
The Molecular and Cellular Strategies of Glioblastoma and Non-Small-Cell Lung Cancer Cells Conferring Radioresistance


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Tumor Heterogeneity
3. Cancer Stem Cells Concept, History, and Properties
3.1. Cancer Stem Cells Related Markers
3.2. Cancer Stem Cells Signaling Pathways
3.3. DNA Damage Repair in Response to Ionizing Radiation
3.4. Cancer Stem Cells in Glioblastoma
3.5. Cancer Stem Cells in Non-Small-Cell Lung Cancer
4. Epithelial-to-Mesenchymal Transition and Migratory Activity in Glioblastoma and Non-Small-Cell Lung Cancer
5. Radiation-Induced Dormancy
5.1. Quiescence-Associated Radioresistance
5.2. Radiation-Induced Senescence
6. Polyploid Giant Cancer Cells/Multinucleated Giant Cancer Cells in Glioblastoma and Non-Small-Cell Lung Cancer
7. Tumor Microenvironment (TME)
8. The Potential Treatment of Radioresistance in Glioblastoma and Non-Small-Cell Lung Cancer
9. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Berger, F.; Gay, E.; Pelletier, L.; Tropel, P.; Wion, D. Development of gliomas: Potential role of asymmetrical cell division of neural stem cells. Lancet Oncol. 2004, 5, 511–514. [Google Scholar] [CrossRef]
- Brandes, A.A. State-of-the-art treatment of high-grade brain tumors. Semin. Oncol. 2003, 30, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Kleihues, P.; Louis, D.N.; Scheithauer, B.W.; Rorke, L.B.; Reifenberger, G.; Burger, P.C.; Cavenee, W.K. The WHO classification of tumors of the nervous system. J. Neuropathol. Exp. Neurol. 2002, 61, 215–225; discussion 219–226. [Google Scholar] [CrossRef] [PubMed]
- Keles, G.E.; Anderson, B.; Berger, M.S. The effect of extent of resection on time to tumor progression and survival in patients with glioblastoma multiforme of the cerebral hemisphere. Surg. Neurol. 1999, 52, 371–379. [Google Scholar] [CrossRef]
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant astrocytic glioma: Genetics, biology, and paths to treatment. Genes Dev. 2007, 21, 2683–2710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gridelli, C.; Rossi, A.; Carbone, D.P.; Guarize, J.; Karachaliou, N.; Mok, T.; Petrella, F.; Spaggiari, L.; Rosell, R. Non-small-cell lung cancer. Nat. Rev. Dis. Prim. 2015, 1, 15009. [Google Scholar] [CrossRef]
- Ko, E.C.; Raben, D.; Formenti, S.C. The Integration of Radiotherapy with Immunotherapy for the Treatment of Non–Small Cell Lung Cancer. Clin. Cancer Res. 2018, 24, 5792–5806. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Sarkaria, J.N.; Petell, C.A.; Paraskevakou, G.; Zollman, P.J.; Schroeder, M.; Carlson, B.; Decker, P.A.; Wu, W.; James, C.D.; et al. Combination of Measles Virus Virotherapy and Radiation Therapy Has Synergistic Activity in the Treatment of Glioblastoma Multiforme. Clin. Cancer Res. 2007, 13, 7155–7165. [Google Scholar] [CrossRef] [Green Version]
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Hamou, M.-F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMTGene Silencing and Benefit from Temozolomide in Glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [Green Version]
- Rich, J.N. Cancer Stem Cells in Radiation Resistance. Cancer Res. 2007, 67, 8980–8984. [Google Scholar] [CrossRef]
- Perry, J.R.; Laperriere, N.; O’Callaghan, C.J.; Brandes, A.A.; Menten, J.; Phillips, C.; Fay, M.; Nishikawa, R.; Cairncross, J.G.; Roa, W.; et al. Short-Course Radiation plus Temozolomide in Elderly Patients with Glioblastoma. N. Engl. J. Med. 2017, 376, 1027–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wisnivesky, J.P.; Bonomi, M.; Henschke, C.; Iannuzzi, M.; McGinn, T. Radiation Therapy for the Treatment of Unresected Stage I-II Non-small Cell Lung Cancer. Chest 2005, 128, 1461–1467. [Google Scholar] [CrossRef] [PubMed]
- Weihua, Z.; Lin, Q.; Ramoth, A.J.; Fan, D.; Fidler, I.J. Formation of solid tumors by a single multinucleated cancer cell. Cancer 2011, 117, 4092–4099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, H.; Iwatsuki, M.; Ieta, K.; Ohta, D.; Haraguchi, N.; Mimori, K.; Mori, M. Cancer stem cells and chemoradiation resistance. Cancer Sci. 2008, 99, 1871–1877. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.-M.; Lin, S.-H.; Logothetis, C.J. Stem-cell origin of metastasis and heterogeneity in solid tumours. Lancet Oncol. 2002, 3, 508–513. [Google Scholar] [CrossRef]
- Passegué, E.; Jamieson, C.H.M.; Ailles, L.E.; Weissman, I.L. Normal and leukemic hematopoiesis: Are leukemias a stem cell disorder or a reacquisition of stem cell characteristics? Proc. Natl. Acad. Sci. USA 2003, 100, 11842–11849. [Google Scholar] [CrossRef] [Green Version]
- Maugeri-Saccà, M.; Vigneri, P.; De Maria, R. Cancer Stem Cells and Chemosensitivity. Clin. Cancer Res. 2011, 17, 4942–4947. [Google Scholar] [CrossRef] [Green Version]
- Arnold, C.R.; Mangesius, J.; Skvortsova, I.-I.; Ganswindt, U. The Role of Cancer Stem Cells in Radiation Resistance. Front. Oncol. 2020, 10, 164. [Google Scholar] [CrossRef] [Green Version]
- Jordan, C.T.; Guzman, M.L.; Noble, M. Cancer Stem Cells. N. Engl. J. Med. 2006, 355, 1253–1261. [Google Scholar] [CrossRef]
- Creighton, C.J.; Li, X.; Landis, M.; Dixon, J.M.; Neumeister, V.M.; Sjolund, A.; Rimm, D.L.; Wong, H.; Rodriguez, A.; Herschkowitz, J.I.; et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc. Natl. Acad. Sci. USA 2009, 106, 13820–13825. [Google Scholar] [CrossRef]
- Han, J.; Won, M.; Kim, J.H.; Jung, E.; Min, K.; Jangili, P.; Kim, J.S. Cancer stem cell-targeted bio-imaging and chemotherapeutic perspective. Chem. Soc. Rev. 2020, 49, 7856–7878. [Google Scholar] [CrossRef] [PubMed]
- Plaks, V.; Kong, N.; Werb, Z. The Cancer Stem Cell Niche: How Essential Is the Niche in Regulating Stemness of Tumor Cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talukdar, S.; Bhoopathi, P.; Emdad, L.; Das, S.; Sarkar, D.; Fisher, P.B. Dormancy and cancer stem cells: An enigma for cancer therapeutic targeting. Adv. Cancer Res. 2019, 141, 43–84. [Google Scholar] [PubMed]
- Liang, H.; Deng, L.; Burnette, B.; Weichselbaum, R.R.; Fu, Y.X. Radiation-induced tumor dormancy reflects an equilibrium between the proliferation and T lymphocyte-mediated death of malignant cells. Oncoimmunology 2013, 2, e25668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vollmann-Zwerenz, A.; Leidgens, V.; Feliciello, G.; Klein, C.A.; Hau, P. Tumor Cell Invasion in Glioblastoma. Int. J. Mol. Sci. 2020, 21, 1932. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Xiong, Y.; Ding, X.; Wang, L.; Zhao, Y.; Fei, Y.; Zhu, Y.; Shen, X.; Tan, C.; Liang, Z. Cathepsin L activated by mutant p53 and Egr-1 promotes ionizing radiation-induced EMT in human NSCLC. J. Exp. Clin. Cancer Res. 2019, 38, 61. [Google Scholar] [CrossRef]
- Iser, I.C.; Pereira, M.B.; Lenz, G.; Wink, M.R. The Epithelial-to-Mesenchymal Transition-Like Process in Glioblastoma: An Updated Systematic Review and In Silico Investigation. Med. Res. Rev. 2017, 37, 271–313. [Google Scholar] [CrossRef]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [Green Version]
- Loh, C.-Y.; Chai, J.; Tang, T.; Wong, W.; Sethi, G.; Shanmugam, M.; Chong, P.; Looi, C. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef] [Green Version]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Zhang, F.; Zhang, T.; Teng, Z.-h.; Zhang, R.; Wang, J.-B.; Mei, Q.-B. Sensitization to γ-irradiation-induced cell cycle arrest and apoptosis by the histone deacetylase inhibitor trichostatin A in non-small cell lung cancer (NSCLC) cells. Cancer Biol. Ther. 2014, 8, 823–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madhusudan, S.; Middleton, M.R. The emerging role of DNA repair proteins as predictive, prognostic and therapeutic targets in cancer. Cancer Treat. Rev. 2005, 31, 603–617. [Google Scholar] [CrossRef] [PubMed]
- Karagiannis, T.C.; El-Osta, A. Double-strand breaks: Signaling pathways and repair mechanisms. Cell Mol. Life Sci. 2004, 61, 2137–2147. [Google Scholar] [CrossRef] [PubMed]
- Sikora, E.; Czarnecka-Herok, J.; Bojko, A.; Sunderland, P. Therapy-induced polyploidization and senescence: Coincidence or interconnection? Semin. Cancer Biol. 2022, 81, 83–95. [Google Scholar] [CrossRef]
- d’Adda di Fagagna, F.; Reaper, P.M.; Clay-Farrace, L.; Fiegler, H.; Carr, P.; Von Zglinicki, T.; Saretzki, G.; Carter, N.P.; Jackson, S.P. A DNA damage checkpoint response in telomere-initiated senescence. Nature 2003, 426, 194–198. [Google Scholar] [CrossRef]
- Shen, Z.; Huhn, S.C.; Haffty, B.G. Escaping death to quiescence: Avoiding mitotic catastrophe after DNA damage. Cell Cycle 2014, 12, 1664. [Google Scholar] [CrossRef] [Green Version]
- Kodym, E.; Kodym, R.; Reis, A.E.; Habib, A.A.; Story, M.D.; Saha, D. The small-molecule CDK inhibitor, SNS-032, enhances cellular radiosensitivity in quiescent and hypoxic non-small cell lung cancer cells. Lung Cancer 2009, 66, 37–47. [Google Scholar] [CrossRef]
- Ivanov, A.; Cragg, M.S.; Erenpreisa, J.; Emzinsh, D.; Lukman, H.; Illidge, T.M. Endopolyploid cells produced after severe genotoxic damage have the potential to repair DNA double strand breaks. J. Cell Sci. 2003, 116, 4095–4106. [Google Scholar] [CrossRef] [Green Version]
- Erenpreisa, J.; Wheatley, D. Endopolyploidy in development and cancer; “survival of the fattest?”. Cell Biology Int. 2005, 29, 981–982. [Google Scholar] [CrossRef] [Green Version]
- Mirzayans, R.; Andrais, B.; Murray, D. Roles of Polyploid/Multinucleated Giant Cancer Cells in Metastasis and Disease Relapse Following Anticancer Treatment. Cancers 2018, 10, 118. [Google Scholar] [CrossRef]
- Illidge, T. Polyploid giant cells provide a survival mechanism for p53 mutant cells after dna damage. Cell Biol. Int. 2000, 24, 621–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puig, P.-E.; Guilly, M.-N.; Bouchot, A.; Droin, N.; Cathelin, D.; Bouyer, F.; Favier, L.; Ghiringhelli, F.; Kroemer, G.; Solary, E. Tumor cells can escape DNA-damaging cisplatin through DNA endoreduplication and reversible polyploidy. Cell Biol. Int. 2008, 32, 1031–1043. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Wang, H.; Liang, Q.; Chen, L.; Ren, J. Radiotherapy for glioblastoma: Clinical issues and nanotechnology strategies. Biomater. Sci. 2022, 10, 892–908. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zong, Y.; Li, K.; Jie, X.; Hong, J.; Zhou, X.; Wu, B.; Li, Z.; Zhang, S.; Wu, G.; et al. Involvement of endothelial CK2 in the radiation induced perivascular resistant niche (PVRN) and the induction of radioresistance for non-small cell lung cancer (NSCLC) cells. Biol. Res. 2019, 52, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedmann-Morvinski, D. Glioblastoma heterogeneity and cancer cell plasticity. Crit. Rev. Oncog. 2014, 19, 327–336. [Google Scholar] [CrossRef]
- Chen, Z.; Fillmore, C.M.; Hammerman, P.S.; Kim, C.F.; Wong, K.K. Non-small-cell lung cancers: A heterogeneous set of diseases. Nat. Rev. Cancer 2014, 14, 535–546. [Google Scholar] [CrossRef]
- Yaes, R.J. Tumor heterogeneity, tumor size, and radioresistance. Int. J. Radiat. Oncol. Biol. Phys. 1989, 17, 993–1005. [Google Scholar] [CrossRef]
- Lim, Z.F.; Ma, P.C. Emerging insights of tumor heterogeneity and drug resistance mechanisms in lung cancer targeted therapy. J. Hematol. Oncol. 2019, 12, 134. [Google Scholar] [CrossRef] [Green Version]
- Soubannier, V.; Stifani, S. NF-kappaB Signalling in Glioblastoma. Biomedicines 2017, 5, 29. [Google Scholar] [CrossRef] [Green Version]
- Iwadate, Y. Epithelial-mesenchymal transition in glioblastoma progression. Oncol. Lett. 2016, 11, 1615–1620. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2017, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; Weinberg, R.A.; Marjanovic, N.D. Cell Plasticity and Heterogeneity in Cancer. Clin. Chem. 2013, 59, 168–179. [Google Scholar] [CrossRef] [Green Version]
- Nowell, P.C. The Clonal Evolution of Tumor Cell Populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Lovly, C.M.; Salama, A.K.; Salgia, R. Tumor Heterogeneity and Therapeutic Resistance. Am. Soc. Clin. Oncol. Educ. Book 2016, 35, e585–e593. [Google Scholar] [CrossRef]
- Rich, J.N. Cancer stem cells. Medicine 2016, 95, S2–S7. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Jin, D.-Y.; Leung, E.L.-H.; Fiscus, R.R.; Tung, J.W.; Tin, V.P.-C.; Cheng, L.C.; Sihoe, A.D.-L.; Fink, L.M.; Ma, Y.; Wong, M.P. Non-Small Cell Lung Cancer Cells Expressing CD44 Are Enriched for Stem Cell-Like Properties. PLoS ONE 2010, 5, e14062. [Google Scholar] [CrossRef] [Green Version]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar] [CrossRef]
