 15, e009405. [Google Scholar] [CrossRef] [PubMed]
- Tsao, C.W.; Aday, A.W.; Almarzooq, Z.I.; Alonso, A.; Beaton, A.Z.; Bittencourt, M.S.; Boehme, A.K.; Buxton, A.E.; Carson, A.P.; Commodore-Mensah, Y.; et al. Heart disease and stroke statistics-2022 update: A report from the American Heart Association. Circulation 2022, 145, e153–e639. [Google Scholar] [CrossRef] [PubMed]
- Savarese, G.; D’Amario, D. Sex differences in heart failure. Adv. Exp. Med. Biol. 2018, 1065, 529–544. [Google Scholar] [PubMed]
- Elgendy, I.Y.; Mahtta, D.; Pepine, C.J. Medical therapy for heart failure caused by ischemic heart disease. Circ. Res. 2019, 124, 1520–1535. [Google Scholar] [CrossRef]
- He, J.; Ogden, L.G.; Bazzano, L.A.; Vupputuri, S.; Loria, C.; Whelton, P.K. Risk factors for congestive heart failure in US men and women: NHANESIepidemiologic follow-up study. Arch. Intern. Med. 2001, 161, 996–1002. [Google Scholar] [CrossRef] [Green Version]
- Velagaleti, R.S.; Vasan, R.S. Heart failure in the twenty-first century: Is it a coronary artery disease or hypertension problem? Cardiol. Clin. 2007, 25, 487–495. [Google Scholar] [CrossRef] [Green Version]
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N.; et al. Heart disease and stroke statistics-2021 update: A report from the American Heart Association. Circulation 2021, 143, e254–e743. [Google Scholar] [CrossRef]
- Emmons-Bell, S.; Johnson, C.; Roth, G. Prevalence, incidence and survival of heart failure: A systematic review. Heart 2022, 108, 1351–1360. [Google Scholar] [CrossRef]
- Savarese, G.; Becher, P.M.; Lund, L.H.; Seferovic, P.; Rosano, G.M.C.; Coats, A. Global burden of heart failure: A comprehensive and updated review of epidemiology. Cardiovasc. Res. 2022; cvac013, online ahead of print. [Google Scholar]
- Creager, M.A.; Faxon, D.P.; Cutler, S.S.; Kohlmann, O.; Ryan, T.J.; Gavras, H. Contribution of vasopressin to vasoconstriction in patients with congestive heart failure: Comparison with the renin-angiotensin system and the sympathetic nervous system. J. Am. Coll. Cardiol. 1986, 7, 758–765. [Google Scholar] [CrossRef] [Green Version]
- Floras, J.S. Sympathetic nervous system activation in human heart failure: Clinical implications of an updated model. J. Am. Coll. Cardiol. 2009, 54, 375–385. [Google Scholar] [CrossRef]
- Saul, J.P.; Arai, Y.; Berger, R.D.; Lilly, L.S.; Colucci, W.S.; Cohen, R.J. Assessment of autonomic regulation in chronic congestive heart failure by heart rate spectral analysis. Am. J. Cardiol. 1988, 61, 1292–1299. [Google Scholar] [CrossRef]
- Schwartz, P.J.; De Ferrari, G.M. Sympathetic-parasympathetic interaction in health and disease: Abnormalities and relevance in heart failure. Heart Fail. Rev. 2011, 16, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Triposkiadis, F.; Karayannis, G.; Giamouzis, G.; Skoularigis, J.; Louridas, G.; Butler, J. The sympathetic nervous system in heart failure physiology, pathophysiology, and clinical implications. J. Am. Coll. Cardiol. 2009, 54, 1747–1762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.J.; Cox, H.S.; Dart, A.M.; Esler, M.D. Sympathetic activation triggers ventricular arrhythmias in rat heart with chronic infarction and failure. Cardiovasc. Res. 1999, 43, 919–929. [Google Scholar] [CrossRef] [Green Version]
- Gilmour, R.F. Life out of balance: The sympathetic nervous system and cardiac arrhythmias. Cardiovasc. Res. 2001, 51, 625–626. [Google Scholar] [CrossRef] [Green Version]
- Kalla, M.; Herring, N.; Paterson, D.J. Cardiac sympatho-vagal balance and ventricular arrhythmia. Auton. Neurosci. 2016, 199, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Podrid, P.J.; Fuchs, T.; Candinas, R. Role of the sympathetic nervous system in the genesis of ventricular arrhythmia. Circulation 1990, 82, I103–I113. [Google Scholar]
- Schwartz, P.J. Cardiac sympathetic denervation to prevent life-threatening arrhythmias. Nat. Rev. Cardiol. 2014, 11, 346–353. [Google Scholar] [CrossRef]
- Thompson, B.S. Sudden cardiac death and heart failure. AACN Adv. Crit. Care 2009, 20, 356–365. [Google Scholar]
- Tomaselli, G.F.; Zipes, D.P. What causes sudden death in heart failure? Circ. Res. 2004, 95, 754–763. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Jung, B.C.; Tan, A.Y.; Trang, V.Q.; Gholmieh, G.; Han, S.W.; Lin, S.F.; Fishbein, M.C.; Chen, P.S.; Chen, L.S. Spontaneous stellate ganglion nerve activity and ventricular arrhythmia in a canine model of sudden death. Heart Rhythm 2008, 5, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Zipes, D.P. Heart-brain interactions in cardiac arrhythmias: Role of the autonomic nervous system. Cleve. Clin. J. Med. 2008, 75, S94–S96. [Google Scholar] [CrossRef] [PubMed]
- Carson, P.; Anand, I.; O’Connor, C.; Jaski, B.; Steinberg, J.; Lwin, A.; Lindenfeld, J.; Ghali, J.; Barnet, J.H.; Feldman, A.M.; et al. Mode of death in advanced heart failure: The Comparison of Medical, Pacing, and Defibrillation Therapies in Heart Failure (COMPANION) trial. J. Am. Coll. Cardiol. 2005, 46, 2329–2334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cygankiewicz, I.; Zareba, W.; Vazquez, R.; Vallverdu, M.; Gonzalez-Juanatey, J.R.; Valdes, M.; Almendral, J.; Cinca, J.; Caminal, P.; de Luna, A.B. Heart rate turbulence predicts all-cause mortality and sudden death in congestive heart failure patients. Heart Rhythm 2008, 5, 1095–1102. [Google Scholar] [CrossRef] [PubMed]
- Doval, H.C.; Nul, D.R.; Grancelli, H.O.; Varini, S.D.; Soifer, S.; Corrado, G.; Dubner, S.; Scapin, O.; Perrone, S.V. Nonsustained ventricular tachycardia in severe heart failure. Independent marker of increased mortality due to sudden death. GESICA-GEMA Investigators. Circulation 1996, 94, 3198–3203. [Google Scholar] [CrossRef] [PubMed]
- Engelstein, E.D.; Zipes, D.P. Sudden cardiac death. In Hurst’s The Heart; Alexander, R.W., Schlant, R.C., Fuster, V., Eds.; McGraw Hill: New York, NY, USA, 1998; Volume 9, pp. 1081–1112. [Google Scholar]
- Huikuri, H.V.; Castellanos, A.; Myerburg, R.J. Sudden death due to cardiac arrhythmias. N. Engl. J. Med. 2001, 345, 1473–1482. [Google Scholar] [CrossRef]
