
Signal Peptide Variants in Inherited Retinal Diseases: A Multi-Institutional Case Series

Abstract
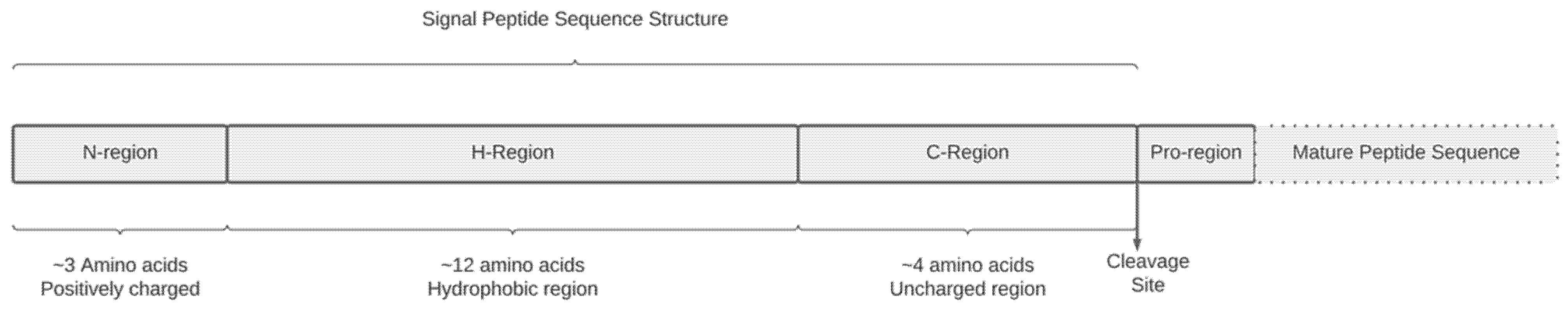
:1. Introduction
2. Results
3. Discussion
4. Materials and Methods
4.1. Identification of Signal Peptide Possessing Genes and Cases of Interest
4.2. In Silico Bioinformatic Evaluation of Signal Peptide Variants
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Clinical Summaries of Cases in Our Patient Cohort
- Case 1
- Case 2
- Case 3
- Case 4
- Case 5
- Case 6
References
- Von Heijne, G. The signal peptide. J. Membr. Biol. 1990, 115, 195–201. [Google Scholar] [CrossRef] [PubMed]
- UniProt. Available online: https://www.uniprot.org/ (accessed on 14 July 2021).
- Jarjanazi, H.; Savas, S.; Pabalan, N.; Dennis, J.; Ozelik, H. Biological implications of SNPs in signal peptide domains of human proteins. Proteins 2008, 70, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Inglehearn, C.F. Molecular genetics of human retinal dystrophies. Eye 1998, 12, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Berger, W.; Kloeckener-Gruissem, B.; Neidhardt, J. The molecular basis of human retinal and vitreoretinal diseases. Prog. Retin. Eye Res. 2010, 29, 335–375. [Google Scholar] [CrossRef] [Green Version]
- Hiraoka, M.; Takahashi, H.; Orimo, H.; Hiraoka, M.; Ogata, T.; Azuma, N. Genetic screening of Wnt signaling factors in advanced retinopathy of prematurity. Mol. Vis. 2010, 16, 2572–2577. [Google Scholar]
- Vijayasarathy, C.; Sui, R.; Zeng, Y.; Yang, G.; Xu, F.; Caruso, R.C.; Lewis, R.A.; Ziccardi, L.; Sieving, P.A. Molecular mechanisms leading to null-protein product from retinoschisin (RS1) signal-sequence mutants in X-Linked Retinoschisis (XLRS) disease. Hum. Mutat. 2010, 31, 1251–1260. [Google Scholar] [CrossRef] [Green Version]
- Hosono, K.; Nishina, S.; Yokoi, T.; Katagiri, S.; Saitsu, H.; Kurata, K.; Miyamichi, D.; Hikoya, A.; Mizobuchi, K.; Nakano, T.; et al. Molecular diagnosis of 34 Japanese families with leber congenital amaurosis using targeted next generation sequencing. Sci. Rep. 2018, 8, 8279. [Google Scholar] [CrossRef] [Green Version]
- Glöckle, N.; Kohl, S.; Mohr, J.; Scheurenbrand, T.; Sprecher, A.; Weisschuh, N.; Bernd, A.; Rudolph, G.; Schubach, M.; Poloschek, C.; et al. Panel-based next generation sequencing as a reliable and efficient technique to detect mutations in unselected patients with retinal dystrophies. Eur. J. Hum. Genet. 2014, 22, 99–104. [Google Scholar] [CrossRef] [Green Version]
- McGuigan, D.B.; Heon, E.; Cideciyan, A.V.; Ratnapriya, R.; Lu, M.; Sumaroka, A.; Roman, A.J.; Batmanabane, V.; Garafalo, A.V.; Stone, E.M.; et al. EYS mutations causing autosomal recessive retinitis pigmentosa: Changes of retinal structure and function with disease progression. Genes 2017, 8, 178. [Google Scholar] [CrossRef]
- Stone, E.M.; Andorf, J.L.; Whitmore, S.S.; DeLuca, A.P.; Giacalone, J.C.; Streb, L.M.; Braun, T.A.; Mullins, R.F.; Scheetz, T.E.; Sheffield, V.C.; et al. Clinically Focused Molecular Investigation of 1000 Consecutive Families with Inherited Retinal Disease. Ophthalmology 2017, 124, 1314–1331. [Google Scholar] [CrossRef]