- Alamgeer, M.; Peacock, C.D.; Matsui, W.; Ganju, V.; Watkins, D.N. Cancer stem cells in lung cancer: Evidence and controversies. Respirology 2013, 18, 757–764. [Google Scholar] [CrossRef] [Green Version]
- Ghisolfi, L.; Keates, A.C.; Hu, X.; Lee, D.K.; Li, C.J. Ionizing radiation induces stemness in cancer cells. PLoS ONE 2012, 7, e43628. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, M.; Luo, J.; Zhou, H. Radiotherapy targeting cancer stem cells "awakens" them to induce tumour relapse and metastasis in oral cancer. Int. J. Oral Sci. 2020, 12, 19. [Google Scholar] [CrossRef] [PubMed]
- Koury, J.; Zhong, L.; Hao, J. Targeting Signaling Pathways in Cancer Stem Cells for Cancer Treatment. Stem Cells Int. 2017, 2017, 2925869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najafi, M.; Farhood, B.; Mortezaee, K. Cancer stem cells (CSCs) in cancer progression and therapy. J. Cell. Physiol. 2018, 234, 8381–8395. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Yuan, X.; Zeng, Z.; Tunici, P.; Ng, H.; Abdulkadir, I.R.; Lu, L.; Irvin, D.; Black, K.L.; Yu, J.S. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol. Cancer 2006, 5, 67. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.M.; Hu, G.Y.; Zhao, X.Q.; Lu, T.; Zhu, F.; Yu, S.Y.; Xiong, H. Hypoxia-induced autophagy contributes to radioresistance via c-Jun-mediated Beclin1 expression in lung cancer cells. J. Huazhong Univ. Sci. Technol. Med. Sci. 2014, 34, 761–767. [Google Scholar] [CrossRef]
- Li, F.; Zhou, K.; Gao, L.; Zhang, B.; Li, W.; Yan, W.; Song, X.; Yu, H.; Wang, S.; Yu, N.; et al. Radiation induces the generation of cancer stem cells: A novel mechanism for cancer radioresistance. Oncol. Lett. 2016, 12, 3059–3065. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Pestell, T.G.; Lisanti, M.P.; Pestell, R.G. Cancer stem cells. Int. J. Biochem. Cell Biol. 2012, 44, 2144–2151. [Google Scholar] [CrossRef] [Green Version]
- Tabu, K.; Kimura, T.; Sasai, K.; Wang, L.; Bizen, N.; Nishihara, H.; Taga, T.; Tanaka, S. Analysis of an alternative human CD133 promoter reveals the implication of Ras/ERK pathway in tumor stem-like hallmarks. Mol. Cancer 2010, 9, 39. [Google Scholar] [CrossRef] [Green Version]
- Isobe, T.; Hisamori, S.; Hogan, D.J.; Zabala, M.; Hendrickson, D.G.; Dalerba, P.; Cai, S.; Scheeren, F.; Kuo, A.H.; Sikandar, S.S.; et al. miR-142 regulates the tumorigenicity of human breast cancer stem cells through the canonical WNT signaling pathway. eLife 2014, 3, e01977. [Google Scholar] [CrossRef] [Green Version]
- Shimono, Y.; Zabala, M.; Cho, R.W.; Lobo, N.; Dalerba, P.; Qian, D.; Diehn, M.; Liu, H.; Panula, S.P.; Chiao, E.; et al. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell 2009, 138, 592–603. [Google Scholar] [CrossRef]
- Huntly, B.J.; Gilliland, D.G. Leukaemia stem cells and the evolution of cancer-stem-cell research. Nat. Rev. Cancer 2005, 5, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Fiala, S. The cancer cell as a stem cell unable to differentiate. A theory of carcinogenesis. Neoplasma 1968, 15, 607–622. [Google Scholar] [PubMed]
- Clarkson, B.; Fried, J.; Strife, A.; Sakai, Y.; Ota, K.; Ohkita, T. Studies of cellular proliferation in human leukemia.III. Behavior of leukemic cells in three adults with acute leukemia given continuous infusions of3H-thymidine for 8 or 10 days. Cancer 1970, 25, 1237–1260. [Google Scholar] [CrossRef]
- Clarkson, B.D.; Dowling, M.D.; Gee, T.S.; Cunningham, I.B.; Burchenal, J.H. Treatment of acute leukemia in adults. Cancer 1975, 36, 775–795. [Google Scholar] [CrossRef]
- Hamburger, A.W.; Salmon, S.E. Primary Bioassay of Human Tumor Stem Cells. Science 1977, 197, 461–463. [Google Scholar] [CrossRef]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef]
- Lessard, J.; Sauvageau, G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature 2003, 423, 255–260. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [Green Version]
- Ailles, L.E.; Weissman, I.L. Cancer stem cells in solid tumors. Curr. Opin. Biotechnol. 2007, 18, 460–466. [Google Scholar] [CrossRef]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar]
- Matsui, W.; Huff, C.A.; Wang, Q.; Malehorn, M.T.; Barber, J.; Tanhehco, Y.; Smith, B.D.; Civin, C.I.; Jones, R.J. Characterization of clonogenic multiple myeloma cells. Blood 2004, 103, 2332–2336. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.F.; Dick, J.E.; Dirks, P.B.; Eaves, C.J.; Jamieson, C.H.; Jones, D.L.; Visvader, J.; Weissman, I.L.; Wahl, G.M. Cancer stem cells--perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006, 66, 9339–9344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.C.; Shyh-Chang, N.; Yang, H.; Rai, A.; Umashankar, S.; Ma, S.; Soh, B.S.; Sun, L.L.; Tai, B.C.; Nga, M.E.; et al. Glycine decarboxylase activity drives non-small cell lung cancer tumor-initiating cells and tumorigenesis. Cell 2012, 148, 259–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef] [Green Version]
- Collins, A.T.; Berry, P.A.; Hyde, C.; Stower, M.J.; Maitland, N.J. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005, 65, 10946–10951. [Google Scholar] [CrossRef] [Green Version]
- Aguglia, U.; Gambarelli, D.; Farnarier, G.; Quattrone, A. Different susceptibilities of the geniculate and extrageniculate visual pathways to human Creutzfeldt-Jakob disease (a combined neurophysiological-neuropathological study). Electroencephalogr. Clin. Neurophysiol. 1991, 78, 413–423. [Google Scholar] [CrossRef]
- Terris, B.; Cavard, C.; Perret, C. EpCAM, a new marker for cancer stem cells in hepatocellular carcinoma. J. Hepatol. 2010, 52, 280–281. [Google Scholar] [CrossRef] [Green Version]
- Zou, G.M. Cancer stem cells in leukemia, recent advances. J. Cell Physiol. 2007, 213, 440–444. [Google Scholar] [CrossRef]
- Boiko, A.D.; Razorenova, O.V.; van de Rijn, M.; Swetter, S.M.; Johnson, D.L.; Ly, D.P.; Butler, P.D.; Yang, G.P.; Joshua, B.; Kaplan, M.J.; et al. Human melanoma-initiating cells express neural crest nerve growth factor receptor CD271. Nature 2010, 466, 133–137. [Google Scholar] [CrossRef]
- Liang, M.H.; Robb-Nicholson, C. Health status and utility measurement viewed from the right brain: Experience from the rheumatic diseases. J. Chronic. Dis. 1987, 40, 579–583. [Google Scholar] [CrossRef]
- Organista-Nava, J.; Gomez-Gomez, Y.; Garibay-Cerdenares, O.L.; Leyva-Vazquez, M.A.; Illades-Aguiar, B. Cervical cancer stem cell-associated genes: Prognostic implications in cervical cancer. Oncol. Lett. 2019, 18, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Curley, M.D.; Therrien, V.A.; Cummings, C.L.; Sergent, P.A.; Koulouris, C.R.; Friel, A.M.; Roberts, D.J.; Seiden, M.V.; Scadden, D.T.; Rueda, B.R.; et al. CD133 expression defines a tumor initiating cell population in primary human ovarian cancer. Stem Cells 2009, 27, 2875–2883. [Google Scholar] [CrossRef] [PubMed]
- Takaishi, S.; Okumura, T.; Tu, S.; Wang, S.S.; Shibata, W.; Vigneshwaran, R.; Gordon, S.A.; Shimada, Y.; Wang, T.C. Identification of gastric cancer stem cells using the cell surface marker CD44. Stem Cells 2009, 27, 1006–1020. [Google Scholar] [CrossRef] [Green Version]
- Eramo, A.; Lotti, F.; Sette, G.; Pilozzi, E.; Biffoni, M.; Di Virgilio, A.; Conticello, C.; Ruco, L.; Peschle, C.; De Maria, R. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2007, 15, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Dubrovska, A. Report on the International Workshop ‘Cancer stem cells: The mechanisms of radioresistance and biomarker discovery’. Int. J. Radiat. Biol. 2014, 90, 607–614. [Google Scholar] [CrossRef]
- Peitzsch, C.; Nathansen, J.; Schniewind, S.I.; Schwarz, F.; Dubrovska, A. Cancer Stem Cells in Head and Neck Squamous Cell Carcinoma: Identification, Characterization and Clinical Implications. Cancers 2019, 11, 616. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, C.A.; Ross, A.H. Cancer stem cells: Cell culture, markers, and targets for new therapies. J. Cell Biochem. 2009, 108, 1031–1038. [Google Scholar] [CrossRef] [Green Version]
- Vlashi, E.; Pajonk, F. The metabolic state of cancer stem cells—A valid target for cancer therapy? Free Radic. Biol. Med. 2015, 79, 264–268. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [Green Version]
- Prieto-Vila, M.; Takahashi, R.U.; Usuba, W.; Kohama, I.; Ochiya, T. Drug Resistance Driven by Cancer Stem Cells and Their Niche. Int. J. Mol. Sci. 2017, 18, 2574. [Google Scholar] [CrossRef] [Green Version]
- Rycaj, K.; Tang, D.G. Cancer stem cells and radioresistance. Int. J. Radiat. Biol. 2014, 90, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Eyler, C.E.; Rich, J.N. Survival of the fittest: Cancer stem cells in therapeutic resistance and angiogenesis. J. Clin. Oncol. 2008, 26, 2839–2845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borah, A.; Raveendran, S.; Rochani, A.; Maekawa, T.; Kumar, D.S. Targeting self-renewal pathways in cancer stem cells: Clinical implications for cancer therapy. Oncogenesis 2015, 4, e177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Atkinson, R.L.; Rosen, J.M. Selective targeting of radiation-resistant tumor-initiating cells. Proc. Natl. Acad. Sci. USA 2010, 107, 3522–3527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talukdar, S.; Pradhan, A.K.; Bhoopathi, P.; Shen, X.-N.; August, L.A.; Windle, J.J.; Sarkar, D.; Furnari, F.B.; Cavenee, W.K.; Das, S.K.; et al. MDA-9/Syntenin regulates protective autophagy in anoikis-resistant glioma stem cells. Proc. Natl. Acad. Sci. USA 2018, 115, 5768–5773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talukdar, S.; Emdad, L.; Das, S.K.; Sarkar, D.; Fisher, P.B. Evolving Strategies for Therapeutically Targeting Cancer Stem Cells. Adv. Cancer Res. 2016, 131, 159–191. [Google Scholar]
- Lyakhovich, A.; Lleonart, M.E. Bypassing Mechanisms of Mitochondria-Mediated Cancer Stem Cells Resistance to Chemo- and Radiotherapy. Oxidative Med. Cell. Longev. 2016, 2016, 1716341. [Google Scholar] [CrossRef] [Green Version]
- Kurth, I.; Hein, L.; Mabert, K.; Peitzsch, C.; Koi, L.; Cojoc, M.; Kunz-Schughart, L.; Baumann, M.; Dubrovska, A. Cancer stem cell related markers of radioresistance in head and neck squamous cell carcinoma. Oncotarget 2015, 6, 34494–34509. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.; Graham, P.; Hao, J.; Ni, J.; Deng, J.; Bucci, J.; Malouf, D.; Gillatt, D.; Li, Y. Cancer stem cells and signaling pathways in radioresistance. Oncotarget 2016, 7, 11002–11017. [Google Scholar] [CrossRef] [Green Version]
- Cojoc, M.; Mabert, K.; Muders, M.H.; Dubrovska, A. A role for cancer stem cells in therapy resistance: Cellular and molecular mechanisms. Semin. Cancer Biol. 2015, 31, 16–27. [Google Scholar] [CrossRef]
- Shin, M.K.; Cheong, J.H. Mitochondria-centric bioenergetic characteristics in cancer stem-like cells. Arch. Pharm. Res. 2019, 42, 113–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dayem, A.A.; Choi, H.Y.; Kim, J.H.; Cho, S.G. Role of oxidative stress in stem, cancer, and cancer stem cells. Cancers 2010, 2, 859–884. [Google Scholar] [CrossRef] [PubMed]
- Dando, I.; Cordani, M.; Dalla Pozza, E.; Biondani, G.; Donadelli, M.; Palmieri, M. Antioxidant Mechanisms and ROS-Related MicroRNAs in Cancer Stem Cells. Oxid. Med. Cell Longev. 2015, 2015, 425708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef] [Green Version]
- Mondal, S.; Bhattacharya, K.; Mandal, C. Nutritional stress reprograms dedifferention in glioblastoma multiforme driven by PTEN/Wnt/Hedgehog axis: A stochastic model of cancer stem cells. Cell Death Discov. 2018, 4, 110. [Google Scholar] [CrossRef] [Green Version]
- Szotek, P.P.; Pieretti-Vanmarcke, R.; Masiakos, P.T.; Dinulescu, D.M.; Connolly, D.; Foster, R.; Dombkowski, D.; Preffer, F.; Maclaughlin, D.T.; Donahoe, P.K. Ovarian cancer side population defines cells with stem cell-like characteristics and Mullerian Inhibiting Substance responsiveness. Proc. Natl. Acad. Sci. USA 2006, 103, 11154–11159. [Google Scholar] [CrossRef] [Green Version]
- Müller, E.; Ansorge, M.; Werner, C.; Pompe, T. Mimicking the Hematopoietic Stem Cell Niche by Biomaterials. In Bio-Inspired Materials for Biomedical Engineering; John Wiley & Sons: Hoboken, NJ, USA, 2014; pp. 309–326. [Google Scholar]
- Bissell, M.J.; Labarge, M.A. Context, tissue plasticity, and cancer: Are tumor stem cells also regulated by the microenvironment? Cancer Cell 2005, 7, 17–23. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Bao, S.; Wu, Q.; Wang, H.; Eyler, C.; Sathornsumetee, S.; Shi, Q.; Cao, Y.; Lathia, J.; McLendon, R.E.; et al. Hypoxia-Inducible Factors Regulate Tumorigenic Capacity of Glioma Stem Cells. Cancer Cell 2009, 15, 501–513. [Google Scholar] [CrossRef] [Green Version]