- Jost, A.; Rauch, B.; Hochadel, M.; Winkler, R.; Schneider, S.; Jacobs, M.; Kilkowski, C.; Kilkowski, A.; Lorenz, H.; Muth, K.; et al. Beta-blocker treatment of chronic systolic heart failure improves prognosis even in patients meeting one or more exclusion criteria of the MERIT-HF study. Eur. Heart J. 2005, 26, 2689–2697. [Google Scholar] [CrossRef]
- Maskin, C.S.; Siskind, S.J.; LeJemtel, T.H. High prevalence of nonsustained ventricular tachycardia in severe congestive heart failure. Am. Heart J. 1984, 107, 896–901. [Google Scholar] [CrossRef]
- Podrid, P.J.; Fogel, R.I.; Fuchs, T.T. Ventricular arrhythmia in congestive heart failure. Am. J. Cardiol. 1992, 69, 82G–95G. [Google Scholar] [CrossRef]
- Sami, M.H. Sudden death in congestive heart failure. J. Clin. Pharmacol. 1991, 31, 1081–1084. [Google Scholar] [CrossRef]
- Singh, B.N. Significance and control of cardiac arrhythmias in patients with congestive cardiac failure. Heart Fail. Rev. 2002, 7, 285–300. [Google Scholar] [CrossRef]
- Singh, S.N.; Carson, P.E.; Fisher, S.G. Nonsustained ventricular tachycardia in severe heart failure. Circulation 1997, 96, 3794–3795. [Google Scholar] [PubMed]
- Teerlink, J.R.; Jalaluddin, M.; Anderson, S.; Kukin, M.L.; Eichhorn, E.J.; Francis, G.; Packer, M.; Massie, B.M. Ambulatory ventricular arrhythmias in patients with heart failure do not specifically predict an increased risk of sudden death. PROMISE (Prospective Randomized Milrinone Survival Evaluation) Investigators. Circulation 2000, 101, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Al-Gobari, M.; El, K.C.; Pillon, F.; Gueyffier, F. Beta-blockers for the prevention of sudden cardiac death in heart failure patients: A meta-analysis of randomized controlled trials. BMC Cardiovasc. Disord. 2013, 13, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babick, A.; Elimban, V.; Zieroth, S.; Dhalla, N.S. Reversal of cardiac dysfunction and subcellular alterations by metoprolol in heart failure due to myocardial infarction. J. Cell. Physiol. 2013, 228, 2063–2070. [Google Scholar] [CrossRef]
- De Ferrari, G.M.; Schwartz, P.J. Left cardiac sympathetic denervation in patients with heart failure: A new indication for an old intervention? J. Cardiovasc. Transl. Res. 2014, 7, 338–346. [Google Scholar] [CrossRef]
- Fiuzat, M.; Wojdyla, D.; Kitzman, D.; Fleg, J.; Keteyian, S.J.; Kraus, W.E.; Pina, I.L.; Whellan, D.; O’Connor, C.M. Relationship of beta-blocker dose with outcomes in ambulatory heart failure patients with systolic dysfunction: Results from the HF-ACTION (Heart Failure: A Controlled Trial Investigating Outcomes of Exercise Training) trial. J. Am. Coll. Cardiol. 2012, 60, 208–215. [Google Scholar] [CrossRef] [Green Version]
- Gheorghiade, M.; Colucci, W.S.; Swedberg, K. Beta-blockers in chronic heart failure. Circulation 2003, 107, 1570–1575. [Google Scholar] [CrossRef]
- Nevzorov, R.; Porath, A.; Henkin, Y.; Kobal, S.L.; Jotkowitz, A.; Novack, V. Effect of beta blocker therapy on survival of patients with heart failure and preserved systolic function following hospitalization with acute decompensated heart failure. Eur. J. Intern. Med. 2012, 23, 374–378. [Google Scholar] [CrossRef]
- Shah, R.; Assis, F.; Alugubelli, N.; Okada, D.R.; Cardoso, R.; Shivkumar, K.; Tandri, H. Cardiac sympathetic denervation for refractory ventricular arrhythmias in patients with structural heart disease: A systematic review. Heart Rhythm 2019, 16, 1499–1505. [Google Scholar] [CrossRef]
- Vaseghi, M.; Barwad, P.; Malavassi Corrales, F.J.; Tandri, H.; Mathuria, N.; Shah, R.; Sorg, J.M.; Gima, J.; Mandal, K.; Saenz Morales, L.C.; et al. Cardiac sympathetic denervation for refractory ventricular arrhythmias. J. Am. Coll. Cardiol. 2017, 69, 3070–3080. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, A.S.; DeVore, A.D.; DeWald, T.A.; Swedberg, K.; Mentz, R.J. Achieving a maximally tolerated beta-blocker dose in heart failure patients: Is there room for improvement? J. Am. Coll. Cardiol. 2017, 69, 2542–2550. [Google Scholar] [CrossRef] [PubMed]
- Bos, J.M.; Bos, K.M.; Johnson, J.N.; Moir, C.; Ackerman, M.J. Left cardiac sympathetic denervation in long QT syndrome: Analysis of therapeutic nonresponders. Circ. Arrhythm. Electrophysiol. 2013, 6, 705–711. [Google Scholar] [CrossRef] [Green Version]
- Coleman, M.A.; Bos, J.M.; Johnson, J.N.; Owen, H.J.; Deschamps, C.; Moir, C.; Ackerman, M.J. Videoscopic left cardiac sympathetic denervation for patients with recurrent ventricular fibrillation/malignant ventricular arrhythmia syndromes besides congenital long-QT syndrome. Circ. Arrhythm. Electrophysiol. 2012, 5, 782–788. [Google Scholar] [CrossRef] [Green Version]
- Moss, A.J.; Zareba, W.; Hall, W.J.; Schwartz, P.J.; Crampton, R.S.; Benhorin, J.; Vincent, G.M.; Locati, E.H.; Priori, S.G.; Napolitano, C.; et al. Effectiveness and limitations of beta-blocker therapy in congenital long-QT syndrome. Circulation 2000, 101, 616–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napolitano, C.; Priori, S.G. Diagnosis and treatment of catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 2007, 4, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.V.; Dunning, J.R.; Fischer, C.M.; MacLean, T.E.; Bosque-Hamilton, J.W.; Fera, L.E.; Grant, J.Y.; Zelle, D.J.; Matta, L.; Gaziano, T.A.; et al. Evaluation of the usage and dosing of guideline-directed medical therapy for heart failure with reduced ejection fraction patients in clinical practice. J. Pharm. Pract. 2022, 35, 747–751. [Google Scholar] [CrossRef]
- Veenis, J.F.; Rocca, H.B.; Linssen, G.C.M.; Erol-Yilmaz, A.; Pronk, A.C.B.; Engelen, D.J.M.; van Tooren, R.M.; Koornstra-Wortel, H.J.J.; de Boer, R.A.; van der Meer, P.; et al. Impact of sex-specific target dose in chronic heart failure patients with reduced ejection fraction. Eur. J. Prev. Cardiol. 2021, 28, 957–965. [Google Scholar] [CrossRef]
- Hofferberth, S.C.; Cecchin, F.; Loberman, D.; Fynn-Thompson, F. Left thoracoscopic sympathectomy for cardiac denervation in patients with life-threatening ventricular arrhythmias. J. Thorac. Cardiovasc. Surg. 2014, 147, 404–409. [Google Scholar] [CrossRef] [Green Version]