- Andrew, D.W.; Young, J.C.; Mirelsq, L.F.; Czarnota, G.J. The Role of the N Region in Signal Sequence and Signal-anchor Function*. J. Biol. Chem. 1992, 267, 7761–7769. [Google Scholar] [CrossRef]
- Guo, H.; Sun, J.; Li, X.; Xiong, Y.; Wang, H.; Shu, H.; Zhu, R.; Liu, Q.; Huang, Y.; Madley, R.; et al. Positive charge in the n-region of the signal peptide contributes to efficient post-translational translocation of small secretory preproteins. J. Biol. Chem. 2018, 293, 1899–1907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Sun, J.; Cui, J.; Chen, W.; Guo, H.; Barbetti, F.; Arvan, P. INS-gene mutations: From genetics and beta cell biology to clinical disease. Mol. Asp. Med. 2016, 176, 139–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abad-Navarro, F.; De La Morena-Barrio, M.E.; Fernández-Breis, J.T.; Corral, J. Lost in translation: Bioinformatic analysis of variations affecting the translation initiation codon in the human genome. Bioinformatics 2018, 34, 3788–3794. [Google Scholar] [CrossRef]
- Kearse, M.G.; Wilusz, J.E. Non-AUG translation: A new start for protein synthesis in eukaryotes. Genes Dev. 2017, 31, 1717–1731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Zhang, J.; Chen, N.; Wang, L.; Zhang, F.; Ma, Z.; Li, G.; Yang, L. Application of whole exome and targeted panel sequencing in the clinical molecular diagnosis of 319 Chinese families with inherited retinal dystrophy and comparison study. Genes 2018, 9, 360. [Google Scholar] [CrossRef] [Green Version]
- Navarro-Fernández, J.; Dybedal, I.; Águila, S.; Bohdam, N.; Corrales, F.; Miqueo, C.; Andresen, M.; Ferrer, F.; Tjønnfjord, G.E.; Martínez-Martínez, I.; et al. C0380: Clinical and Biochemical Consequences of Met1Ileu Mutation in Serpinc1 Gene: Generation of a Small Non-Inhibitory Antithrombin Variant without the N-Terminal Region by Use of an Alternative Initiation Codon that Has a Strong Gain-Of-Function Associ. Thromb. Res. 2014, 133, S11. [Google Scholar] [CrossRef]
- Owji, H.; Nezafat, N.; Negahdaripour, M.; Hajiebrahimi, A.; Ghasemi, Y. A comprehensive review of signal peptides: Structure, roles, and applications. Eur. J. Cell Biol. 2018, 97, 422–441. [Google Scholar] [CrossRef]
- Chung, B.D.; Kayserili, H.; Ai, M.; Freudenberg, J.; Üzümcü, A.; Uyguner, O.; Bartels, C.F.; Höning, S.; Ramirez, A.; Hanisch, F.G.; et al. A mutation in the signal sequence of LRP5 in a family with an osteoporosis-pseudoglioma syndrome(OPPG)-like phenotype indicates a novel disease mechanism for trinucleotide repeats. Hum. Mutat. 2009, 30, 641–648. [Google Scholar] [CrossRef]
- Hughes, A.E.; Ralston, S.H.; Marken, J.; Bell, C.; MacPherson, H.; Wallace, R.G.H.; Van Hul, W.; Whyte, M.P.; Nakatsuka, K.; Hovy, L.; et al. Mutations in TNFRSF11A, affecting the signal peptide of RANK, cause familial expansile osteolysis. Nat. Genet. 2000, 24, 45–48. [Google Scholar] [CrossRef]