- Korkaya, H.; Liu, S.; Wicha, M.S. Regulation of cancer stem cells by cytokine networks: Attacking cancer’s inflammatory roots. Clin. Cancer Res. 2011, 17, 6125–6129. [Google Scholar] [CrossRef] [Green Version]
- Fukumura, D.; Xu, L.; Chen, Y.; Gohongi, T.; Seed, B.; Jain, R.K. Hypoxia and acidosis independently up-regulate vascular endothelial growth factor transcription in brain tumors in vivo. Cancer Res. 2001, 61, 6020–6024. [Google Scholar]
- Hjelmeland, A.B.; Wu, Q.; Heddleston, J.M.; Choudhary, G.S.; MacSwords, J.; Lathia, J.D.; McLendon, R.; Lindner, D.; Sloan, A.; Rich, J.N. Acidic stress promotes a glioma stem cell phenotype. Cell Death Differ. 2010, 18, 829–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Xiao, Z.; Wong, S.K.; Tin, V.P.; Ho, K.Y.; Wang, J.; Sham, M.H.; Wong, M.P. Lung cancer tumorigenicity and drug resistance are maintained through ALDH(hi)CD44(hi) tumor initiating cells. Oncotarget 2013, 4, 1698–1711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.J.; Li, C.; Simeone, D.M. Human Pancreatic Cancer Stem Cells: Implications for How We Treat Pancreatic Cancer. Transl. Oncol. 2008, 1, 14–18. [Google Scholar] [CrossRef]
- Vail, D.M. (Ed.) Withrow and MacEwen’s Small Animal Clinical Oncology; Elsevier: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Ho, M.M.; Ng, A.V.; Lam, S.; Hung, J.Y. Side population in human lung cancer cell lines and tumors is enriched with stem-like cancer cells. Cancer Res. 2007, 67, 4827–4833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, W.T.; Ryu, C.J. Cancer stem cell surface markers on normal stem cells. BMB Rep. 2017, 50, 285–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, D.; Roth, I.; Wickremesekera, A.; Davis, P.; Kaye, A.; Mantamadiotis, T.; Stylli, S.; Tan, S. Therapeutic Targeting of Cancer Stem Cells in Human Glioblastoma by Manipulating the Renin-Angiotensin System. Cells 2019, 8, 1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Zhao, W.; Li, Y. Stemness-related markers in cancer. Cancer Transl. Med. 2017, 3, 87–95. [Google Scholar] [CrossRef] [Green Version]
- Klonisch, T.; Wiechec, E.; Hombach-Klonisch, S.; Ande, S.R.; Wesselborg, S.; Schulze-Osthoff, K.; Los, M. Cancer stem cell markers in common cancers—Therapeutic implications. Trends Mol. Med. 2008, 14, 450–460. [Google Scholar] [CrossRef] [Green Version]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Shackleton, M.; Vaillant, F.; Simpson, K.J.; Stingl, J.; Smyth, G.K.; Asselin-Labat, M.-L.; Wu, L.; Lindeman, G.J.; Visvader, J.E. Generation of a functional mammary gland from a single stem cell. Nature 2006, 439, 84–88. [Google Scholar] [CrossRef]
- Redmer, T.; Walz, I.; Klinger, B.; Khouja, S.; Welte, Y.; Schäfer, R.; Regenbrecht, C. The role of the cancer stem cell marker CD271 in DNA damage response and drug resistance of melanoma cells. Oncogenesis 2017, 6, e291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lupia, M.; Angiolini, F.; Bertalot, G.; Freddi, S.; Sachsenmeier, K.F.; Chisci, E.; Kutryb-Zajac, B.; Confalonieri, S.; Smolenski, R.T.; Giovannoni, R.; et al. CD73 Regulates Stemness and Epithelial-Mesenchymal Transition in Ovarian Cancer-Initiating Cells. Stem Cell Rep. 2018, 10, 1412–1425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taddei, A.; Giampietro, C.; Conti, A.; Orsenigo, F.; Breviario, F.; Pirazzoli, V.; Potente, M.; Daly, C.; Dimmeler, S.; Dejana, E. Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat. Cell Biol. 2008, 10, 923–934. [Google Scholar] [CrossRef] [PubMed]
- Medema, J.P. Cancer stem cells: The challenges ahead. Nat. Cell Biol. 2013, 15, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Begicevic, R.-R.; Falasca, M. ABC Transporters in Cancer Stem Cells: Beyond Chemoresistance. Int. J. Mol. Sci. 2017, 18, 2362. [Google Scholar] [CrossRef] [Green Version]
- Padmanabhan, R.; Chen, K.G.; Gottesman, M.M. Lost in Translation: Regulation of ABCG2 Expression in Human Embryonic Stem Cells. J. Stem Cell Res. 2014, 4, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markovsky, E.; Vax, E.; Ben-Shushan, D.; Eldar-Boock, A.; Shukrun, R.; Yeini, E.; Barshack, I.; Caspi, R.; Harari-Steinberg, O.; Pode-Shakked, N.; et al. Wilms Tumor NCAM-Expressing Cancer Stem Cells as Potential Therapeutic Target for Polymeric Nanomedicine. Mol. Cancer Ther. 2017, 16, 2462–2472. [Google Scholar] [CrossRef] [Green Version]
- Pustovalova, M.; Blokhina, T.; Alhaddad, L.; Chigasova, A.; Chuprov-Netochin, R.; Veviorskiy, A.; Filkov, G.; Osipov, A.N.; Leonov, S. CD44+ and CD133+ Non-Small Cell Lung Cancer Cells Exhibit DNA Damage Response Pathways and Dormant Polyploid Giant Cancer Cell Enrichment Relating to Their p53 Status. Int. J. Mol. Sci. 2022, 23, 4922. [Google Scholar] [CrossRef]
- Roy, S.K.; Shrivastava, A.; Srivastav, S.; Shankar, S.; Srivastava, R.K. SATB2 is a novel biomarker and therapeutic target for cancer. J. Cell. Mol. Med. 2020, 24, 11064–11069. [Google Scholar] [CrossRef]
- Bao, B.; Wang, Z.; Ali, S.; Kong, D.; Banerjee, S.; Ahmad, A.; Li, Y.; Azmi, A.S.; Miele, L.; Sarkar, F.H. Over-expression of FoxM1 leads to epithelial-mesenchymal transition and cancer stem cell phenotype in pancreatic cancer cells. J. Cell. Biochem. 2011, 112, 2296–2306. [Google Scholar] [CrossRef] [Green Version]
- Qi, Y.; Wei, J.; Zhang, X. Requirement of transcription factor NME2 for the maintenance of the stemness of gastric cancer stem-like cells. Cell Death Dis. 2021, 12, 924. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Han, Z.; Zhu, Y.; Chen, J.; Li, W. Role of hypoxia inducible factor-1 in cancer stem cells (Review). Mol. Med. Rep. 2021, 23, 17. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Yu, X.; Liu, S. Pluripotency transcription factors and cancer stem cells: Small genes make a big difference. Chin. J. Cancer 2013. [Google Scholar] [CrossRef] [PubMed]
- Pandit, H.; Li, Y.; Li, X.; Zhang, W.; Li, S.; Martin, R.C.G. Enrichment of cancer stem cells via beta-catenin contributing to the tumorigenesis of hepatocellular carcinoma. BMC Cancer 2018, 18, 783. [Google Scholar] [CrossRef] [PubMed]
- Wurdak, H.; Zhu, S.; Romero, A.; Lorger, M.; Watson, J.; Chiang, C.Y.; Zhang, J.; Natu, V.S.; Lairson, L.L.; Walker, J.R.; et al. An RNAi screen identifies TRRAP as a regulator of brain tumor-initiating cell differentiation. Cell Stem Cell 2010, 6, 37–47. [Google Scholar] [CrossRef] [Green Version]
- Hao, J.; Zhang, Y.; Jing, D.; Li, Y.; Li, J.; Zhao, Z. Role of Hippo Signaling in Cancer Stem Cells. J. Cell. Physiol. 2014, 229, 266–270. [Google Scholar] [CrossRef]
- Safa, A.R. Resistance to Cell Death and Its Modulation in Cancer Stem Cells. Crit. Rev. Oncog. 2016, 21, 203–219. [Google Scholar] [CrossRef] [Green Version]
- Hong, M.; Tan, H.; Li, S.; Cheung, F.; Wang, N.; Nagamatsu, T.; Feng, Y. Cancer Stem Cells: The Potential Targets of Chinese Medicines and Their Active Compounds. Int. J. Mol. Sci. 2016, 17, 893. [Google Scholar] [CrossRef] [Green Version]
- Zakaria, N.; Mohd Yusoff, N.; Zakaria, Z.; Widera, D.; Yahaya, B.H. Inhibition of NF-kappaB Signaling Reduces the Stemness Characteristics of Lung Cancer Stem Cells. Front. Oncol. 2018, 8, 166. [Google Scholar] [CrossRef] [Green Version]
- Kelly, P.N.; Strasser, A. The role of Bcl-2 and its pro-survival relatives in tumourigenesis and cancer therapy. Cell Death Differ. 2011, 18, 1414–1424. [Google Scholar] [CrossRef] [Green Version]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells: Current status and evolving complexities. Cell Stem Cell 2012, 10, 717–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badve, S.; Nakshatri, H. Breast-cancer stem cells-beyond semantics. Lancet Oncol. 2012, 13, e43–e48. [Google Scholar] [CrossRef]
- Liu, S.; Cong, Y.; Wang, D.; Sun, Y.; Deng, L.; Liu, Y.; Martin-Trevino, R.; Shang, L.; McDermott, S.P.; Landis, M.D.; et al. Breast Cancer Stem Cells Transition between Epithelial and Mesenchymal States Reflective of their Normal Counterparts. Stem Cell Rep. 2014, 2, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Aghajani, M.; Mansoori, B.; Mohammadi, A.; Asadzadeh, Z.; Baradaran, B. New emerging roles of CD133 in cancer stem cell: Signaling pathway and miRNA regulation. J. Cell. Physiol. 2019, 234, 21642–21661. [Google Scholar] [CrossRef]
- Desai, A.; Webb, B.; Gerson, S.L. CD133+ cells contribute to radioresistance via altered regulation of DNA repair genes in human lung cancer cells. Radiother. Oncol. 2014, 110, 538–545. [Google Scholar] [CrossRef] [Green Version]
- Levina, V.; Marrangoni, A.; Wang, T.; Parikh, S.; Su, Y.; Herberman, R.; Lokshin, A.; Gorelik, E. Elimination of Human Lung Cancer Stem Cells through Targeting of the Stem Cell Factor–c-kit Autocrine Signaling Loop. Cancer Res. 2010, 70, 338–346. [Google Scholar] [CrossRef] [Green Version]
- Saya, H. MS 26.01 Therapeutic Strategies Targeting Cancer Stem Cells. J. Thorac. Oncol. 2017, 12, S1725–S1726. [Google Scholar] [CrossRef]
- Tsubouchi, K.; Minami, K.; Hayashi, N.; Yokoyama, Y.; Mori, S.; Yamamoto, H.; Koizumi, M. The CD44 standard isoform contributes to radioresistance of pancreatic cancer cells. J. Radiat. Res. 2017, 58, 816–826. [Google Scholar] [CrossRef] [Green Version]
- Kaidi, A.; Williams, A.C.; Paraskeva, C. Interaction between β-catenin and HIF-1 promotes cellular adaptation to hypoxia. Nat. Cell Biol. 2007, 9, 210–217. [Google Scholar] [CrossRef]
- Moeller, B.J.; Dreher, M.R.; Rabbani, Z.N.; Schroeder, T.; Cao, Y.; Li, C.Y.; Dewhirst, M.W. Pleiotropic effects of HIF-1 blockade on tumor radiosensitivity. Cancer Cell 2005, 8, 99–110. [Google Scholar] [CrossRef] [Green Version]
- Dimri, G.P.; Martinez, J.L.; Jacobs, J.J.; Keblusek, P.; Itahana, K.; Van Lohuizen, M.; Campisi, J.; Wazer, D.E.; Band, V. The Bmi-1 oncogene induces telomerase activity and immortalizes human mammary epithelial cells. Cancer Res. 2002, 62, 4736–4745. [Google Scholar] [PubMed]
- Tao, W.; Zhang, A.; Zhai, K.; Huang, Z.; Huang, H.; Zhou, W.; Huang, Q.; Fang, X.; Prager, B.C.; Wang, X.; et al. SATB2 drives glioblastoma growth by recruiting CBP to promote FOXM1 expression in glioma stem cells. EMBO Mol. Med. 2020, 12, e12291. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Copsel, S.; Garcia, C.; Diez, F.; Vermeulem, M.; Baldi, A.; Bianciotti, L.G.; Russel, F.G.; Shayo, C.; Davio, C. Multidrug resistance protein 4 (MRP4/ABCC4) regulates cAMP cellular levels and controls human leukemia cell proliferation and differentiation. J. Biol. Chem. 2011, 286, 6979–6988. [Google Scholar] [CrossRef] [Green Version]
- Glavinas, H.; Krajcsi, P.; Cserepes, J.; Sarkadi, B. The role of ABC transporters in drug resistance, metabolism and toxicity. Curr. Drug Deliv. 2004, 1, 27–42. [Google Scholar] [CrossRef]
- Bleau, A.-M.; Hambardzumyan, D.; Ozawa, T.; Fomchenko, E.I.; Huse, J.T.; Brennan, C.W.; Holland, E.C. PTEN/PI3K/Akt Pathway Regulates the Side Population Phenotype and ABCG2 Activity in Glioma Tumor Stem-like Cells. Cell Stem Cell 2009, 4, 226–235. [Google Scholar] [CrossRef] [Green Version]
- Moitra, K. Overcoming Multidrug Resistance in Cancer Stem Cells. BioMed Res. Int. 2015, 2015, 635745. [Google Scholar] [CrossRef] [Green Version]
- Pustovalova, M.; Alhaddad, L.; Smetanina, N.; Chigasova, A.; Blokhina, T.; Chuprov-Netochin, R.; Osipov, A.N.; Leonov, S. The p53-53BP1-Related Survival of A549 and H1299 Human Lung Cancer Cells after Multifractionated Radiotherapy Demonstrated Different Response to Additional Acute X-ray Exposure. Int. J. Mol. Sci. 2020, 21, 3342. [Google Scholar] [CrossRef]
- Castellan, M.; Guarnieri, A.; Fujimura, A.; Zanconato, F.; Battilana, G.; Panciera, T.; Sladitschek, H.L.; Contessotto, P.; Citron, A.; Grilli, A.; et al. Single-cell analyses reveal YAP/TAZ as regulators of stemness and cell plasticity in glioblastoma. Nat. Cancer 2020, 2, 174–188. [Google Scholar] [CrossRef]
- Benham-Pyle, B.W.; Pruitt, B.L.; Nelson, W.J. Cell adhesion. Mechanical strain induces E-cadherin-dependent Yap1 and beta-catenin activation to drive cell cycle entry. Science 2015, 348, 1024–1027. [Google Scholar] [CrossRef] [Green Version]
- Shreberk-Shaked, M.; Dassa, B.; Sinha, S.; Di Agostino, S.; Azuri, I.; Mukherjee, S.; Aylon, Y.; Blandino, G.; Ruppin, E.; Oren, M. A Division of Labor between YAP and TAZ in Non-Small Cell Lung Cancer. Cancer Res. 2020, 80, 4145–4157. [Google Scholar] [CrossRef] [PubMed]