- Schneider, H.E.; Steinmetz, M.; Krause, U.; Kriebel, T.; Ruschewski, W.; Paul, T. Left cardiac sympathetic denervation for the management of life-threatening ventricular tachyarrhythmias in young patients with catecholaminergic polymorphic ventricular tachycardia and long QT syndrome. Clin. Res. Cardiol. 2013, 102, 33–42. [Google Scholar] [CrossRef] [Green Version]
- Rathinam, S.; Nanjaiah, P.; Sivalingam, S.; Rajesh, P.B. Excision of sympathetic ganglia and the rami communicantes with histological confirmation offers better early and late outcomes in Video assisted thoracoscopic sympathectomy. J. Cardiothorac. Surg. 2008, 3, 50–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webster, G.; Monge, M.C. Left cardiac sympathetic denervation: Should we sweat the side effects? Circ. Arrhythm. Electrophysiol. 2015, 8, 1007–1009. [Google Scholar] [CrossRef] [PubMed]
- Lathro, D.A.; Spooner, P.M. On the neural connection. J. Cardiovasc. Electrophysiol. 2001, 12, 841–844. [Google Scholar] [CrossRef] [PubMed]
- Verrier, R.L.; Antzelevitch, C. Autonomic aspects of arrhythmogenesis: The enduring and the new. Curr. Opin. Cardiol. 2004, 19, 2–11. [Google Scholar] [CrossRef] [Green Version]
- Zucker, I.H.; Xiao, L.; Haack, K.K. The central renin-angiotensin system and sympathetic nerve activity in chronic heart failure. Clin. Sci. 2014, 126, 695–706. [Google Scholar] [CrossRef] [Green Version]
- Zucker, I.H.; Schultz, H.D.; Patel, K.P.; Wang, W.; Gao, L. Regulation of central angiotensin type 1 receptors and sympathetic outflow in heart failure. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1557–H1566. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Liu, J.; Tu, H.; Melleman, R.L.; Cornish, K.G.; Li, Y.L. In-vivo transfection of manganese superoxide dismutase gene or NFkB shRNA in nodose ganglia improves aortic baroreceptor function in heart failure rats. Hypertension 2014, 63, 88–95. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Chen, J.S.; Zucker, I.H. Carotid sinus baroreceptor sensitivity in experimental heart failure. Circulation 1990, 81, 1959–1966. [Google Scholar] [CrossRef] [Green Version]
- Schultz, H.D.; Li, Y.L.; Ding, Y. Arterial chemoreceptors and sympathetic nerve activity: Implications for hypertension and heart failure. Hypertension 2007, 50, 6–13. [Google Scholar] [CrossRef] [Green Version]
- Patel, K.P.; Zheng, H. Central neural control of sympathetic nerve activity in heart failure following exercise training. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H527–H537. [Google Scholar] [CrossRef] [Green Version]
- May, C.N.; Yao, S.T.; Booth, L.C.; Ramchandra, R. Cardiac sympathoexcitation in heart failure. Auton. Neurosci. 2013, 175, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, A.; Rengo, G.; Koch, W.J. Adrenergic nervous system in heart failure: Pathophysiology and therapy. Circ. Res. 2013, 113, 739–753. [Google Scholar] [CrossRef] [PubMed]
- Felder, R.B. Mineralocorticoid receptors, inflammation and sympathetic drive in a rat model of systolic heart failure. Exp. Physiol. 2010, 95, 19–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, B.; Li, H. Brain mechanisms of sympathetic activation in heart failure: Roles of the renin-angiotensin system, nitric oxide and pro-inflammatory cytokines (Review). Mol. Med. Rep. 2015, 12, 7823–7829. [Google Scholar] [CrossRef] [Green Version]
- Doehner, W.; Ural, D.; Haeusler, K.G.; Čelutkienė, J.; Bestetti, R.; Cavusoglu, Y.; Peña-Duque, M.A.; Glavas, D.; Iacoviello, M.; Laufs, U.; et al. Heart and brain interaction in patients with heart failure: Overview and proposal for a taxonomy. A position paper from the Study Group on Heart and Brain Interaction of the Heart Failure Association. Eur. J. Heart Fail. 2018, 20, 199–215. [Google Scholar] [CrossRef] [Green Version]
- Cuevas, J. Molecular mechanisms of dysautonomia during heart failure. Focus on “Heart failure-induced changes of voltage-gated Ca2+ channels and cell excitability in rat cardiac postganglionic neurons”. Am. J. Physiol. Cell Physiol. 2014, 306, C121–C122. [Google Scholar] [CrossRef] [Green Version]
- Wallis, D.; Watson, A.H.; Mo, N. Cardiac neurons of autonomic ganglia. Microsc. Res. Tech. 1996, 35, 69–79. [Google Scholar] [CrossRef]
- Wehrwein, E.A.; Orer, H.S.; Barman, S.M. Overview of the anatomy, physiology, and pharmacology of the autonomic nervous system. Compr. Physiol. 2016, 6, 1239–1278. [Google Scholar]
- Wink, J.; van Delft, R.; Notenboom, R.G.E.; Wouters, P.F.; DeRuiter, M.C.; Plevier, J.W.M.; Jongbloed, M.R.M. Human adult cardiac autonomic innervation: Controversies in anatomical knowledge and relevance for cardiac neuromodulation. Auton. Neurosci. 2020, 227, 102674. [Google Scholar] [CrossRef]
- Hasan, W. Autonomic cardiac innervation: Development and adult plasticity. Organogenesis 2013, 9, 176–193. [Google Scholar] [CrossRef] [Green Version]
- Pardini, B.J.; Lund, D.D.; Schmid, P.G. Organization of the sympathetic postganglionic innervation of the rat heart. J. Auton. Nerv. Syst. 1989, 28, 193–201. [Google Scholar] [CrossRef]
- Hoang, J.D.; Salavatian, S.; Yamaguchi, N.; Swid, M.A.; David, H.; Vaseghi, M. Cardiac sympathetic activation circumvents high-dose beta blocker therapy in part through release of neuropeptide Y. JCI Insight 2020, 5, e135519. [Google Scholar] [CrossRef] [PubMed]
- Borovac, J.A.; D’Amario, D.; Bozic, J.; Glavas, D. Sympathetic nervous system activation and heart failure: Current state of evidence and the pathophysiology in the light of novel biomarkers. World J. Cardiol. 2020, 12, 373–408. [Google Scholar] [CrossRef] [PubMed]
- de Lucia, C.; Piedepalumbo, M.; Paolisso, G.; Koch, W.J. Sympathetic nervous system in age–related cardiovascular dysfunction: Pathophysiology and therapeutic perspective. Int. J. Biochem. Cell Biol. 2019, 108, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Coote, J.H.; Chauhan, R.A. The sympathetic innervation of the heart: Important new insights. Auton. Neurosci. 2016, 199, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Shivkumar, K.; Ajijola, O. Autonomic regulation and ventricular arrhythmias. Curr. Treat Options Cardiovasc. Med. 2018, 20, 38. [Google Scholar] [CrossRef]