- Seppen, J.; Steenken, E.; Lindhout, D.; Bosma, P.J.; Oude Elferink, R.P.J. A mutation which disrupts the hydrophobic core of the signal peptide of bilirubin UDP-glucuronosyltransferase, an endoplasmic reticulum membrane protein, causes Crigler-Najjar type II. FEBS Lett. 1996, 390, 294–298. [Google Scholar] [CrossRef]
- Vezzoli, V.; Duminuco, P.; Vottero, A.; Kleinau, G.; Schülein, R.; Minari, R.; Bassi, I.; Bernasconi, S.; Persani, L.; Bonomi, M. A newvariant in signal peptide of the human luteinizing hormone receptor (LHCGR) affects receptor biogenesis causing leydig cell hypoplasia. Hum. Mol. Genet. 2015, 24, 6003–6012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef] [Green Version]
- Karamyshev, A.L.; Karamysheva, Z.N. Lost in translation: Ribosome-associated mRNA and protein quality controls. Front. Genet. 2018, 9, 431. [Google Scholar] [CrossRef] [Green Version]
- Noorwez, S.M.; Kuksa, V.; Imanishi, Y.; Zhu, L.; Filipek, S.; Palczewski, K.; Kaushal, S. Pharmacological chaperone-mediated in vivo folding and stabilization of the P23H-opsin mutant associated with autosomal dominant retinitis pigmentosa. J. Biol. Chem. 2003, 278, 14442–14450. [Google Scholar] [CrossRef] [Green Version]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [Green Version]
- Guerriero, C.J.; Brodsky, J.L. The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol. Rev. 2012, 92, 537–576. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.X.; Sanders, E.; Fliesler, S.J.; Wang, J.J. Endoplasmic reticulum stress and the unfolded protein responses in retinal degeneration. Exp. Eye Res. 2014, 125, 30–40. [Google Scholar] [CrossRef] [Green Version]
- Kroeger, H.; Lavail, M.M.; Lin, J.H. Endoplasmic reticulum stress in vertebrate mutant rhodopsin models of retinal degeneration. In Advances in Experimental Medicine and Biology; NIH Public Access: Bethesda, MD, USA, 2014; Volume 801, pp. 585–592. [Google Scholar]
- Datta, R.; Waheed, A.; Shah, G.N.; Sly, W.S. Signal sequence mutation in autosomal dominant form of hypoparathyroidism induces apoptosis that is corrected by a chemical chaperone. Proc. Natl. Acad. Sci. USA 2007, 104, 19989–19994. [Google Scholar] [CrossRef] [Green Version]
- Kosmaoglou, M.; Schwarz, N.; Bett, J.S.; Cheetham, M.E. Molecular chaperones and photoreceptor function. Prog. Retin. Eye Res. 2008, 27, 434–449. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.Q.; Ishii, S.; Asano, N.; Suzuki, Y. Accelerated transport and maturation of lysosomal α-galactosidase A in fabry lymphoblasts by an enzyme inhibitor. Nat. Med. 1999, 5, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Lam, B.L.; Leroy, B.P.; Black, G.; Ong, T.; Yoon, D.; Trzupek, K. Genetic testing and diagnosis of inherited retinal diseases. Orphanet J. Rare Dis. 2021, 16, 514. [Google Scholar] [CrossRef] [PubMed]
- Teufel, F.; Juan, J.; Armenteros, A.; Johansen, A.R.; Gíslason, M.H.; Pihl, S.I.; Tsirigos, K.D.; Winther, O.; Brunak, S.; Von Heijne, G.; et al. SignalP 6.0 achieves signal peptide prediction across all types using protein language models. Nat. Biotechnol. 2021, 40, 1023–1025. [Google Scholar] [CrossRef] [PubMed]