- Overholtzer, M.; Zhang, J.; Smolen, G.A.; Muir, B.; Li, W.; Sgroi, D.C.; Deng, C.X.; Brugge, J.S.; Haber, D.A. Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc. Natl. Acad. Sci. USA 2006, 103, 12405–12410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neradil, J.; Veselska, R. Nestin as a marker of cancer stem cells. Cancer Sci. 2015, 106, 803–811. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.A.; Kreso, A.; Jamieson, C.H. Cancer stem cells and self-renewal. Clin. Cancer Res. 2010, 16, 3113–3120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stahl, S.; Fung, E.; Adams, C.; Lengqvist, J.; Mork, B.; Stenerlow, B.; Lewensohn, R.; Lehtio, J.; Zubarev, R.; Viktorsson, K. Proteomics and pathway analysis identifies JNK signaling as critical for high linear energy transfer radiation-induced apoptosis in non-small lung cancer cells. Mol. Cell Proteom. 2009, 8, 1117–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct. Target. Ther. 2020, 5, 8. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.Q.; Ahmed, E.I.; Elareer, N.R.; Junejo, K.; Steinhoff, M.; Uddin, S. Role of miRNA-Regulated Cancer Stem Cells in the Pathogenesis of Human Malignancies. Cells 2019, 8, 840. [Google Scholar] [CrossRef] [Green Version]
- Correnti, M.; Booijink, R.; Di Maira, G.; Raggi, C.; Marra, F. Stemness features in liver cancer. Hepatoma Res. 2018, 4, 69. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.W.; Kim, J.Y.; Qiao, J.; Clark, R.A.; Powers, C.M.; Correa, H.; Chung, D.H. Dual-Targeting AKT2 and ERK in cancer stem-like cells in neuroblastoma. Oncotarget 2019, 10, 5645–5659. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wakeman, T.P.; Lathia, J.D.; Hjelmeland, A.B.; Wang, X.F.; White, R.R.; Rich, J.N.; Sullenger, B.A. Notch promotes radioresistance of glioma stem cells. Stem Cells 2010, 28, 17–28. [Google Scholar] [CrossRef] [Green Version]
- Stewart, D.J. Wnt signaling pathway in non-small cell lung cancer. J. Natl. Cancer Inst. 2014, 106, djt356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsui, W.H. Cancer stem cell signaling pathways. Medicine 2016, 95, S8–S19. [Google Scholar] [CrossRef] [PubMed]
- Schreck, K.C.; Taylor, P.; Marchionni, L.; Gopalakrishnan, V.; Bar, E.E.; Gaiano, N.; Eberhart, C.G. The Notch target Hes1 directly modulates Gli1 expression and Hedgehog signaling: A potential mechanism of therapeutic resistance. Clin. Cancer Res. 2010, 16, 6060–6070. [Google Scholar] [CrossRef] [PubMed]
- Vlashi, E.; Pajonk, F. Cancer stem cells, cancer cell plasticity and radiation therapy. Semin. Cancer Biol. 2015, 31, 28–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Zhou, H.; Zhang, G.; Xue, X. Targeting the canonical Wnt/beta-catenin pathway in cancer radioresistance: Updates on the molecular mechanisms. J. Cancer Res. Ther. 2019, 15, 272–277. [Google Scholar] [CrossRef]
- Liu, C.; Wang, R. The Roles of Hedgehog Signaling Pathway in Radioresistance of Cervical Cancer. Dose-Response 2019, 17, 1559325819885293. [Google Scholar] [CrossRef] [Green Version]
- Kim, R.K.; Kaushik, N.; Suh, Y.; Yoo, K.C.; Cui, Y.H.; Kim, M.J.; Lee, H.J.; Kim, I.G.; Lee, S.J. Radiation driven epithelial-mesenchymal transition is mediated by Notch signaling in breast cancer. Oncotarget 2016, 7, 53430–53442. [Google Scholar] [CrossRef] [Green Version]
- Kong, D.; Banerjee, S.; Ahmad, A.; Li, Y.; Wang, Z.; Sethi, S.; Sarkar, F.H. Epithelial to mesenchymal transition is mechanistically linked with stem cell signatures in prostate cancer cells. PLoS ONE 2010, 5, e12445. [Google Scholar] [CrossRef]
- Hassan, K.A.; Wang, L.; Korkaya, H.; Chen, G.; Maillard, I.; Beer, D.G.; Kalemkerian, G.P.; Wicha, M.S. Notch pathway activity identifies cells with cancer stem cell-like properties and correlates with worse survival in lung adenocarcinoma. Clin. Cancer Res. 2013, 19, 1972–1980. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Shin, J.E.; Park, H.W. The Role of Hippo Pathway in Cancer Stem Cell Biology. Mol. Cells 2018, 41, 83–92. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Y.; Zhou, D.; Wang, K.; Wang, X.; Wang, X.; Jiang, Y.; Zhao, M.; Yu, R.; Zhou, X. Radiation-induced YAP activation confers glioma radioresistance via promoting FGF2 transcription and DNA damage repair. Oncogene 2021, 40, 4580–4591. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Zhao, Y.; Du, Y.; Tang, R. Evaluation of Hippo Pathway and CD133 in Radiation Resistance in Small-Cell Lung Cancer. J. Oncol. 2021, 2021, 8842554. [Google Scholar] [CrossRef] [PubMed]
- Krause, M.; Dubrovska, A.; Linge, A.; Baumann, M. Cancer stem cells: Radioresistance, prediction of radiotherapy outcome and specific targets for combined treatments. Adv. Drug Deliv. Rev. 2017, 109, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Maugeri-Saccà, M.; Bartucci, M.; De Maria, R. DNA Damage Repair Pathways in Cancer Stem Cells. Mol. Cancer Ther. 2012, 11, 1627–1636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eriksson, D.; Stigbrand, T. Radiation-induced cell death mechanisms. Tumor Biol. 2010, 31, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Babayan, N.; Vorobyeva, N.; Grigoryan, B.; Grekhova, A.; Pustovalova, M.; Rodneva, S.; Fedotov, Y.; Tsakanova, G.; Aroutiounian, R.; Osipov, A. Low Repair Capacity of DNA Double-Strand Breaks Induced by Laser-Driven Ultrashort Electron Beams in Cancer Cells. Int. J. Mol. Sci. 2020, 21, 9488. [Google Scholar] [CrossRef] [PubMed]
- Mladenov, E.; Magin, S.; Soni, A.; Iliakis, G. DNA Double-Strand Break Repair as Determinant of Cellular Radiosensitivity to Killing and Target in Radiation Therapy. Front. Oncol. 2013, 3, 113. [Google Scholar] [CrossRef] [Green Version]
- Bushmanov, A.; Vorobyeva, N.; Molodtsova, D.; Osipov, A.N. Utilization of DNA double-strand breaks for biodosimetry of ionizing radiation exposure. Environ. Adv. 2022, 8, 100207. [Google Scholar] [CrossRef]
- Biau, J.; Chautard, E.; Berthault, N.; de Koning, L.; Court, F.; Pereira, B.; Verrelle, P.; Dutreix, M. Combining the DNA Repair Inhibitor Dbait With Radiotherapy for the Treatment of High Grade Glioma: Efficacy and Protein Biomarkers of Resistance in Preclinical Models. Front. Oncol. 2019, 9, 549. [Google Scholar] [CrossRef]
- Wyman, C.; Kanaar, R. DNA double-strand break repair: All’s well that ends well. Annu. Rev. Genet. 2006, 40, 363–383. [Google Scholar] [CrossRef]
- Morgan, M.A.; Lawrence, T.S. Molecular Pathways: Overcoming Radiation Resistance by Targeting DNA Damage Response Pathways. Clin. Cancer Res. 2015, 21, 2898–2904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biau, J.; Chautard, E.; Verrelle, P.; Dutreix, M. Altering DNA Repair to Improve Radiation Therapy: Specific and Multiple Pathway Targeting. Front. Oncol. 2019, 9, 1009. [Google Scholar] [CrossRef] [PubMed]
- Ulyanenko, S.; Pustovalova, M.; Koryakin, S.; Beketov, E.; Lychagin, A.; Ulyanenko, L.; Kaprin, A.; Grekhova, A.; Ozerova, A.M.; Ozerov, I.V.; et al. Formation of gammaH2AX and pATM Foci in Human Mesenchymal Stem Cells Exposed to Low Dose-Rate Gamma-Radiation. Int. J. Mol. Sci. 2019, 20, 2645. [Google Scholar] [CrossRef] [PubMed]
- Britton, S.; Coates, J.; Jackson, S.P. A new method for high-resolution imaging of Ku foci to decipher mechanisms of DNA double-strand break repair. J. Cell Biol. 2013, 202, 579–595. [Google Scholar] [CrossRef] [Green Version]
- Park, E.J.; Chan, D.W.; Park, J.H.; Oettinger, M.A.; Kwon, J. DNA-PK is activated by nucleosomes and phosphorylates H2AX within the nucleosomes in an acetylation-dependent manner. Nucleic Acids Res. 2003, 31, 6819–6827. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Pannicke, U.; Schwarz, K.; Lieber, M.R. Hairpin Opening and Overhang Processing by an Artemis/DNA-Dependent Protein Kinase Complex in Nonhomologous End Joining and V(D)J Recombination. Cell 2002, 108, 781–794. [Google Scholar] [CrossRef] [Green Version]
- Nick McElhinny, S.A.; Snowden, C.M.; McCarville, J.; Ramsden, D.A. Ku Recruits the XRCC4-Ligase IV Complex to DNA Ends. Mol. Cell. Biol. 2000, 20, 2996–3003. [Google Scholar] [CrossRef] [Green Version]
- Ahnesorg, P.; Smith, P.; Jackson, S.P. XLF Interacts with the XRCC4-DNA Ligase IV Complex to Promote DNA Nonhomologous End-Joining. Cell 2006, 124, 301–313. [Google Scholar] [CrossRef] [Green Version]
- Belyaev, I.Y. Radiation-induced DNA repair foci: Spatio-temporal aspects of formation, application for assessment of radiosensitivity and biological dosimetry. Mutat. Res./Rev. Mutat. Res. 2010, 704, 132–141. [Google Scholar] [CrossRef]
- Tsvetkova, A.; Ozerov, I.V.; Pustovalova, M.; Grekhova, A.; Eremin, P.; Vorobyeva, N.; Eremin, I.; Pulin, A.; Zorin, V.; Kopnin, P.; et al. γH2AX, 53BP1 and Rad51 protein foci changes in mesenchymal stem cells during prolonged X-ray irradiation. Oncotarget 2017, 8, 64317–64329. [Google Scholar] [CrossRef] [Green Version]
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle 2014, 7, 2902–2906. [Google Scholar] [CrossRef] [PubMed]
- Kakarougkas, A.; Jeggo, P.A. DNA DSB repair pathway choice: An orchestrated handover mechanism. Br. J. Radiol. 2014, 87, 20130685. [Google Scholar] [CrossRef] [PubMed]
- Safa, A.R.; Saadatzadeh, M.R.; Cohen-Gadol, A.A.; Pollok, K.E.; Bijangi-Vishehsaraei, K. Glioblastoma stem cells (GSCs) epigenetic plasticity and interconversion between differentiated non-GSCs and GSCs. Genes Dis. 2015, 2, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Vlashi, E.; Lagadec, C.; Vergnes, L.; Matsutani, T.; Masui, K.; Poulou, M.; Popescu, R.; Della Donna, L.; Evers, P.; Dekmezian, C.; et al. Metabolic state of glioma stem cells and nontumorigenic cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16062–16067. [Google Scholar] [CrossRef] [Green Version]
- Ogden, A.T.; Waziri, A.E.; Lochhead, R.A.; Fusco, D.; Lopez, K.; Ellis, J.A.; Kang, J.; Assanah, M.; McKhann, G.M.; Sisti, M.B.; et al. Identification of A2b5+Cd133− Tumor-Initiating Cells in Adult Human Gliomas. Neurosurgery 2008, 62, 505–515. [Google Scholar] [CrossRef] [Green Version]
- Aum, D.J.; Kim, D.H.; Beaumont, T.L.; Leuthardt, E.C.; Dunn, G.P.; Kim, A.H. Molecular and cellular heterogeneity: The hallmark of glioblastoma. Neurosurg. Focus 2014, 37, E11. [Google Scholar] [CrossRef] [Green Version]
- Nakano, I. Stem cell signature in glioblastoma: Therapeutic development for a moving target. J. Neurosurg. 2015, 122, 324–330. [Google Scholar] [CrossRef] [Green Version]
- Jackson, E.L.; Alvarez-Buylla, A. Characterization of adult neural stem cells and their relation to brain tumors. Cells Tissues Organs 2008, 188, 212–224. [Google Scholar] [CrossRef]
- Zhu, Y.; Guignard, F.; Zhao, D.; Liu, L.; Burns, D.K.; Mason, R.P.; Messing, A.; Parada, L.F. Early inactivation of p53 tumor suppressor gene cooperating with NF1 loss induces malignant astrocytoma. Cancer Cell 2005, 8, 119–130. [Google Scholar] [CrossRef] [Green Version]
- Kondo, T.; Setoguchi, T.; Taga, T. Persistence of a small subpopulation of cancer stem-like cells in the C6 glioma cell line. Proc. Natl. Acad. Sci. USA 2004, 101, 781–786. [Google Scholar] [CrossRef] [Green Version]
- Goodell, M.A.; Brose, K.; Paradis, G.; Conner, A.S.; Mulligan, R.C. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J. Exp. Med. 1996, 183, 1797–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.C.; Roberts, T.L.; Day, B.W.; Harding, A.; Kozlov, S.; Kijas, A.W.; Ensbey, K.S.; Walker, D.G.; Lavin, M.F. A role for homologous recombination and abnormal cell-cycle progression in radioresistance of glioma-initiating cells. Mol. Cancer Ther. 2012, 11, 1863–1872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, M.J.; Woolard, K.; Nam, D.H.; Lee, J.; Fine, H.A. SSEA-1 is an enrichment marker for tumor-initiating cells in human glioblastoma. Cell Stem Cell 2009, 4, 440–452. [Google Scholar] [CrossRef] [Green Version]
- Patru, C.; Romao, L.; Varlet, P.; Coulombel, L.; Raponi, E.; Cadusseau, J.; Renault-Mihara, F.; Thirant, C.; Leonard, N.; Berhneim, A.; et al. CD133, CD15/SSEA-1, CD34 or side populations do not resume tumor-initiating properties of long-term cultured cancer stem cells from human malignant glio-neuronal tumors. BMC Cancer 2010, 10, 66. [Google Scholar] [CrossRef]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Huang, Z.; Cheng, L.; Guryanova, O.A.; Wu, Q.; Bao, S. Cancer stem cells in glioblastoma--molecular signaling and therapeutic targeting. Protein Cell 2010, 1, 638–655. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.G.; Zhang, X.; Xue, X.Y.; Guo, G.; Wang, P.; Zhang, W.; Fei, Z.; Zhen, H.N.; You, S.W.; Yang, H. Brain Tumor Stem-Like Cells Identified by Neural Stem Cell Marker CD15. Transl. Oncol. 2009, 2, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Jijiwa, M.; Demir, H.; Gupta, S.; Leung, C.; Joshi, K.; Orozco, N.; Huang, T.; Yildiz, V.O.; Shibahara, I.; de Jesus, J.A.; et al. CD44v6 regulates growth of brain tumor stem cells partially through the AKT-mediated pathway. PLoS ONE 2011, 6, e24217. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; Li, Z.; Sathornsumetee, S.; Wang, H.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. Targeting cancer stem cells through L1CAM suppresses glioma growth. Cancer Res. 2008, 68, 6043–6048. [Google Scholar] [CrossRef] [Green Version]