- Chen, P.S.; Chen, L.S.; Cao, J.M.; Sharifi, B.; Karagueuzian, H.S.; Fishbein, M.C. Sympathetic nerve sprouting, electrical remodeling and the mechanisms of sudden cardiac death. Cardiovasc. Res. 2001, 50, 409–416. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.A.; Boyle, N.G.; Vaseghi, M. Cardiac innervation and the autonomic nervous system in sudden cardiac death. Card. Electrophysiol. Clin. 2017, 9, 665–679. [Google Scholar] [CrossRef]
- Kimura, K.; Ieda, M.; Fukuda, K. Development, maturation, and transdifferentiation of cardiac sympathetic nerves. Circ. Res. 2012, 110, 325–336. [Google Scholar] [CrossRef]
- Backs, J.; Haunstetter, A.; Gerber, S.H.; Metz, J.; Borst, M.M.; Strasser, R.H.; Kübler, W.; Haass, M. The neuronal norepinephrine transporter in experimental heart failure: Evidence for a posttranscriptional downregulation. J. Mol. Cell. Cardiol. 2001, 33, 461–472. [Google Scholar] [CrossRef]
- Eisenhofer, G.; Friberg, P.; Rundqvist, B.; Quyyumi, A.A.; Lambert, G.; Kaye, D.M.; Kopin, I.J.; Goldstein, D.S.; Esler, M.D. Cardiac sympathetic nerve function in congestive heart failure. Circulation 1996, 93, 1667–1676. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.S. Cardiac sympathetic nerve terminal function in congestive heart failure. Acta Pharmacol. Sin. 2007, 28, 921–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanani, M.; Spray, D.C. Emerging importance of satellite glia in nervous system function and dysfunction. Nat. Rev. Neurosci. 2020, 21, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Hanani, M. Satellite glial cells in sympathetic and parasympathetic ganglia: In search of function. Brain Res. Rev. 2010, 64, 304–327. [Google Scholar] [CrossRef]
- Hanani, M.; Verkhratsky, A. Satellite glial cells and astrocytes, a comparative review. Neurochem. Res. 2021, 46, 2525–2537. [Google Scholar] [CrossRef]
- Durham, P.L.; Garrett, F.G. Development of functional units within trigeminal ganglia correlates with increased expression of proteins involved in neuron-glia interactions. Neuron Glia Biol. 2010, 6, 171–181. [Google Scholar] [CrossRef] [Green Version]
- Grubišić, V.; McClain, J.L.; Fried, D.E.; Grants, I.; Rajasekhar, P.; Csizmadia, E.; Ajijola, O.A.; Watson, R.E.; Poole, D.P.; Robson, S.C.; et al. Enteric glia modulate macrophage phenotype and visceral sensitivity following inflammation. Cell Rep. 2020, 32, 108100. [Google Scholar] [CrossRef]
- Kurata, S.; Goto, T.; Gunjigake, K.K.; Kataoka, S.; Kuroishi, K.N.; Ono, K.; Toyono, T.; Kobayashi, S.; Yamaguchi, K. Nerve growth factor involves mutual interaction between neurons and satellite glial cells in the rat trigeminal ganglion. Acta Histochem. Cytochem. 2013, 46, 65–73. [Google Scholar] [CrossRef] [Green Version]
- van Weperen, V.Y.H.; Littman, R.J.; Arneson, D.V.; Contreras, J.; Yang, X.; Ajijola, O.A. Single-cell transcriptomic profiling of satellite glial cells in stellate ganglia reveals developmental and functional axial dynamics. Glia 2021, 69, 1281–1291. [Google Scholar] [CrossRef]
- Nguyen, B.L.; Li, H.; Fishbein, M.C.; Lin, S.F.; Gaudio, C.; Chen, P.S.; Chen, L.S. Acute myocardial infarction induces bilateral stellate ganglia neural remodeling in rabbits. Cardiovasc. Pathol. 2012, 21, 143–148. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Ajijola, O.A.; Aliotta, E.; Armour, J.A.; Ardell, J.L.; Shivkumar, K. Pathological effects of chronic myocardial infarction on peripheral neurons mediating cardiac neurotransmission. Auton. Neurosci. 2016, 197, 34–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.; Kobayashi, K.; Joung, B.; Piccirillo, G.; Maruyama, M.; Vinters, H.V.; March, K.; Lin, S.F.; Shen, C.; Fishbein, M.C.; et al. Electroanatomic remodeling of the left stellate ganglion after myocardial infarction. J. Am. Coll. Cardiol. 2012, 59, 954–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, A.Y.; Elharrif, K.; Cardona-Guarache, R.; Mankad, P.; Ayers, O.; Joslyn, M.; Das, A.; Kaszala, K.; Lin, S.F.; Ellenbogen, K.A.; et al. Persistent proarrhythmic neural remodeling despite recovery from premature ventricular contraction-induced cardiomyopathy. J. Am. Coll. Cardiol. 2020, 75, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ajijola, O.A.; Yagishita, D.; Reddy, N.K.; Yamakawa, K.; Vaseghi, M.; Downs, A.M.; Hoover, D.B.; Ardell, J.L.; Shivkumar, K. Remodeling of stellate ganglion neurons after spatially targeted myocardial infarction: Neuropeptide and morphologic changes. Heart Rhythm 2015, 12, 1027–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ajijola, O.A.; Wisco, J.J.; Lambert, H.W.; Mahajan, A.; Stark, E.; Fishbein, M.C.; Shivkumar, K. Extracardiac neural remodeling in humans with cardiomyopathy. Circ. Arrhythm. Electrophysiol. 2012, 5, 1010–1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, M.; Zhou, S.; Tan, A.Y.; Song, J.; Gholmieh, G.; Fishbein, M.C.; Luo, H.; Siegel, R.J.; Karagueuzian, H.S.; Chen, L.S.; et al. Left stellate ganglion and vagal nerve activity and cardiac arrhythmias in ambulatory dogs with pacing-induced congestive heart failure. J. Am. Coll. Cardiol. 2007, 50, 335–343. [Google Scholar] [CrossRef] [Green Version]
- Tu, H.; Liu, J.; Zhang, D.; Zheng, H.; Patel, K.P.; Cornish, K.G.; Wang, W.Z.; Muelleman, R.L.; Li, Y.L. Heart failure-induced changes of voltage-gated Ca2+ channels and cell excitability in rat cardiac postganglionic neurons. Am. J. Physiol. Cell Physiol. 2014, 306, C132–C142. [Google Scholar] [CrossRef] [Green Version]
- Augustine, G.J. How does calcium trigger neurotransmitter release? Curr. Opin. Neurobiol. 2001, 11, 320–326. [Google Scholar] [CrossRef]
- Borst, J.G.; Sakmann, B. Calcium influx and transmitter release in a fast CNS synapse. Nature 1996, 383, 431–434. [Google Scholar] [CrossRef]
- Zucker, R.S. Calcium and transmitter release. J. Physiol. Paris 1993, 87, 25–36. [Google Scholar] [CrossRef]