- Pejaver, V.; Urresti, J.; Lugo-Martinez, J.; Pagel, K.A.; Lin, G.N.; Nam, H.J.; Mort, M.; Cooper, D.N.; Sebat, J.; Iakoucheva, L.M.; et al. Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat. Commun. 2020, 11, 5918. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Case | Gene | NM | Signal Peptide Mutation | Non-SP Allele Mutation | Diagnosis | Inheritance Pattern | Affected SP Region | Likelihood Change (%) a | Cleavage Site Loss | MutPred ^ | MutationTaster | Mutation References | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Sp Allele |
Non-SP Allele |
SP Allele |
Non-SP Allele | |||||||||||
| 1 | CRB1 | 201,253.3 | c.2T>C (p.Met1*) | c.2056C>T (p.R686C) | retinitis pigmentosa | AR | N-terminal | N/A * | N/A * | 0.793 | 0.465 | Disease causing | Polymorphism | Hosono et al. [8] |
| 2 | NDP | _000266 | c.37_57del21 (p.L13_M19del) | N/A | familial exudative vitreoretinopathy | XL | H-core | 0.991 → 0 (−100%) | Yes | 0.619 | N/A | Disease causing | N/A | Novel |
| 3 | FZD4 | _012193.4 | c.23deIC (p.P8Rfs*53) | None | familial exudative vitreoretinopathy | AD | H-core | 0.999 → 0 (−100%) | Yes | 0.557 | N/A | Disease causing | N/A | Novel |
| 4 | EYS | _198283.2 | c.32dupT (p. M12Dfs*14) | c.95G>T (p.W32L) | retinitis pigmentosa | AR | H-core | 0.999 → 0 (−100%) | Yes | Not performed † | 0.872 | Disease causing | Polymorphism | Glockle et al. [9] McGuian et al. [10] |
| 5 | RS1 | _000330.4 | c.52+1 G>C | N/A | X-linked juvenile retinoschisis | XL | C-terminal | 0.998 → 0.998 (0%) | No | Not performed ± | N/A | Disease causing | N/A | Vijayasarathy et al. [7] |
| 6 | RS1 | _000330.4 | c.(52+1_53-1)_(78+1_79-1),del (p.A18Pfs*108) | N/A | X-linked juvenile retinoschisis | XL | C-terminal | 0.998 → 0.114 (−88.50%) | Yes | 0.529 | N/A | Disease causing | N/A | Stone et al. [11] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jimenez, H.J.; Procopio, R.A.; Thuma, T.B.T.; Marra, M.H.; Izquierdo, N.; Klufas, M.A.; Nagiel, A.; Pennesi, M.E.; Pulido, J.S. Signal Peptide Variants in Inherited Retinal Diseases: A Multi-Institutional Case Series. Int. J. Mol. Sci. 2022, 23, 13361. https://doi.org/10.3390/ijms232113361
Jimenez HJ, Procopio RA, Thuma TBT, Marra MH, Izquierdo N, Klufas MA, Nagiel A, Pennesi ME, Pulido JS. Signal Peptide Variants in Inherited Retinal Diseases: A Multi-Institutional Case Series. International Journal of Molecular Sciences. 2022; 23(21):13361. https://doi.org/10.3390/ijms232113361
Chicago/Turabian StyleJimenez, Hiram J., Rebecca A. Procopio, Tobin B. T. Thuma, Molly H. Marra, Natalio Izquierdo, Michael A. Klufas, Aaron Nagiel, Mark E. Pennesi, and Jose S. Pulido. 2022. "Signal Peptide Variants in Inherited Retinal Diseases: A Multi-Institutional Case Series" International Journal of Molecular Sciences 23, no. 21: 13361. https://doi.org/10.3390/ijms232113361
APA StyleJimenez, H. J., Procopio, R. A., Thuma, T. B. T., Marra, M. H., Izquierdo, N., Klufas, M. A., Nagiel, A., Pennesi, M. E., & Pulido, J. S. (2022). Signal Peptide Variants in Inherited Retinal Diseases: A Multi-Institutional Case Series. International Journal of Molecular Sciences, 23(21), 13361. https://doi.org/10.3390/ijms232113361