- Lathia, J.D.; Gallagher, J.; Heddleston, J.M.; Wang, J.; Eyler, C.E.; Macswords, J.; Wu, Q.; Vasanji, A.; McLendon, R.E.; Hjelmeland, A.B.; et al. Integrin alpha 6 regulates glioblastoma stem cells. Cell Stem Cell 2010, 6, 421–432. [Google Scholar] [CrossRef] [Green Version]
- Yoo, K.C.; Kang, J.H.; Choi, M.Y.; Suh, Y.; Zhao, Y.; Kim, M.J.; Chang, J.H.; Shim, J.K.; Yoon, S.J.; Kang, S.G.; et al. Soluble ICAM-1 a Pivotal Communicator between Tumors and Macrophages, Promotes Mesenchymal Shift of Glioblastoma. Adv. Sci. 2022, 9, e2102768. [Google Scholar] [CrossRef]
- Reifenberger, G.; Szymas, J.; Wechsler, W. Differential expression of glial- and neuronal-associated antigens in human tumors of the central and peripheral nervous system. Acta Neuropathol. 1987, 74, 105–123. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, H.; Aotsuka, Y.; Yamamoto, Y.; Miyahara, J.; Mitoh, Y. Determination of the antigen/epitope that is recognized by human monoclonal antibody CLN-IgG. Hum. Antibodies 2001, 10, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Babic, B.; Matthias Corvinus, F.; Hadjijusufovic, E.; Tagkalos, E.; Lang, H.; Grimminger, P. Ps01.162: Is There a Difference in Survival between Younger and Older Gastric Cancer (Including Aeg Ii and Aeg Iii) Patients after Gastrectomy? Dis. Esophagus 2018, 31, 95. [Google Scholar] [CrossRef]
- Hugwil, A.V. The meaning of the anti-cancer antibody CLN-IgG (Pritumumab) generated by human×human hybridoma technology against the cyto-skeletal protein, vimentin, in the course of the treatment of malignancy. Med. Hypotheses 2013, 81, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Clement, V.; Sanchez, P.; de Tribolet, N.; Radovanovic, I.; Ruiz i Altaba, A. HEDGEHOG-GLI1 Signaling Regulates Human Glioma Growth, Cancer Stem Cell Self-Renewal, and Tumorigenicity. Curr. Biol. 2007, 17, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Wang, H.; Sun, Q.; Ji, X.; Zhu, L.; Cong, Z.; Zhou, Y.; Liu, H.; Zhou, M. Nrf2 is required to maintain the self-renewal of glioma stem cells. BMC Cancer 2013, 13, 380. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Wang, H.; Fan, Y.; Lin, Y.; Zhang, L.; Ji, X.; Zhou, M. Targeting the NF-E2-related factor 2 pathway: A novel strategy for glioblastoma (review). Oncol. Rep. 2014, 32, 443–450. [Google Scholar] [CrossRef] [Green Version]
- Leelatian, N.; Ihrie, R.A. Head of the Class: OLIG2 and Glioblastoma Phenotype. Cancer Cell 2016, 29, 613–615. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Dai, B.; Kang, S.H.; Ban, K.; Huang, F.J.; Lang, F.F.; Aldape, K.D.; Xie, T.X.; Pelloski, C.E.; Xie, K.; et al. FoxM1B is overexpressed in human glioblastomas and critically regulates the tumorigenicity of glioma cells. Cancer Res. 2006, 66, 3593–3602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.T.; Li, J.C.; Feng, M.; Zhou, Y.X.; Du, Z.W. Novel lncRNA-ZNF281 regulates cell growth, stemness and invasion of glioma stem-like U251s cells. Neoplasma 2019, 66, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Suva, M.L.; Rheinbay, E.; Gillespie, S.M.; Patel, A.P.; Wakimoto, H.; Rabkin, S.D.; Riggi, N.; Chi, A.S.; Cahill, D.P.; Nahed, B.V.; et al. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell 2014, 157, 580–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiang, L.; Wu, T.; Zhang, H.W.; Lu, N.; Hu, R.; Wang, Y.J.; Zhao, L.; Chen, F.H.; Wang, X.T.; You, Q.D.; et al. HIF-1alpha is critical for hypoxia-mediated maintenance of glioblastoma stem cells by activating Notch signaling pathway. Cell Death Differ. 2012, 19, 284–294. [Google Scholar] [CrossRef] [Green Version]
- Dahlrot, R.H.; Hermansen, S.K.; Hansen, S.; Kristensen, B.W. What is the clinical value of cancer stem cell markers in gliomas? Int. J. Clin. Exp. Pathol. 2013, 6, 334–348. [Google Scholar]
- Banelli, B.; Forlani, A.; Allemanni, G.; Morabito, A.; Pistillo, M.P.; Romani, M. MicroRNA in Glioblastoma: An Overview. Int. J. Genom. 2017, 2017, 7639084. [Google Scholar] [CrossRef] [Green Version]
- Charles, N.A.; Holland, E.C. TRRAP and the maintenance of stemness in gliomas. Cell Stem Cell 2010, 6, 6–7. [Google Scholar] [CrossRef] [Green Version]
- Spehalski, E.I.; Peters, C.; Camphausen, K.A.; Tofilon, P. Distinctions Between the Metabolic Changes in Glioblastoma Cells and Glioma Stem-like Cells Following Irradiation. Int. J. Radiat. Oncol. Biol. Phys. 2017, 99, e617. [Google Scholar] [CrossRef]
- Godoy, P.R.D.V.; Mello, S.S.; Magalhães, D.A.R.; Donaires, F.S.; Nicolucci, P.; Donadi, E.A.; Passos, G.A.; Sakamoto-Hojo, E.T. Ionizing radiation-induced gene expression changes in TP53 proficient and deficient glioblastoma cell lines. Mutat. Res./Genet. Toxicol. Environ. Mutagen. 2013, 756, 46–55. [Google Scholar] [CrossRef]
- Camphausen, K.; Purow, B.; Sproull, M.; Scott, T.; Ozawa, T.; Deen, D.F.; Tofilon, P.J. Orthotopic Growth of Human Glioma Cells Quantitatively and Qualitatively Influences Radiation-Induced Changes in Gene Expression. Cancer Res. 2005, 65, 10389–10393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, M.-H.; Cook, J.A.; Chandramouli, G.V.R.; DeGraff, W.; Yan, H.; Zhao, S.; Coleman, C.N.; Mitchell, J.B.; Chuang, E.Y. Gene Expression Profiling of Breast, Prostate, and Glioma Cells following Single versus Fractionated Doses of Radiation. Cancer Res. 2007, 67, 3845–3852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Cheng, F.; Wei, Y.; Zhang, L.; Guo, D.; Wang, B.; Li, W. Inhibition of TAZ contributes radiation-induced senescence and growth arrest in glioma cells. Oncogene 2018, 38, 2788–2799. [Google Scholar] [CrossRef] [PubMed]
- Behrooz, A.B.; Syahir, A. Could We Address the Interplay Between CD133, Wnt/beta-Catenin, and TERT Signaling Pathways as a Potential Target for Glioblastoma Therapy? Front. Oncol. 2021, 11, 642719. [Google Scholar] [CrossRef] [PubMed]
- Olive, P.L. DNA damage and repair in individual cells: Applications of the comet assay in radiobiology. Int. J. Radiat. Biol. 1999, 75, 395–405. [Google Scholar] [CrossRef]
- Legler, J.M.; Ries, L.A.; Smith, M.A.; Warren, J.L.; Heineman, E.F.; Kaplan, R.S.; Linet, M.S. Cancer surveillance series [corrected]: Brain and other central nervous system cancers: Recent trends in incidence and mortality. J. Natl. Cancer Inst. 1999, 91, 1382–1390. [Google Scholar] [CrossRef] [Green Version]
- Beier, D.; Hau, P.; Proescholdt, M.; Lohmeier, A.; Wischhusen, J.; Oefner, P.J.; Aigner, L.; Brawanski, A.; Bogdahn, U.; Beier, C.P. CD133(+) and CD133(-) glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res. 2007, 67, 4010–4015. [Google Scholar] [CrossRef] [Green Version]
- Campos, B.; Zeng, L.; Daotrong, P.H.; Eckstein, V.; Unterberg, A.; Mairbaurl, H.; Herold-Mende, C. Expression and regulation of AC133 and CD133 in glioblastoma. Glia 2011, 59, 1974–1986. [Google Scholar] [CrossRef]
- Gambelli, F.; Sasdelli, F.; Manini, I.; Gambarana, C.; Oliveri, G.; Miracco, C.; Sorrentino, V. Identification of cancer stem cells from human glioblastomas: Growth and differentiation capabilities and CD133/prominin-1 expression. Cell Biol. Int. 2012, 36, 29–38. [Google Scholar] [CrossRef]
- Brown, D.V.; Filiz, G.; Daniel, P.M.; Hollande, F.; Dworkin, S.; Amiridis, S.; Kountouri, N.; Ng, W.; Morokoff, A.P.; Mantamadiotis, T. Expression of CD133 and CD44 in glioblastoma stem cells correlates with cell proliferation, phenotype stability and intra-tumor heterogeneity. PLoS ONE 2017, 12, e0172791. [Google Scholar] [CrossRef] [Green Version]
- Kowalski-Chauvel, A.; Modesto, A.; Gouaze-Andersson, V.; Baricault, L.; Gilhodes, J.; Delmas, C.; Lemarie, A.; Toulas, C.; Cohen-Jonathan-Moyal, E.; Seva, C. Alpha-6 integrin promotes radioresistance of glioblastoma by modulating DNA damage response and the transcription factor Zeb1. Cell Death Dis. 2018, 9, 872. [Google Scholar] [CrossRef] [PubMed]
- Halliday, J.; Helmy, K.; Pattwell, S.S.; Pitter, K.L.; LaPlant, Q.; Ozawa, T.; Holland, E.C. In vivo radiation response of proneural glioma characterized by protective p53 transcriptional program and proneural-mesenchymal shift. Proc. Natl. Acad. Sci. USA 2014, 111, 5248–5253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trog, D.; Moenkemann, H.; Haertel, N.; Schuller, H.; Golubnitschaja, O. Expression of ABC-1 transporter is elevated in human glioma cells under irradiation and temozolomide treatment. Amino Acids 2005, 28, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.K. The Transcription Regulator Kruppel-Like Factor 4 and Its Dual Roles of Oncogene in Glioblastoma and Tumor Suppressor in Neuroblastoma. Immunopathol. Dis Ther. 2016, 7, 127–139. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef]
- Dreger, H.; Westphal, K.; Weller, A.; Baumann, G.; Stangl, V.; Meiners, S.; Stangl, K. Nrf2-dependent upregulation of antioxidative enzymes: A novel pathway for proteasome inhibitor-mediated cardioprotection. Cardiovasc. Res. 2009, 83, 354–361. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Bodas, M.; Wakabayashi, N.; Bunz, F.; Biswal, S. Gain of Nrf2 function in non-small-cell lung cancer cells confers radioresistance. Antioxid. Redox. Signal. 2010, 13, 1627–1637. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Yamamoto, M. Molecular basis of the Keap1-Nrf2 system. Free Radic. Biol. Med. 2015, 88, 93–100. [Google Scholar] [CrossRef] [Green Version]
- Shibata, T.; Ohta, T.; Tong, K.I.; Kokubu, A.; Odogawa, R.; Tsuta, K.; Asamura, H.; Yamamoto, M.; Hirohashi, S. Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proc. Natl. Acad. Sci. USA 2008, 105, 13568–13573. [Google Scholar] [CrossRef] [Green Version]
- Facchino, S.; Abdouh, M.; Chatoo, W.; Bernier, G. BMI1 confers radioresistance to normal and cancerous neural stem cells through recruitment of the DNA damage response machinery. J. Neurosci. 2010, 30, 10096–10111. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, B.; McEllin, B.; Camacho, C.V.; Tomimatsu, N.; Sirasanagandala, S.; Nannepaga, S.; Hatanpaa, K.J.; Mickey, B.; Madden, C.; Maher, E.; et al. EGFRvIII and DNA double-strand break repair: A molecular mechanism for radioresistance in glioblastoma. Cancer Res. 2009, 69, 4252–4259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majc, B.; Sever, T.; Zaric, M.; Breznik, B.; Turk, B.; Lah, T.T. Epithelial-to-mesenchymal transition as the driver of changing carcinoma and glioblastoma microenvironment. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118782. [Google Scholar] [CrossRef] [PubMed]
- Hovinga, K.E.; Stalpers, L.J.; van Bree, C.; Donker, M.; Verhoeff, J.J.; Rodermond, H.M.; Bosch, D.A.; van Furth, W.R. Radiation-enhanced vascular endothelial growth factor (VEGF) secretion in glioblastoma multiforme cell lines--a clue to radioresistance? J. Neurooncol. 2005, 74, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.G.; Heijn, M.; di Tomaso, E.; Griffon-Etienne, G.; Ancukiewicz, M.; Koike, C.; Park, K.R.; Ferrara, N.; Jain, R.K.; Suit, H.D.; et al. Anti-Vascular endothelial growth factor treatment augments tumor radiation response under normoxic or hypoxic conditions. Cancer Res. 2000, 60, 5565–5570. [Google Scholar] [PubMed]
- Maachani, U.B.; Shankavaram, U.; Kramp, T.; Tofilon, P.J.; Camphausen, K.; Tandle, A.T. FOXM1 and STAT3 interaction confers radioresistance in glioblastoma cells. Oncotarget 2016, 7, 77365–77377. [Google Scholar] [CrossRef] [Green Version]
- Povlsen, L.K.; Beli, P.; Wagner, S.A.; Poulsen, S.L.; Sylvestersen, K.B.; Poulsen, J.W.; Nielsen, M.L.; Bekker-Jensen, S.; Mailand, N.; Choudhary, C. Systems-wide analysis of ubiquitylation dynamics reveals a key role for PAF15 ubiquitylation in DNA-damage bypass. Nat. Cell Biol. 2012, 14, 1089–1098. [Google Scholar] [CrossRef]
- Lee, H.C.; Kim, D.W.; Jung, K.Y.; Park, I.C.; Park, M.J.; Kim, M.S.; Woo, S.H.; Rhee, C.H.; Yoo, H.; Lee, S.H.; et al. Increased expression of antioxidant enzymes in radioresistant variant from U251 human glioblastoma cell line. Int. J. Mol. Med. 2004, 13, 883–887. [Google Scholar] [CrossRef]
- Flor, S.; Oliva, C.R.; Ali, M.Y.; Coleman, K.L.; Greenlee, J.D.; Jones, K.A.; Monga, V.; Griguer, C.E. Catalase Overexpression Drives an Aggressive Phenotype in Glioblastoma. Antioxidants 2021, 10, 1988. [Google Scholar] [CrossRef]