- Tsien, R.W.; Lipscombe, D.; Madison, D.; Bley, K.; Fox, A. Reflections on Ca(2+)-channel diversity, 1988–1994. Trends Neurosci. 1995, 18, 52–54. [Google Scholar] [PubMed]
- Tsien, R.W.; Lipscombe, D.; Madison, D.V.; Bley, K.R.; Fox, A.P. Multiple types of neuronal calcium channels and their selective modulation. Trends Neurosci. 1988, 11, 431–438. [Google Scholar] [CrossRef]
- Benarroch, E.E. Neuronal voltage-gated calcium channels: Brief overview of their function and clinical implications in neurology. Neurology 2010, 74, 1310–1315. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A. Structure and regulation of voltage-gated Ca2+ channels. Annu. Rev. Cell Dev. Biol. 2000, 16, 521–555. [Google Scholar] [CrossRef] [PubMed]
- Ino, M.; Yoshinaga, T.; Wakamori, M.; Miyamoto, N.; Takahashi, E.; Sonoda, J.; Kagaya, T.; Oki, T.; Nagasu, T.; Nishizawa, Y.; et al. Functional disorders of the sympathetic nervous system in mice lacking the alpha 1B subunit (Cav 2.2) of N-type calcium channels. Proc. Natl. Acad. Sci. USA 2001, 98, 5323–5328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molderings, G.J.; Likungu, J.; Gothert, M. N-Type calcium channels control sympathetic neurotransmission in human heart atrium. Circulation 2000, 101, 403–407. [Google Scholar] [CrossRef] [Green Version]
- Yamada, Y.; Kinoshita, H.; Kuwahara, K.; Nakagawa, Y.; Kuwabara, Y.; Minami, T.; Yamada, C.; Shibata, J.; Nakao, K.; Cho, K.; et al. Inhibition of N-type Ca2+ channels ameliorates an imbalance in cardiac autonomic nerve activity and prevents lethal arrhythmias in mice with heart failure. Cardiovasc. Res. 2014, 104, 183–193. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Tu, H.; Wang, C.; Cao, L.; Hu, W.; Hackfort, B.T.; Muelleman, R.L.; Wadman, M.C.; Li, Y.L. Inhibition of N-type calcium channels in cardiac sympathetic neurons attenuates ventricular arrhythmogenesis in heart failure. Cardiovasc. Res. 2021, 117, 137–148. [Google Scholar] [CrossRef]
- Barber, M.J.; Mueller, T.M.; Henry, D.P.; Felten, S.Y.; Zipes, D.P. Transmural myocardial infarction in the dog produces sympathectomy in noninfarcted myocardium. Circulation 1983, 67, 787–796. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Knowlton, D.; Van Winkle, D.M.; Habecker, B.A. Infarction alters both the distribution and noradrenergic properties of cardiac sympathetic neurons. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H2229–H2236. [Google Scholar] [CrossRef] [Green Version]
- Zipes, D.P. Influence of myocardial ischemia and infarction on autonomic innervation of heart. Circulation 1990, 82, 1095–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, J.M.; Fishbein, M.C.; Han, J.B.; Lai, W.W.; Lai, A.C.; Wu, T.J.; Czer, L.; Wolf, P.L.; Denton, T.A.; Shintaku, I.P.; et al. Relationship between regional cardiac hyperinnervation and ventricular arrhythmia. Circulation 2000, 101, 1960–1969. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.S.; Jong, A.Y.; Kim, D.T.; Li, H.; Wang, C.; Zemljic-Harpf, A.; Ross, R.S.; Fishbein, M.C.; Chen, P.S.; Chen, L.S. Spatial distribution of nerve sprouting after myocardial infarction in mice. Heart Rhythm 2006, 3, 728–736. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Chen, L.S.; Miyauchi, Y.; Miyauchi, M.; Kar, S.; Kangavari, S.; Fishbein, M.C.; Sharifi, B.; Chen, P.S. Mechanisms of cardiac nerve sprouting after myocardial infarction in dogs. Circ. Res. 2004, 95, 76–83. [Google Scholar] [CrossRef] [Green Version]
- Kimura, K.; Kanazawa, H.; Ieda, M.; Kawaguchi-Manabe, H.; Miyake, Y.; Yagi, T.; Arai, T.; Sano, M.; Fukuda, K. Norepinephrine-induced nerve growth factor depletion causes cardiac sympathetic denervation in severe heart failure. Auton. Neurosci. 2010, 156, 27–35. [Google Scholar] [CrossRef]
- Lorentz, C.U.; Parrish, D.C.; Alston, E.N.; Pellegrino, M.J.; Woodward, W.R.; Hempstead, B.L.; Habecker, B.A. Sympathetic denervation of peri-infarct myocardium requires the p75 neurotrophin receptor. Exp. Neurol. 2013, 249, 111–119. [Google Scholar] [CrossRef] [Green Version]
- Clyburn, C.; Sepe, J.J.; Habecker, B.A. What gets on the nerves of cardiac patients? Pathophysiological changes in cardiac innervation. J. Physiol. 2022, 600, 451–461. [Google Scholar] [CrossRef]
- Herring, N.; Kalla, M.; Paterson, D.J. The autonomic nervous system and cardiac arrhythmias: Current concepts and emerging therapies. Nat. Rev. Cardiol. 2019, 16, 707–726. [Google Scholar] [CrossRef]
- Grkovski, M.; Zanzonico, P.B.; Modak, S.; Humm, J.L.; Narula, J.; Pandit-Taskar, N. F-18 meta-fluorobenzylguanidine PET imaging of myocardial sympathetic innervation. J. Nucl. Cardiol. 2022; online ahead of print. [Google Scholar]
- Li, J.; Zheng, L. The mechanism of cardiac sympathetic activity assessment methods: Current knowledge. Front. Cardiovasc. Med. 2022, 9, 931219. [Google Scholar] [CrossRef]
- Orimo, S.; Yogo, M.; Nakamura, T.; Suzuki, M.; Watanabe, H. (123)I-meta-iodobenzylguanidine (MIBG) cardiac scintigraphy in α-synucleinopathies. Ageing Res. Rev. 2016, 30, 122–133. [Google Scholar] [CrossRef]
- Rubart, M.; Zipes, D.P. Mechanisms of sudden cardiac death. J. Clin. Investig. 2005, 115, 2305–2315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoyama, T.; Lee, J.K.; Miwa, K.; Opthof, T.; Tomoyama, S.; Nakanishi, H.; Yoshida, A.; Yasui, H.; Iida, T.; Miyagawa, S.; et al. Quantification of sympathetic hyperinnervation and denervation after myocardial infarction by three-dimensional assessment of the cardiac sympathetic network in cleared transparent murine hearts. PLoS ONE 2017, 12, e0182072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, A.; Elser, M.D.; Socratous, F.; Kaye, D.M. Evidence for functional presynaptic alpha-2 adrenoceptors and their down-regulation in human heart failure. J. Am. Coll. Cardiol. 2001, 37, 1246–1251. [Google Scholar] [CrossRef]
- Kiuchi, M.G.; Nolde, J.M.; Villacorta, H.; Carnagarin, R.; Chan, J.J.S.; Lugo-Gavidia, L.M.; Ho, J.K.; Matthews, V.B.; Dwivedi, G.; Schlaich, M.P. New approaches in the management of sudden cardiac death in patients with heart failure-targeting the sympathetic nervous system. Int. J. Mol. Sci. 2019, 20, 2430. [Google Scholar] [CrossRef] [Green Version]