- Pang, L.Y.; Hurst, E.A.; Argyle, D.J. Cyclooxygenase-2: A Role in Cancer Stem Cell Survival and Repopulation of Cancer Cells during Therapy. Stem Cells Int. 2016, 2016, 2048731. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.I.; Chiou, S.H.; Hueng, D.Y.; Tai, L.K.; Huang, P.I.; Kao, C.L.; Chen, Y.W.; Sytwu, H.K. Celecoxib and radioresistant glioblastoma-derived CD133+ cells: Improvement in radiotherapeutic effects. Laboratory investigation. J. Neurosurg. 2011, 114, 651–662. [Google Scholar] [CrossRef]
- Wang, W.; Long, L.; Wang, L.; Tan, C.; Fei, X.; Chen, L.; Huang, Q.; Liang, Z. Knockdown of Cathepsin L promotes radiosensitivity of glioma stem cells both in vivo and in vitro. Cancer Lett. 2016, 371, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yang, T.; Xu, G.; Liu, H.; Ren, C.; Xie, W.; Wang, M. Cyclin-Dependent Kinase 2 Promotes Tumor Proliferation and Induces Radio Resistance in Glioblastoma. Transl. Oncol. 2016, 9, 548–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marampon, F.; Megiorni, F.; Camero, S.; Crescioli, C.; McDowell, H.P.; Sferra, R.; Vetuschi, A.; Pompili, S.; Ventura, L.; De Felice, F.; et al. HDAC4 and HDAC6 sustain DNA double strand break repair and stem-like phenotype by promoting radioresistance in glioblastoma cells. Cancer Lett. 2017, 397, 1–11. [Google Scholar] [CrossRef]
- Deng, X.; Ma, L.; Wu, M.; Zhang, G.; Jin, C.; Guo, Y.; Liu, R. miR-124 radiosensitizes human glioma cells by targeting CDK4. J. Neurooncol. 2013, 114, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Moskwa, P.; Zinn, P.O.; Choi, Y.E.; Shukla, S.A.; Fendler, W.; Chen, C.C.; Lu, J.; Golub, T.R.; Hjelmeland, A.; Chowdhury, D. A functional screen identifies miRs that induce radioresistance in glioblastomas. Mol. Cancer Res. 2014, 12, 1767–1778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alkhaibary, A.; Alassiri, A.H.; AlSufiani, F.; Alharbi, M.A. Ki-67 labeling index in glioblastoma; does it really matter? Hematol./Oncol. Stem Cell Ther. 2019, 12, 82–88. [Google Scholar] [CrossRef]
- Mastronardi, L.; Guiducci, A.; Puzzilli, F.; Ruggeri, A. Relationship between Ki-67 labeling index and survival in high-grade glioma patients treated after surgery with tamoxifen. J. Neurosurg. Sci. 1999, 43, 263–270. [Google Scholar]
- Tamura, K.; Aoyagi, M.; Ando, N.; Ogishima, T.; Wakimoto, H.; Yamamoto, M.; Ohno, K. Expansion of CD133-positive glioma cells in recurrent de novo glioblastomas after radiotherapy and chemotherapy. J. Neurosurg. 2013, 119, 1145–1155. [Google Scholar] [CrossRef] [Green Version]
- Zottel, A.; Jovcevska, I.; Samec, N.; Komel, R. Cytoskeletal proteins as glioblastoma biomarkers and targets for therapy: A systematic review. Crit. Rev. Oncol. Hematol. 2021, 160, 103283. [Google Scholar] [CrossRef]
- Nguemgo Kouam, P.; Rezniczek, G.A.; Kochanneck, A.; Priesch-Grzeszkowiak, B.; Hero, T.; Adamietz, I.A.; Buhler, H. Robo1 and vimentin regulate radiation-induced motility of human glioblastoma cells. PLoS ONE 2018, 13, e0198508. [Google Scholar] [CrossRef] [Green Version]
- Chakravarti, A.; Zhai, G.G.; Zhang, M.; Malhotra, R.; Latham, D.E.; Delaney, M.A.; Robe, P.; Nestler, U.; Song, Q.; Loeffler, J. Survivin enhances radiation resistance in primary human glioblastoma cells via caspase-independent mechanisms. Oncogene 2004, 23, 7494–7506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Zhang, S.; Ibrahim, A.N.; Wang, J.; Deng, Z.; Wang, M. RCC2 promotes proliferation and radio-resistance in glioblastoma via activating transcription of DNMT1. Biochem. Biophys. Res. Commun. 2019, 516, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Xue, X.; Zhou, H.; Zhang, G. A molecular view of the radioresistance of gliomas. Oncotarget 2017, 8, 100931–100941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marampon, F.; Gravina, G.L.; Zani, B.M.; Popov, V.M.; Fratticci, A.; Cerasani, M.; Di Genova, D.; Mancini, M.; Ciccarelli, C.; Ficorella, C.; et al. Hypoxia sustains glioblastoma radioresistance through ERKs/DNA-PKcs/HIF-1alpha functional interplay. Int. J. Oncol. 2014, 44, 2121–2131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marwick, C. Pharmaceutical industry commission, government agencies seek to expedite new addiction therapy. JAMA 1991, 265, 841. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Ezhilarasan, R.; Phillips, E.; Gallego-Perez, D.; Sparks, A.; Taylor, D.; Ladner, K.; Furuta, T.; Sabit, H.; Chhipa, R.; et al. Serine/Threonine Kinase MLK4 Determines Mesenchymal Identity in Glioma Stem Cells in an NF-kappaB-dependent Manner. Cancer Cell 2016, 29, 201–213. [Google Scholar] [CrossRef] [Green Version]
- Bar, E.E.; Chaudhry, A.; Lin, A.; Fan, X.; Schreck, K.; Matsui, W.; Piccirillo, S.; Vescovi, A.L.; DiMeco, F.; Olivi, A.; et al. Cyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem Cells 2007, 25, 2524–2533. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Wei, P.; Gong, A.; Chiu, W.T.; Lee, H.T.; Colman, H.; Huang, H.; Xue, J.; Liu, M.; Wang, Y.; et al. FoxM1 promotes beta-catenin nuclear localization and controls Wnt target-gene expression and glioma tumorigenesis. Cancer Cell 2011, 20, 427–442. [Google Scholar] [CrossRef] [Green Version]
- Burris, H.A., III. Overcoming acquired resistance to anticancer therapy: Focus on the PI3K/AKT/mTOR pathway. Cancer Chemother. Pharm. 2013, 71, 829–842. [Google Scholar] [CrossRef]
- Ashton, C.H.; Rawlins, M.D.; Tyrer, S.P. Buspirone in benzodiazepine withdrawal. Br. J. Psychiatry 1991, 158, 283–284. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Lathia, J.D.; Eyler, C.E.; Wu, Q.; Li, Z.; Wang, H.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. Erythropoietin Receptor Signaling Through STAT3 Is Required for Glioma Stem Cell Maintenance. Genes Cancer 2010, 1, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.Y.; Oliva, C.R.; Noman, A.S.M.; Allen, B.G.; Goswami, P.C.; Zakharia, Y.; Monga, V.; Spitz, D.R.; Buatti, J.M.; Griguer, C.E. Radioresistance in Glioblastoma and the Development of Radiosensitizers. Cancers 2020, 12, 2511. [Google Scholar] [CrossRef]
- Farooqi, A.A.; Siddik, Z.H. Platelet-derived growth factor (PDGF) signalling in cancer: Rapidly emerging signalling landscape. Cell Biochem. Funct. 2015, 33, 257–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godlewski, J.; Nowicki, M.O.; Bronisz, A.; Williams, S.; Otsuki, A.; Nuovo, G.; Raychaudhury, A.; Newton, H.B.; Chiocca, E.A.; Lawler, S. Targeting of the Bmi-1 oncogene/stem cell renewal factor by microRNA-128 inhibits glioma proliferation and self-renewal. Cancer Res. 2008, 68, 9125–9130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koul, D. PTEN signaling pathways in glioblastoma. Cancer Biol. Ther. 2008, 7, 1321–1325. [Google Scholar] [CrossRef] [PubMed]
- Morgenroth, A.; Vogg, A.T.; Ermert, K.; Zlatopolskiy, B.; Mottaghy, F.M. Hedgehog signaling sensitizes glioma stem cells to endogenous nano-irradiation. Oncotarget 2014, 5, 5483–5493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, N.; Hu, G.; Shi, L.; Long, G.; Yang, L.; Xi, Q.; Guo, Q.; Wang, J.; Dong, Z.; Zhang, M. Notch1 ablation radiosensitizes glioblastoma cells. Oncotarget 2017, 8, 88059–88068. [Google Scholar] [CrossRef] [Green Version]
- Bhat, K.P.L.; Balasubramaniyan, V.; Vaillant, B.; Ezhilarasan, R.; Hummelink, K.; Hollingsworth, F.; Wani, K.; Heathcock, L.; James, J.D.; Goodman, L.D.; et al. Mesenchymal differentiation mediated by NF-kappaB promotes radiation resistance in glioblastoma. Cancer Cell 2013, 24, 331–346. [Google Scholar] [CrossRef] [Green Version]
- Xu, R.X.; Liu, R.Y.; Wu, C.M.; Zhao, Y.S.; Li, Y.; Yao, Y.Q.; Xu, Y.H. DNA damage-induced NF-kappaB activation in human glioblastoma cells promotes miR-181b expression and cell proliferation. Cell Physiol. Biochem. 2015, 35, 913–925. [Google Scholar] [CrossRef]
- Huang, T.T.; Wuerzberger-Davis, S.M.; Seufzer, B.J.; Shumway, S.D.; Kurama, T.; Boothman, D.A.; Miyamoto, S. NF-kappaB activation by camptothecin. A linkage between nuclear DNA damage and cytoplasmic signaling events. J. Biol. Chem. 2000, 275, 9501–9509. [Google Scholar] [CrossRef] [Green Version]
- McCool, K.W.; Miyamoto, S. DNA damage-dependent NF-kappaB activation: NEMO turns nuclear signaling inside out. Immunol. Rev. 2012, 246, 311–326. [Google Scholar] [CrossRef] [PubMed]
- Volcic, M.; Karl, S.; Baumann, B.; Salles, D.; Daniel, P.; Fulda, S.; Wiesmuller, L. NF-kappaB regulates DNA double-strand break repair in conjunction with BRCA1-CtIP complexes. Nucleic Acids Res. 2012, 40, 181–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, K.; Jiang, S.W.; Thangaraju, M.; Wu, G.; Couch, F.J. Induction of the BRCA2 promoter by nuclear factor-kappa B. J. Biol. Chem. 2000, 275, 35548–35556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyamoto, S. Nuclear initiated NF-kappaB signaling: NEMO and ATM take center stage. Cell Res. 2011, 21, 116–130. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.K.; Ye, Z.; Darras, B.T. Frequency of p53 tumor suppressor gene mutations in human primary brain tumors. Neurosurgery 1993, 33, 824–830; discussion 821–830. [Google Scholar] [CrossRef]
- Chen, P.; Iavarone, A.; Fick, J.; Edwards, M.; Prados, M.; Israel, M.A. Constitutional p53 mutations associated with brain tumors in young adults. Cancer Genet. Cytogenet. 1995, 82, 106–115. [Google Scholar] [CrossRef]
- Shu, H.K.; Kim, M.M.; Chen, P.; Furman, F.; Julin, C.M.; Israel, M.A. The intrinsic radioresistance of glioblastoma-derived cell lines is associated with a failure of p53 to induce p21(BAX) expression. Proc. Natl. Acad. Sci. USA 1998, 95, 14453–14458. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.; Pore, N.; Cerniglia, G.J.; Mick, R.; Georgescu, M.M.; Bernhard, E.J.; Hahn, S.M.; Gupta, A.K.; Maity, A. Phosphatase and tensin homologue deficiency in glioblastoma confers resistance to radiation and temozolomide that is reversed by the protease inhibitor nelfinavir. Cancer Res. 2007, 67, 4467–4473. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Liu, F.; Salmonsen, R.A.; Turner, T.K.; Litofsky, N.S.; Di Cristofano, A.; Pandolfi, P.P.; Jones, S.N.; Recht, L.D.; Ross, A.H. PTEN in neural precursor cells: Regulation of migration, apoptosis, and proliferation. Mol. Cell Neurosci. 2002, 20, 21–29. [Google Scholar] [CrossRef]
- Lomonaco, S.L.; Finniss, S.; Xiang, C.; Decarvalho, A.; Umansky, F.; Kalkanis, S.N.; Mikkelsen, T.; Brodie, C. The induction of autophagy by gamma-radiation contributes to the radioresistance of glioma stem cells. Int. J. Cancer 2009, 125, 717–722. [Google Scholar] [CrossRef]
- Zhuang, W.; Qin, Z.; Liang, Z. The role of autophagy in sensitizing malignant glioma cells to radiation therapy. Acta Biochim. Biophys. Sin. 2009, 41, 341–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Casal, R.; Bhattacharya, C.; Ganesh, N.; Bailey, L.; Basse, P.; Gibson, M.; Epperly, M.; Levina, V. Non-small cell lung cancer cells survived ionizing radiation treatment display cancer stem cell and epithelial-mesenchymal transition phenotypes. Mol. Cancer 2013, 12, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shien, K.; Toyooka, S.; Ichimura, K.; Soh, J.; Furukawa, M.; Maki, Y.; Muraoka, T.; Tanaka, N.; Ueno, T.; Asano, H.; et al. Prognostic impact of cancer stem cellTransporters in Cancer non-small cell lung cancer patients treated with induction chemoradiotherapy. Lung Cancer 2012, 77, 162–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakaria, N.; Yusoff, N.M.; Zakaria, Z.; Lim, M.N.; Baharuddin, P.J.; Fakiruddin, K.S.; Yahaya, B. Human non-small cell lung cancer expresses putative cancer stem cell markers and exhibits the transcriptomic profile of multipotent cells. BMC Cancer 2015, 15, 84. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.J.; Choe, G.; Jheon, S.; Sung, S.W.; Lee, C.T.; Chung, J.H. CD24, a novel cancer biomarker, predicting disease-free survival of non-small cell lung carcinomas: A retrospective study of prognostic factor analysis from the viewpoint of forthcoming (seventh) new TNM classification. J. Thorac. Oncol. 2010, 5, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Karimi-Busheri, F.; Rasouli-Nia, A.; Zadorozhny, V.; Fakhrai, H. CD24+/CD38- as new prognostic marker for non-small cell lung cancer. Multidiscip. Respir. Med. 2013, 8, 65. [Google Scholar] [CrossRef] [Green Version]
- Summer, R.; Kotton, D.N.; Sun, X.; Ma, B.; Fitzsimmons, K.; Fine, A. Side population cells and Bcrp1 expression in lung. Am. J. Physiol. Lung Cell Mol. Physiol. 2003, 285, L97–L104. [Google Scholar] [CrossRef] [Green Version]