- Latini, R.; Masson, S.; Jeremic, G.; LuvarAÿ, G.; Fiordaliso, F.; Calvillo, L.; Bernasconi, R.; Torri, M.; Rondelli, I.; Razzetti, R.; et al. Comparative efficacy of a DA2/alpha2 agonist and a beta-blocker in reducing adrenergic drive and cardiac fibrosis in an experimental model of left ventricular dysfunction after coronary artery occlusion. J. Cardiovasc. Pharmacol. 1998, 31, 601–608. [Google Scholar] [CrossRef]
- Minatoguchi, S. Heart failure and its treatment from the perspective of sympathetic nerve activity. J. Cardiol. 2022, 79, 691–697. [Google Scholar] [CrossRef] [PubMed]
- Ramchandra, R.; Hood, S.G.; Xing, D.; Lambert, G.W.; May, C.N. Mechanisms underlying the increased cardiac norepinephrine spillover in heart failure. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H340–H347. [Google Scholar] [CrossRef] [PubMed]
- Tygesen, H.; Rundqvist, B.; Waagstein, F.; Wennerblom, B. Heart rate variability measurement correlates with cardiac norepin ephrine spillover in congestive heart failure. Am. J. Cardiol. 2001, 87, 1308–1311. [Google Scholar] [CrossRef]
- Meredith, I.T.; Eisenhofer, G.; Lambert, G.W.; Dewar, E.M.; Jennings, G.L.; Esler, M.D. Cardiac sympathetic nervous activity in congestive heart failure. Evidence for increased neuronal norepinephrine release and preserved neuronal uptake. Circulation 1993, 88, 136–145. [Google Scholar] [CrossRef] [Green Version]
- Bateman, T.M.; Ananthasubramaniam, K.; Berman, D.S.; Gerson, M.; Gropler, R.; Henzlova, M.; Mendoza, F.; Miyamoto, M.; Shah, M.; Weiland, F. Reliability of the (123)I-mIBG heart/mediastinum ratio: Results of a multicenter test-retest reproducibility study. J. Nucl. Cardiol. 2019, 26, 1555–1565. [Google Scholar] [CrossRef]
- Jacobson, A.F.; Senior, R.; Cerqueira, M.D.; Wong, N.D.; Thomas, G.S.; Lopez, V.A.; Agostini, D.; Weiland, F.; Chandna, H.; Narula, J. Myocardial iodine-123 meta-iodobenzylguanidine imaging and cardiac events in heart failure. Results of the prospective ADMIRE-HF (AdreView Myocardial Imaging for Risk Evaluation in Heart Failure) study. J. Am. Coll. Cardiol. 2010, 55, 2212–2221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merlet, P.; Valette, H.; Dubois-Randé, J.L.; Moyse, D.; Duboc, D.; Dove, P.; Bourguignon, M.H.; Benvenuti, C.; Duval, A.M.; Agostini, D.; et al. Prognostic value of cardiac metaiodobenzylguanidine imaging in patients with heart failure. J. Nucl. Med. 1992, 33, 471–477. [Google Scholar] [PubMed]
- Rajapreyar, I.; Pamboukian, S.V. Cardiac sympathetic imaging in heart failure: Is revival possible? J. Nucl. Cardiol. 2021, 28, 86–89. [Google Scholar] [CrossRef]
- Silverio, A.; Polito, M.V.; Pace, L.; D’Auria, F.; Vitulano, G.; Scarano, M.; Citro, R.; Galasso, G.; Piscione, F. Predictors of outcome in patients with de novo diagnosis of heart failure with reduced ejection fraction: Role of combined myocardial and lung Iodine-123 Meta–Iodobenzylguanidine imaging. J. Nucl. Cardiol. 2021, 28, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Ardell, J.L.; Foreman, R.D.; Armour, J.A.; Shivkumar, K. Cardiac sympathectomy and spinal cord stimulation attenuate reflex-mediated norepinephrine release during ischemia preventing ventricular fibrillation. JCI Insight 2019, 4, e131648. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.A.; Vaseghi, M.; Kluge, N.; Shivkumar, K.; Ardell, J.L.; Smith, C. Fast in vivo detection of myocardial norepinephrine levels in the beating porcine heart. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H1091–H1099. [Google Scholar] [CrossRef]
- Gilinsky, M.A.; Faibushevish, A.A.; Lunte, C.E. Determination of myocardial norepinephrine in freely moving rats using in vivo microdialysis sampling and liquid chromatography with dual-electrode amperometric detection. J. Pharm. Biomed. Anal. 2001, 24, 929–935. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Liu, B.; Wu, Q.; Liu, B.; Li, Y.; Sun, S.; Wang, Y.; Wu, X.; Chai, Z.; Jiang, X.; et al. Differential co-release of two neurotransmitters from a vesicle fusion pore in mammalian adrenal chromaffin cells. Neuron 2019, 102, 173–183. [Google Scholar] [CrossRef] [Green Version]
- Yagishita, D.; Chui, R.W.; Yamakawa, K.; Rajendran, P.S.; Ajijola, O.A.; Nakamura, K.; So, E.L.; Mahajan, A.; Shivkumar, K.; Vaseghi, M. Sympathetic nerve stimulation, not circulating norepinephrine, modulates T-peak to T-end interval by increasing global dispersion of repolarization. Circ. Arrhythm. Electrophysiol. 2015, 8, 174–185. [Google Scholar] [CrossRef] [Green Version]
- Zekios, K.C.; Mouchtouri, E.T.; Lekkas, P.; Nikas, D.N.; Kolettis, T.M. Sympathetic activation and arrhythmogenesis after myocardial infarction: Where do we stand? J. Cardiovasc. Dev. Dis. 2021, 8, 57. [Google Scholar] [CrossRef]
- Ajijola, O.A.; Chatterjee, N.A.; Gonzales, M.J.; Gornbein, J.; Liu, K.; Li, D.; Paterson, D.J.; Shivkumar, K.; Singh, J.P.; Herring, N. Coronary sinus neuropeptide Y levels and adverse outcomes in patients with stable chronic heart failure. JAMA Cardiol. 2020, 5, 318–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibbs, T.; Tapoulal, N.; Shanmuganathan, M.; Burrage, M.K.; Borlotti, A.; Banning, A.P.; Choudhury, R.P.; Neubauer, S.; Kharbanda, R.K.; Ferreira, V.M.; et al. Neuropeptide-Y levels in ST-segment-elevation myocardial infarction: Relationship with coronary microvascular function, heart Failure, and mortality. J. Am. Heart Assoc. 2022, 11, e024850. [Google Scholar] [CrossRef] [PubMed]
- Kalla, M.; Hao, G.; Tapoulal, N.; Tomek, J.; Liu, K.; Woodward, L.; Dall’Armellina, E.; Banning, A.P.; Choudhury, R.P.; Neubauer, S.; et al. The cardiac sympathetic co-transmitter neuropeptide Y is pro-arrhythmic following ST-elevation myocardial infarction despite beta-blockade. Eur. Heart J. 2020, 41, 2168–2179. [Google Scholar] [CrossRef] [PubMed]
- Özkaramanli-Gür, D.; Sağbaş, M.; Akyüz, A.; Güzel, S.; Alpsoy, Ş.; Güler, N. Role of sympathetic cotransmitter galanin on autonomic balance in heart failure: An active player or a bystander? Anatol. J. Cardiol. 2017, 18, 281–288. [Google Scholar] [CrossRef]