- Editors, P.O. Retraction: Identification and Characterization of Cells with Cancer Stem Cell Properties in Human Primary Lung Cancer Cell Lines. PLoS ONE 2020, 15, e0232726. [Google Scholar] [CrossRef]
- Jiang, F.; Qiu, Q.; Khanna, A.; Todd, N.W.; Deepak, J.; Xing, L.; Wang, H.; Liu, Z.; Su, Y.; Stass, S.A.; et al. Aldehyde dehydrogenase 1 is a tumor stem cell-associated marker in lung cancer. Mol. Cancer Res. 2009, 7, 330–338. [Google Scholar] [CrossRef] [Green Version]
- Liang, D.; Shi, Y. Aldehyde dehydrogenase-1 is a specific marker for stem cells in human lung adenocarcinoma. Med. Oncol. 2012, 29, 633–639. [Google Scholar] [CrossRef]
- Kiziltunc Ozmen, H.; Simsek, M. Serum IL-23, E-selectin and sICAM levels in non-small cell lung cancer patients before and after radiotherapy. J. Int. Med. Res. 2020, 48, 300060520923493. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zeng, H.; Ying, K. The combination of stem cell markers CD133 and ABCG2 predicts relapse in stage I non-small cell lung carcinomas. Med. Oncol. 2011, 28, 1458–1462. [Google Scholar] [CrossRef] [PubMed]
- Kwa, H.B.; Michalides, R.J.A.M.; Dijkman, J.H.; Mooi, W.J. The prognostic value of NCAM, p53 and cyclin D1 in resected non-small cell lung cancer. Lung Cancer 1996, 14, 207–217. [Google Scholar] [CrossRef]
- Zhang, X.-Y.; Dong, Q.-G.; Huang, J.-S.; Huang, A.-M.; Shi, C.-L.; Jin, B.; Sha, H.-F.; Feng, J.-X.; Geng, Q.; Zhou, J.; et al. The expression of stem cell-related indicators as a prognostic factor in human lung adenocarcinoma. J. Surg. Oncol. 2010, 102, 856–862. [Google Scholar] [CrossRef]
- Shao, W.; Chen, H.; He, J. The role of SOX-2 on the survival of patients with non-small cell lung cancer. J. Thorac. Dis. 2015, 7, 1113–1118. [Google Scholar] [CrossRef]
- Fadous-Khalife, M.C.; Aloulou, N.; Jalbout, M.; Hadchity, J.; Aftimos, G.; Paris, F.; Hadchity, E. Kruppel-like factor 4: A new potential biomarker of lung cancer. Mol. Clin. Oncol. 2016, 5, 35–40. [Google Scholar] [CrossRef] [Green Version]
- Chanvorachote, P.; Sriratanasak, N.; Nonpanya, N. C-myc Contributes to Malignancy of Lung Cancer: A Potential Anticancer Drug Target. Anticancer Res. 2020, 40, 609–618. [Google Scholar] [CrossRef]
- Ma, Y.N.; Zhang, H.Y.; Fei, L.R.; Zhang, M.Y.; Wang, C.C.; Luo, Y.; Han, Y.C. SATB2 suppresses non-small cell lung cancer invasiveness by G9a. Clin. Exp. Med. 2018, 18, 37–44. [Google Scholar] [CrossRef]
- Hsiao, Y.J.; Chang, W.H.; Chen, H.Y.; Hsu, Y.C.; Chiu, S.C.; Chiang, C.C.; Chang, G.C.; Chen, Y.J.; Wang, C.Y.; Chen, Y.M.; et al. MITF functions as a tumor suppressor in non-small cell lung cancer beyond the canonically oncogenic role. Aging 2020, 13, 646–674. [Google Scholar] [CrossRef]
- Harada, D.; Takigawa, N.; Kiura, K. The Role of STAT3 in Non-Small Cell Lung Cancer. Cancers 2014, 6, 708–722. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Xiao, L.; Wang, F.; Yang, J.; Yang, L.; Zhao, Y.; Jin, W. Hypoxia inducible factor-1alpha/B-cell lymphoma 2 signaling impacts radiosensitivity of H1299 non-small cell lung cancer cells in a normoxic environment. Radiat. Environ. Biophys. 2019, 58, 439–448. [Google Scholar] [CrossRef]
- Lu, J.; Zhan, Y.; Feng, J.; Luo, J.; Fan, S. MicroRNAs associated with therapy of non-small cell lung cancer. Int. J. Biol. Sci. 2018, 14, 390–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sone, K.; Maeno, K.; Masaki, A.; Kunii, E.; Takakuwa, O.; Kagawa, Y.; Takeuchi, A.; Fukuda, S.; Uemura, T.; Fukumitsu, K.; et al. Nestin Expression Affects Resistance to Chemotherapy and Clinical Outcome in Small Cell Lung Cancer. Front. Oncol. 2020, 10, 1367. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Tian, T.; Sun, W.; Liu, C.; Fang, X. Bmi-1 overexpression as an efficient prognostic marker in patients with nonsmall cell lung cancer. Medicine 2017, 96, e7346. [Google Scholar] [CrossRef] [PubMed]
- Lang, Y.; Kong, X.; He, C.; Wang, F.; Liu, B.; Zhang, S.; Ning, J.; Zhu, K.; Xu, S. Musashi1 Promotes Non-Small Cell Lung Carcinoma Malignancy and Chemoresistance via Activating the Akt Signaling Pathway. Cell Physiol. Biochem. 2017, 44, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Jaiswal, R.K.; Prasad, R.; Yadav, S.S.; Kumar, A.; Yadava, P.K.; Singh, R.P. PARP-1 induces EMT in non-small cell lung carcinoma cells via modulating the transcription factors Smad4, p65 and ZEB1. Life Sci. 2021, 269, 118994. [Google Scholar] [CrossRef]
- Jafarian, A.H.; Kooshki Forooshani, M.; Reisi, H.; Mohamadian Roshan, N. Matrix metalloproteinase-9 (MMP-9) Expression in Non-Small Cell Lung Carcinoma and Its Association with Clinicopathologic Factors. Iran. J. Pathol. 2020, 15, 326–333. [Google Scholar] [CrossRef]
- Merchant, N.; Nagaraju, G.P.; Rajitha, B.; Lammata, S.; Jella, K.K.; Buchwald, Z.S.; Lakka, S.S.; Ali, A.N. Matrix metalloproteinases: Their functional role in lung cancer. Carcinogenesis 2017, 38, 766–780. [Google Scholar] [CrossRef] [Green Version]
- Byers, L.A.; Heymach, J.V. Dual targeting of the vascular endothelial growth factor and epidermal growth factor receptor pathways: Rationale and clinical applications for non-small-cell lung cancer. Clin. Lung Cancer 2007, 8 (Suppl. 2), S79–S85. [Google Scholar] [CrossRef]
- Otsuka, S.; Bebb, G. The CXCR4/SDF-1 chemokine receptor axis: A new target therapeutic for non-small cell lung cancer. J. Thorac. Oncol. 2008, 3, 1379–1383. [Google Scholar] [CrossRef] [Green Version]
- Kristiansen, G.; Schluns, K.; Yongwei, Y.; Denkert, C.; Dietel, M.; Petersen, I. CD24 is an independent prognostic marker of survival in nonsmall cell lung cancer patients. Br. J. Cancer 2003, 88, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Ma, Y.; Yang, Y.; Zhang, L.; Han, H.; Chen, J. CD44 promotes cell proliferation in non-small cell lung cancer. Oncol. Lett. 2018, 15, 5627–5633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tirino, V.; Camerlingo, R.; Franco, R.; Malanga, D.; La Rocca, A.; Viglietto, G.; Rocco, G.; Pirozzi, G. The role of CD133 in the identification and characterisation of tumour-initiating cells in non-small-cell lung cancer. Eur. J. Cardiothorac. Surg. 2009, 36, 446–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, T.Y.; Kim, J.I.; Lee, S.H. Relationship between Cancer Stem Cell Marker CD133 and Cancer Germline Antigen Genes in NCI-H292 Lung Cancer Cells. Korean J. Thorac. Cardiovasc. Surg. 2020, 53, 22–27. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.Y.; Oh, S.H.; Woo, J.K.; Hong, W.K.; Lee, H.Y. Targeting heat shock protein 90 overrides the resistance of lung cancer cells by blocking radiation-induced stabilization of hypoxia-inducible factor-1alpha. Cancer Res. 2009, 69, 1624–1632. [Google Scholar] [CrossRef]
- Akakura, N.; Kobayashi, M.; Horiuchi, I.; Suzuki, A.; Wang, J.; Chen, J.; Niizeki, H.; Kawamura, K.; Hosokawa, M.; Asaka, M. Constitutive expression of hypoxia-inducible factor-1alpha renders pancreatic cancer cells resistant to apoptosis induced by hypoxia and nutrient deprivation. Cancer Res. 2001, 61, 6548–6554. [Google Scholar]
- Kim, J.Y.; Kim, H.J.; Jung, C.W.; Lee, T.S.; Kim, E.H.; Park, M.J. CXCR4 uses STAT3-mediated slug expression to maintain radioresistance of non-small cell lung cancer cells: Emerges as a potential prognostic biomarker for lung cancer. Cell Death Dis. 2021, 12, 48. [Google Scholar] [CrossRef]
- Loriot, Y.; Mordant, P.; Dorvault, N.; De la motte Rouge, T.; Bourhis, J.; Soria, J.C.; Deutsch, E. BMS-690514, a VEGFR and EGFR tyrosine kinase inhibitor, shows anti-tumoural activity on non-small-cell lung cancer xenografts and induces sequence-dependent synergistic effect with radiation. Br. J. Cancer 2010, 103, 347–353. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Wang, R.; Yan, H.; Jin, L.; Dou, X.; Chen, D. MicroRNA-21 modulates radiation resistance through upregulation of hypoxia-inducible factor-1alpha-promoted glycolysis in non-small cell lung cancer cells. Mol. Med. Rep. 2016, 13, 4101–4107. [Google Scholar] [CrossRef] [Green Version]
- Grosso, S.; Doyen, J.; Parks, S.K.; Bertero, T.; Paye, A.; Cardinaud, B.; Gounon, P.; Lacas-Gervais, S.; Noel, A.; Pouyssegur, J.; et al. MiR-210 promotes a hypoxic phenotype and increases radioresistance in human lung cancer cell lines. Cell Death Dis. 2013, 4, e544. [Google Scholar] [CrossRef] [Green Version]
- He, Z.; Liu, Y.; Xiao, B.; Qian, X. miR-25 modulates NSCLC cell radio-sensitivity through directly inhibiting BTG2 expression. Biochem. Biophys. Res. Commun. 2015, 457, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Han, W.; Ni, T.T.; Lu, L.; Huang, M.; Zhang, Y.; Cao, H.; Zhang, H.Q.; Luo, W.; Li, H. Knockdown of microRNA-1323 restores sensitivity to radiation by suppression of PRKDC activity in radiation-resistant lung cancer cells. Oncol. Rep. 2015, 33, 2821–2828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.S.; Oh, J.H.; Yoon, S.; Kwon, M.S.; Song, C.W.; Kim, K.H.; Cho, M.J.; Mollah, M.L.; Je, Y.J.; Kim, Y.D.; et al. Differential gene expression profiles of radioresistant non-small-cell lung cancer cell lines established by fractionated irradiation: Tumor protein p53-inducible protein 3 confers sensitivity to ionizing radiation. Int. J. Radiat. Oncol. Biol. Phys. 2010, 77, 858–866. [Google Scholar] [CrossRef] [PubMed]
- Davidson, B.; Goldberg, I.; Kopolovic, J.; Lerner-Geva, L.; Gotlieb, W.H.; Ben-Baruch, G.; Reich, R. MMP-2 and TIMP-2 expression correlates with poor prognosis in cervical carcinoma--a clinicopathologic study using immunohistochemistry and mRNA in situ hybridization. Gynecol. Oncol. 1999, 73, 372–382. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, K.; Tsuda, M.; Yazawa, N.; Nakamura, H.; Ishihara, S.; Haga, H.; Yasuda, M.; Yamazaki, R.; Shirato, H.; Kawaguchi, H.; et al. Increased motility and invasiveness in tumor cells that survive 10 Gy irradiation. Cell Struct. Funct. 2009, 34, 89–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, W.; Lee, Y.S.; Son, C.H.; Yang, K.; Park, Y.S.; Bae, J. Radiation-induced matrix metalloproteinases limit natural killer cell-mediated anticancer immunity in NCI-H23 lung cancer cells. Mol. Med. Rep. 2015, 11, 1800–1806. [Google Scholar] [CrossRef]
- Zhuang, X.; Qiao, T.; Xu, G.; Yuan, S.; Zhang, Q.; Chen, X. Combination of nadroparin with radiotherapy results in powerful synergistic antitumor effects in lung adenocarcinoma A549 cells. Oncol. Rep. 2016, 36, 2200–2206. [Google Scholar] [CrossRef] [Green Version]
- Swinson, D.E.; Jones, J.L.; Cox, G.; Richardson, D.; Harris, A.L.; O’Byrne, K.J. Hypoxia-inducible factor-1 alpha in non small cell lung cancer: Relation to growth factor, protease and apoptosis pathways. Int. J. Cancer 2004, 111, 43–50. [Google Scholar] [CrossRef]
- Hussenet, T.; Dali, S.; Exinger, J.; Monga, B.; Jost, B.; Dembele, D.; Martinet, N.; Thibault, C.; Huelsken, J.; Brambilla, E.; et al. SOX2 is an oncogene activated by recurrent 3q26.3 amplifications in human lung squamous cell carcinomas. PLoS ONE 2010, 5, e8960. [Google Scholar] [CrossRef] [Green Version]
- Bass, A.J.; Watanabe, H.; Mermel, C.H.; Yu, S.; Perner, S.; Verhaak, R.G.; Kim, S.Y.; Wardwell, L.; Tamayo, P.; Gat-Viks, I.; et al. SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat. Genet. 2009, 41, 1238–1242. [Google Scholar] [CrossRef]
- Chowdhury, P.; Dey, P.; Ghosh, S.; Sarma, A.; Ghosh, U. Reduction of metastatic potential by inhibiting EGFR/Akt/p38/ERK signaling pathway and epithelial-mesenchymal transition after carbon ion exposure is potentiated by PARP-1 inhibition in non-small-cell lung cancer. BMC Cancer 2019, 19, 829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guster, J.D.; Weissleder, S.V.; Busch, C.J.; Kriegs, M.; Petersen, C.; Knecht, R.; Dikomey, E.; Rieckmann, T. The inhibition of PARP but not EGFR results in the radiosensitization of HPV/p16-positive HNSCC cell lines. Radiother. Oncol. 2014, 113, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Toulany, M.; Iida, M.; Keinath, S.; Iyi, F.F.; Mueck, K.; Fehrenbacher, B.; Mansour, W.Y.; Schaller, M.; Wheeler, D.L.; Rodemann, H.P. Dual targeting of PI3K and MEK enhances the radiation response of K-RAS mutated non-small cell lung cancer. Oncotarget 2016, 7, 43746–43761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, J.; Kim, E.; Kim, W.; Seong, K.M.; Youn, H.; Kim, J.W.; Kim, J.; Youn, B. Rhamnetin and cirsiliol induce radiosensitization and inhibition of epithelial-mesenchymal transition (EMT) by miR-34a-mediated suppression of Notch-1 expression in non-small cell lung cancer cell lines. J. Biol. Chem. 2013, 288, 27343–27357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Y.; Liu, Q.; He, X.; Yuan, X.; Chen, Y.; Chu, Q.; Wu, K. Emerging roles of Nrf2 signal in non-small cell lung cancer. J. Hematol. Oncol. 2016, 9, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.; Li, L.; Yan, W. Knockdown of TC-1 enhances radiosensitivity of non-small cell lung cancer via the Wnt/beta-catenin pathway. Biol. Open 2016, 5, 492–498. [Google Scholar] [CrossRef]