- Tan, C.M.J.; Green, P.; Tapoulal, N.; Lewandowski, A.J.; Leeson, P.; Herring, N. The role of neuropeptide Y in cardiovascular health and disease. Front. Physiol. 2018, 9, 1281. [Google Scholar] [CrossRef] [Green Version]
- Gardner, R.T.; Ripplinger, C.M.; Myles, R.C.; Habecker, B.A. Molecular mechanisms of sympathetic remodeling and arrhythmias. Circ. Arrhythm. Electrophysiol. 2016, 9, e001359. [Google Scholar] [CrossRef] [Green Version]
- Pannese, E.; Procacci, P. Ultrastructural localization of NGF receptors in satellite cells of the rat spinal ganglia. J. Neurocytol. 2002, 31, 755–763. [Google Scholar] [CrossRef]
- Korsching, S.; Thoenen, H. Nerve growth factor in sympathetic ganglia and corresponding target organs of the rat: Correlation with density of sympathetic innervation. Proc. Natl. Acad. Sci. USA 1983, 80, 3513–3516. [Google Scholar] [CrossRef] [Green Version]
- Scott-Solomon, E.; Boehm, E.; Kuruvilla, R. The sympathetic nervous system in development and disease. Nat. Rev. Neurosci. 2021, 22, 685–702. [Google Scholar] [CrossRef]
- Hagan, N.; Kane, J.L.; Grover, D.; Woodworth, L.; Madore, C.; Saleh, J.; Sancho, J.; Liu, J.; Li, Y.; Proto, J.; et al. CSF1R signaling is a regulator of pathogenesis in progressive MS. Cell Death Dis. 2020, 11, 904. [Google Scholar] [CrossRef]
- Pixley, F.J.; Stanley, E.R. CSF-1 regulation of the wandering macrophage: Complexity in action. Trends Cell Biol. 2004, 14, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Bacmeister, L.; Schwarzl, M.; Warnke, S.; Stoffers, B.; Blankenberg, S.; Westermann, D.; Lindner, D. Inflammation and fibrosis in murine models of heart failure. Basic Res. Cardiol. 2019, 114, 19. [Google Scholar] [PubMed]
- Dick, S.A.; Epelman, S. Chronic heart failure and inflammation: What do we really know? Circ. Res. 2016, 119, 159–176. [Google Scholar] [CrossRef] [PubMed]
- Thackeray, J.T.; Hupe, H.C.; Wang, Y.; Bankstahl, J.P.; Berding, G.; Ross, T.L.; Bauersachs, J.; Wollert, K.C.; Bengel, F.M. Myocardial inflammation predicts remodeling and neuroinflammation after myocardial infarction. J. Am. Coll. Cardiol. 2018, 71, 263–275. [Google Scholar] [CrossRef]
- Hulsmans, M.; Clauss, S.; Xiao, L.; Aguirre, A.D.; King, K.R.; Hanley, A.; Hucker, W.J.; Walfers, E.M.; Seemann, G.; Courties, G.; et al. Macrophages facilitate electrical conduction in the heart. Cell 2017, 169, 510–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, J.M.; Lopez, E.F.; Lindsey, M.L. Macrophage roles following myocardial infarction. Int. J. Cardiol. 2008, 130, 147–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Hu, W.; Tu, H.; Hackfort, B.T.; Duan, B.; Xiong, W.; Wadman, M.C.; Li, Y.L. Macrophage depletion in stellate ganglia alleviates cardiac sympathetic overactivation and ventricular arrhythmogenesis by attenuating neuroinflammation in heart failure. Basic Res. Cardiol. 2021, 116, 28. [Google Scholar] [CrossRef]
- Nohara, A.; Okada, S.; Ohshima, K.; Pessin, J.E.; Mori, M. Cyclin-dependent kinase-5 is a key molecule in tumor necrosis factor-alpha-induced insulin resistance. J. Biol. Chem. 2011, 286, 33457–33465. [Google Scholar] [CrossRef] [Green Version]
- Rozas, P.; Lazcano, P.; Pina, R.; Cho, A.; Terse, A.; Pertusa, M.; Madrid, R.; Gonzalez-Billault, C.; Kulkarni, A.B.; Utreras, E. Targeted overexpression of tumor necrosis factor-alpha increases cyclin-dependent kinase 5 activity and TRPV1-dependent Ca2+ influx in trigeminal neurons. Pain 2016, 157, 1346–1362. [Google Scholar] [CrossRef] [Green Version]
- Utreras, E.; Futatsugi, A.; Rudrabhatla, P.; Keller, J.; Iadarola, M.J.; Pant, H.C.; Kulkarni, A.B. Tumor necrosis factor-alpha regulates cyclin-dependent kinase 5 activity during pain signaling through transcriptional activation of p35. J. Biol. Chem. 2009, 284, 2275–2284. [Google Scholar] [CrossRef] [Green Version]
- Su, S.C.; Seo, J.; Pan, J.Q.; Samuels, B.A.; Rudenko, A.; Ericsson, M.; Neve, R.L.; Yue, D.T.; Tsai, L.H. Regulation of N-type voltage-gated calcium channels and presynaptic function by cyclin-dependent kinase 5. Neuron 2012, 75, 675–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Tu, H.; Hu, W.; Wadman, M.C.; Li, Y.L. CDK5 promotes ventricular arrhythmogenesis through phosphorylation of N-type calcium channels in cardiac sympathetic postganglionic neurons. FASEB J. 2020, 34, 03088. [Google Scholar] [CrossRef]
- Ajijola, O.A.; Hoover, D.B.; Simerly, T.M.; Brown, T.C.; Yanagawa, J.; Biniwale, R.M.; Lee, J.M.; Sadeghi, A.; Khanlou, N.; Ardell, J.L.; et al. Inflammation, oxidative stress, and glial cell activation characterize stellate ganglia from humans with electrical storm. JCI Insight 2017, 2, e94715. [Google Scholar] [CrossRef] [PubMed]
- Shanks, J.; Gao, L.; Zucker, I.H. Sympathomodulation in heart failure: A role for stellate ganglia Nrf2. FASEB J. 2019, 33, 564–565. [Google Scholar] [CrossRef]
- Singh, S.; Sayers, S.; Walter, J.S.; Thomas, D.; Dieter, R.S.; Nee, L.M.; Wurster, R.D. Hypertrophy of neurons within cardiac ganglia in human, canine, and rat heart failure: The potential role of nerve growth factor. J. Am. Heart. Assoc. 2013, 2, e000210. [Google Scholar] [CrossRef] [Green Version]
- Meloni, M.; Caporali, A.; Graiani, G.; Lagrasta, C.; Katare, R.; Van Linthout, S.; Spillmann, F.; Campesi, I.; Madeddu, P.; Quaini, F.; et al. Nerve growth factor promotes cardiac repair following myocardial infarction. Circ. Res. 2010, 106, 1275–1284. [Google Scholar] [CrossRef] [Green Version]
- Atwal, J.K.; Massie, B.; Miller, F.D.; Kaplan, D.R. The TrkB-Shc site signals neuronal survival and local axon growth via MEK and P13-kinase. Neuron 2000, 27, 265–277. [Google Scholar] [CrossRef]
- Bodmer, D.; Ascaño, M.; Kuruvilla, R. Isoform-specific dephosphorylation of dynamin1 by calcineurin couples neurotrophin receptor endocytosis to axonal growth. Neuron 2011, 70, 1085–1099. [Google Scholar] [CrossRef] [Green Version]
- Spillane, M.; Ketschek, A.; Donnelly, C.J.; Pacheco, A.; Twiss, J.L.; Gallo, G. Nerve growth factor-induced formation of axonal filopodia and collateral branches involves the intra-axonal synthesis of regulators of the actin-nucleating Arp2/3 complex. J. Neurosci. 2012, 32, 17671–17689. [Google Scholar] [CrossRef] [Green Version]
- Kisiswa, L.; Osório, C.; Erice, C.; Vizard, T.; Wyatt, S.; Davies, A.M. TNFα reverse signaling promotes sympathetic axon growth and target innervation. Nat. Neurosci. 2013, 16, 865–873. [Google Scholar] [CrossRef] [Green Version]
- O’Keeffe, G.W.; Gutierrez, H.; Howard, L.; Laurie, C.W.; Osorio, C.; Gavaldà, N.; Wyatt, S.L.; Davies, A.M. Region-specific role of growth differentiation factor-5 in the establishment of sympathetic innervation. Neural Dev. 2016, 11, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellegrino, M.J.; Habecker, B.A. STAT3 integrates cytokine and neurotrophin signals to promote sympathetic axon regeneration. Mol. Cell. Neurosci. 2013, 56, 272–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellegrino, M.J.; McCully, B.H.; Habecker, B.A. Leptin stimulates sympathetic axon outgrowth. Neurosci. Lett. 2014, 566, 1–5. [Google Scholar] [CrossRef]
- Gamage, K.K.; Cheng, I.; Park, R.E.; Karim, M.S.; Edamura, K.; Hughes, C.; Spano, A.J.; Erisir, A.; Deppmann, C.D. Death receptor 6 promotes wallerian degeneration in peripheral axons. Curr. Biol. 2017, 27, 890–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohn, J.; Aloyz, R.S.; Toma, J.G.; Haak-Frendscho, M.; Miller, F.D. Functionally antagonistic interactions between the TrkA and p75 neurotrophin receptors regulate sympathetic neuron growth and target innervation. J. Neurosci. 1999, 19, 5393–5408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majdan, M.; Walsh, G.S.; Aloyz, R.; Miller, F.D. TrkA mediates developmental sympathetic neuron survival in vivo by silencing an ongoing p75NTR-mediated death signal. J. Cell Biol. 2001, 155, 1275–1285. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, A.; McLaughlin, T.; O’Leary, D.D.; Tessier-Lavigne, M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 2009, 457, 981–989. [Google Scholar] [CrossRef] [Green Version]
- Singh, K.K.; Park, K.J.; Hong, E.J.; Kramer, B.M.; Greenberg, M.E.; Kaplan, D.R.; Miller, F.D. Developmental axon pruning mediated by BDNF-p75NTR-dependent axon degeneration. Nat. Neurosci. 2008, 11, 649–658. [Google Scholar] [CrossRef] [Green Version]
- Teng, K.K.; Felice, S.; Kim, T.; Hempstead, B.L. Understanding proneurotrophin actions: Recent advances and challenges. Dev. Neurobiol. 2010, 70, 350–359. [Google Scholar] [CrossRef] [Green Version]
- Yong, Y.; Gamage, K.; Cheng, I.; Barford, K.; Spano, A.; Winckler, B.; Deppmann, C. p75NTR and DR6 regulate distinct phases of axon degeneration demarcated by spheroid rupture. J. Neurosci. 2019, 39, 9503–9520. [Google Scholar] [CrossRef]
- Sepe, J.J.; Gardner, R.T.; Blake, M.R.; Brooks, D.M.; Staffenson, M.A.; Betts, C.B.; Sivagnanam, S.; Larson, W.; Kumar, S.; Bayles, R.G.; et al. Therapeutics that promote sympathetic reinnervation modulate the inflammatory response after myocardial infarction. JACC Basic Transl. Sci. 2022, 7, 915–930. [Google Scholar] [CrossRef]
- Huang, R.; Wang, Y.; Li, J.; Jiang, X.; Li, Y.; Liu, B.; Wu, X.; Du, X.; Hang, Y.; Jin, M.; et al. Ca(2+)-independent but voltage-dependent quantal catecholamine secretion (CiVDS) in the mammalian sympathetic nervous system. Proc. Natl. Acad. Sci. USA 2019, 116, 20201–20209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beau, S.L.; Saffitz, J.E. Transmural heterogeneity of norepinephrine uptake in failing human hearts. J. Am. Coll. Cardiol. 1994, 23, 579–585. [Google Scholar] [CrossRef]
- Böhm, M.; La Rosée, K.; Schwinger, R.H.; Erdmann, E. Evidence for reduction of norepinephrine uptake sites in the failing human heart. J. Am. Coll. Cardiol. 1995, 25, 146–153. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.S.; Fan, T.H.; Sullebarger, J.T.; Sakamoto, S. Decreased adrenergic neuronal uptake activity in experimental right heart failure. A chamber-specific contributor to beta-adrenoceptor downregulation. J. Clin. Investig. 1989, 84, 1267–1275. [Google Scholar] [CrossRef]
- Mao, W.; Iwai, C.; Qin, F.; Liang, C.S. Norepinephrine induces endoplasmic reticulum stress and downregulation of norepinephrine transporter density in PC12 cells via oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2381–H2389. [Google Scholar] [CrossRef]
- Mao, W.; Qin, F.; Iwai, C.; Vulapalli, R.; Keng, P.C.; Liang, C.S. Extracellular norepinephrine reduces neuronal uptake of norepinephrine by oxidative stress in PC12 cells. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H29–H39. [Google Scholar] [CrossRef] [Green Version]
- Vatta, M.S.; Bianciotti, L.G.; Guil, M.J.; Hope, S.I. Regulation of the norepinephrine transporter by endothelins: A potential therapeutic target. Vitam. Horm. 2015, 98, 371–405. [Google Scholar]
- Backs, J.; Bresch, E.; Lutz, M.; Kristen, A.V.; Haass, M. Endothelin-1 inhibits the neuronal norepinephrine transporter in hearts of male rats. Cardiovasc. Res. 2005, 67, 283–290. [Google Scholar] [CrossRef]
- Mapps, A.A.; Thomsen, M.B.; Boehm, E.; Zhao, H.; Hattar, S.; Kuruvilla, R. Diversity of satellite glia in sympathetic and sensory ganglia. Cell Rep. 2022, 38, 110328. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.-L. Stellate Ganglia and Cardiac Sympathetic Overactivation in Heart Failure. Int. J. Mol. Sci. 2022, 23, 13311. https://doi.org/10.3390/ijms232113311
Li Y-L. Stellate Ganglia and Cardiac Sympathetic Overactivation in Heart Failure. International Journal of Molecular Sciences. 2022; 23(21):13311. https://doi.org/10.3390/ijms232113311
Chicago/Turabian StyleLi, Yu-Long. 2022. "Stellate Ganglia and Cardiac Sympathetic Overactivation in Heart Failure" International Journal of Molecular Sciences 23, no. 21: 13311. https://doi.org/10.3390/ijms232113311
APA StyleLi, Y.-L. (2022). Stellate Ganglia and Cardiac Sympathetic Overactivation in Heart Failure. International Journal of Molecular Sciences, 23(21), 13311. https://doi.org/10.3390/ijms232113311