- Zeng, J.; Aziz, K.; Chettiar, S.T.; Aftab, B.T.; Armour, M.; Gajula, R.; Gandhi, N.; Salih, T.; Herman, J.M.; Wong, J.; et al. Hedgehog pathway inhibition radiosensitizes non-small cell lung cancers. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 143–149. [Google Scholar] [CrossRef] [Green Version]
- Heavey, S.; O’Byrne, K.J.; Gately, K. Strategies for co-targeting the PI3K/AKT/mTOR pathway in NSCLC. Cancer Treat. Rev. 2014, 40, 445–456. [Google Scholar] [CrossRef]
- Zhang, T.; Cui, G.B.; Zhang, J.; Zhang, F.; Zhou, Y.A.; Jiang, T.; Li, X.F. Inhibition of PI3 kinases enhances the sensitivity of non-small cell lung cancer cells to ionizing radiation. Oncol. Rep. 2010, 24, 1683–1689. [Google Scholar] [CrossRef] [Green Version]
- Zagouras, P.; Stifani, S.; Blaumueller, C.M.; Carcangiu, M.L.; Artavanis-Tsakonas, S. Alterations in Notch signaling in neoplastic lesions of the human cervix. Proc. Natl. Acad. Sci. USA 1995, 92, 6414–6418. [Google Scholar] [CrossRef] [Green Version]
- Grabher, C.; von Boehmer, H.; Look, A.T. Notch 1 activation in the molecular pathogenesis of T-cell acute lymphoblastic leukaemia. Nat. Rev. Cancer 2006, 6, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Theys, J.; Yahyanejad, S.; Habets, R.; Span, P.; Dubois, L.; Paesmans, K.; Kattenbeld, B.; Cleutjens, J.; Groot, A.J.; Schuurbiers, O.C.J.; et al. High NOTCH activity induces radiation resistance in non small cell lung cancer. Radiother. Oncol. 2013, 108, 440–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, B.; Zhou, X.L.; Lai, S.Q.; Liu, J.C. Notch signaling and non-small cell lung cancer. Oncol. Lett. 2018, 15, 3415–3421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eliasz, S.; Liang, S.; Chen, Y.; De Marco, M.A.; Machek, O.; Skucha, S.; Miele, L.; Bocchetta, M. Notch-1 stimulates survival of lung adenocarcinoma cells during hypoxia by activating the IGF-1R pathway. Oncogene 2010, 29, 2488–2498. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Mao, A.; Yan, J.; Sun, C.; Di, C.; Zhou, X.; Li, H.; Guo, R.; Zhang, H. Downregulation of Nrf2 promotes radiation-induced apoptosis through Nrf2 mediated Notch signaling in non-small cell lung cancer cells. Int. J. Oncol. 2016, 48, 765–773. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.H.; Pan, S.L.; Wang, J.C.; Kuo, S.H.; Cheng, J.C.; Teng, C.M. Radiation-induced VEGF-C expression and endothelial cell proliferation in lung cancer. Strahlenther. Onkol. 2014, 190, 1154–1162. [Google Scholar] [CrossRef]
- Jiang, L.; Huang, J.; Hu, Y.; Lu, P.; Luo, Q.; Wang, L. Gli promotes tumor progression through regulating epithelial-mesenchymal transition in non-small-cell lung cancer. J. Cardiothorac. Surg. 2020, 15, 18. [Google Scholar] [CrossRef] [Green Version]
- Garcia Campelo, M.R.; Alonso Curbera, G.; Aparicio Gallego, G.; Grande Pulido, E.; Anton Aparicio, L.M. Stem cell and lung cancer development: Blaming the Wnt, Hh and Notch signalling pathway. Clin. Transl. Oncol. 2011, 13, 77–83. [Google Scholar] [CrossRef]
- Pustovalova, M.; Alhaddad, L.; Blokhina, T.; Smetanina, N.; Chigasova, A.; Chuprov-Netochin, R.; Eremin, P.; Gilmutdinova, I.; Osipov, A.N.; Leonov, S. The CD44high Subpopulation of Multifraction Irradiation-Surviving NSCLC Cells Exhibits Partial EMT-Program Activation and DNA Damage Response Depending on Their p53 Status. Int. J. Mol. Sci. 2021, 22, 2369. [Google Scholar] [CrossRef]
- Cao, Z.; Livas, T.; Kyprianou, N. Anoikis and EMT: Lethal "Liaisons" during Cancer Progression. Crit. Rev. Oncog. 2016, 21, 155–168. [Google Scholar] [CrossRef] [Green Version]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Lah, T.T.; Novak, M.; Breznik, B. Brain malignancies: Glioblastoma and brain metastases. Semin. Cancer Biol. 2020, 60, 262–273. [Google Scholar] [CrossRef] [PubMed]
- Mittal, V. Epithelial Mesenchymal Transition in Tumor Metastasis. Annu. Rev. Pathol. 2018, 13, 395–412. [Google Scholar] [CrossRef] [PubMed]
- Das, R.; Gregory, P.A.; Hollier, B.G.; Tilley, W.D.; Selth, L.A. Epithelial plasticity in prostate cancer: Principles and clinical perspectives. Trends Mol. Med. 2014, 20, 643–651. [Google Scholar] [CrossRef]
- Ansieau, S.; Bastid, J.; Doreau, A.; Morel, A.P.; Bouchet, B.P.; Thomas, C.; Fauvet, F.; Puisieux, I.; Doglioni, C.; Piccinin, S.; et al. Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell 2008, 14, 79–89. [Google Scholar] [CrossRef] [Green Version]
- Odero-Marah, V.; Hawsawi, O.; Henderson, V.; Sweeney, J. Epithelial-Mesenchymal Transition (EMT) and Prostate Cancer. Adv. Exp. Med. Biol. 2018, 1095, 101–110. [Google Scholar] [CrossRef]
- Radisky, E.S.; Radisky, D.C. Matrix metalloproteinase-induced epithelial-mesenchymal transition in breast cancer. J. Mammary Gland Biol. Neoplasia 2010, 15, 201–212. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.; Kang, Y. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev. Cell 2019, 49, 361–374. [Google Scholar] [CrossRef]
- Jolly, M.K.; Mani, S.A.; Levine, H. Hybrid epithelial/mesenchymal phenotype(s): The ‘fittest’ for metastasis? Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 151–157. [Google Scholar] [CrossRef]
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 2014, 7, re8. [Google Scholar] [CrossRef] [Green Version]
- Huber, M.A.; Kraut, N.; Beug, H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr. Opin. Cell Biol. 2005, 17, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, Zeb and bHLH factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Kuner, R.; Muley, T.; Meister, M.; Ruschhaupt, M.; Buness, A.; Xu, E.C.; Schnabel, P.; Warth, A.; Poustka, A.; Sultmann, H.; et al. Global gene expression analysis reveals specific patterns of cell junctions in non-small cell lung cancer subtypes. Lung Cancer 2009, 63, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Pierdomenico, M.; Palone, F.; Cesi, V.; Vitali, R.; Mancuso, A.B.; Cucchiara, S.; Oliva, S.; Aloi, M.; Stronati, L. Transcription Factor ZNF281: A Novel Player in Intestinal Inflammation and Fibrosis. Front. Immunol. 2018, 9, 2907. [Google Scholar] [CrossRef]
- De Craene, B.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef]
- Boelens, M.C.; van den Berg, A.; Vogelzang, I.; Wesseling, J.; Postma, D.S.; Timens, W.; Groen, H.J. Differential expression and distribution of epithelial adhesion molecules in non-small cell lung cancer and normal bronchus. J. Clin. Pathol. 2007, 60, 608–614. [Google Scholar] [CrossRef]
- Scheel, C.; Weinberg, R.A. Cancer stem cells and epithelial-mesenchymal transition: Concepts and molecular links. Semin. Cancer Biol. 2012, 22, 396–403. [Google Scholar] [CrossRef]
- Kawamoto, A.; Yokoe, T.; Tanaka, K.; Saigusa, S.; Toiyama, Y.; Yasuda, H.; Inoue, Y.; Miki, C.; Kusunoki, M. Radiation induces epithelial-mesenchymal transition in colorectal cancer cells. Oncol. Rep. 2012, 27, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Andarawewa, K.L.; Erickson, A.C.; Chou, W.S.; Costes, S.V.; Gascard, P.; Mott, J.D.; Bissell, M.J.; Barcellos-Hoff, M.H. Ionizing radiation predisposes nonmalignant human mammary epithelial cells to undergo transforming growth factor beta induced epithelial to mesenchymal transition. Cancer Res. 2007, 67, 8662–8670. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.E. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Jin, W. Role of JAK/STAT3 Signaling in the Regulation of Metastasis, the Transition of Cancer Stem Cells, and Chemoresistance of Cancer by Epithelial-Mesenchymal Transition. Cells 2020, 9, 217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smit, M.A.; Peeper, D.S. Epithelial-mesenchymal transition and senescence: Two cancer-related processes are crossing paths. Aging 2010, 2, 735–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheel, C.; Eaton, E.N.; Li, S.H.; Chaffer, C.L.; Reinhardt, F.; Kah, K.J.; Bell, G.; Guo, W.; Rubin, J.; Richardson, A.L.; et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell 2011, 145, 926–940. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.; Graham, P.H.; Hao, J.; Bucci, J.; Cozzi, P.J.; Kearsley, J.H.; Li, Y. Emerging roles of radioresistance in prostate cancer metastasis and radiation therapy. Cancer Metastasis Rev. 2014, 33, 469–496. [Google Scholar] [CrossRef]
- Marie-Egyptienne, D.T.; Lohse, I.; Hill, R.P. Cancer stem cells, the epithelial to mesenchymal transition (EMT) and radioresistance: Potential role of hypoxia. Cancer Lett. 2013, 341, 63–72. [Google Scholar] [CrossRef]
- Yao, Y.H.; Cui, Y.; Qiu, X.N.; Zhang, L.Z.; Zhang, W.; Li, H.; Yu, J.M. Attenuated LKB1-SIK1 signaling promotes epithelial-mesenchymal transition and radioresistance of non-small cell lung cancer cells. Chin. J. Cancer 2016, 35, 50. [Google Scholar] [CrossRef] [Green Version]
- Heddleston, J.M.; Li, Z.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle 2009, 8, 3274–3284. [Google Scholar] [CrossRef] [Green Version]
- Salnikov, A.V.; Liu, L.; Platen, M.; Gladkich, J.; Salnikova, O.; Ryschich, E.; Mattern, J.; Moldenhauer, G.; Werner, J.; Schemmer, P.; et al. Hypoxia induces EMT in low and highly aggressive pancreatic tumor cells but only cells with cancer stem cell characteristics acquire pronounced migratory potential. PLoS ONE 2012, 7, e46391. [Google Scholar] [CrossRef]
- Brabletz, S.; Brabletz, T. The ZEB/miR-200 feedback loop--a motor of cellular plasticity in development and cancer? EMBO Rep. 2010, 11, 670–677. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, S.C.; Peters, H.L.; Taguchi, A.; Katayama, H.; Wang, H.; Momin, A.; Jolly, M.K.; Celiktas, M.; Rodriguez-Canales, J.; Liu, H.; et al. Immunoproteasome deficiency is a feature of non-small cell lung cancer with a mesenchymal phenotype and is associated with a poor outcome. Proc. Natl. Acad. Sci. USA 2016, 113, E1555–E1564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Gibbons, D.L.; Goswami, S.; Cortez, M.A.; Ahn, Y.H.; Byers, L.A.; Zhang, X.; Yi, X.; Dwyer, D.; Lin, W.; et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat. Commun. 2014, 5, 5241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Creighton, C.J.; Chang, J.C.; Rosen, J.M. Epithelial-mesenchymal transition (EMT) in tumor-initiating cells and its clinical implications in breast cancer. J. Mammary Gland Biol. Neoplasia 2010, 15, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Vega, S.; Morales, A.V.; Ocana, O.H.; Valdes, F.; Fabregat, I.; Nieto, M.A. Snail blocks the cell cycle and confers resistance to cell death. Genes Dev. 2004, 18, 1131–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comaills, V.; Kabeche, L.; Morris, R.; Buisson, R.; Yu, M.; Madden, M.W.; LiCausi, J.A.; Boukhali, M.; Tajima, K.; Pan, S.; et al. Genomic Instability Is Induced by Persistent Proliferation of Cells Undergoing Epithelial-to-Mesenchymal Transition. Cell Rep. 2016, 17, 2632–2647. [Google Scholar] [CrossRef]
- Liu, M.; Quek, L.E.; Sultani, G.; Turner, N. Epithelial-mesenchymal transition induction is associated with augmented glucose uptake and lactate production in pancreatic ductal adenocarcinoma. Cancer Metab. 2016, 4, 19. [Google Scholar] [CrossRef]
- Cha, Y.H.; Yook, J.I.; Kim, H.S.; Kim, N.H. Catabolic metabolism during cancer EMT. Arch. Pharm. Res. 2015, 38, 313–320. [Google Scholar] [CrossRef]
- Chamberlain, M.C. Radiographic patterns of relapse in glioblastoma. J. Neurooncol. 2011, 101, 319–323. [Google Scholar] [CrossRef]
- Bremnes, R.M.; Al-Shibli, K.; Donnem, T.; Sirera, R.; Al-Saad, S.; Andersen, S.; Stenvold, H.; Camps, C.; Busund, L.T. The role of tumor-infiltrating immune cells and chronic inflammation at the tumor site on cancer development, progression, and prognosis: Emphasis on non-small cell lung cancer. J. Thorac. Oncol. 2011, 6, 824–833. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.Y.; Jeong, E.K.; Ju, M.K.; Jeon, H.M.; Kim, M.Y.; Kim, C.H.; Park, H.G.; Han, S.I.; Kang, H.S. Induction of metastasis, cancer stem cell phenotype, and oncogenic metabolism in cancer cells by ionizing radiation. Mol. Cancer 2017, 16, 10. [Google Scholar] [CrossRef] [Green Version]
- Cho, J.H.; Hong, W.G.; Jung, Y.J.; Lee, J.; Lee, E.; Hwang, S.G.; Um, H.D.; Park, J.K. Gamma-Ionizing radiation-induced activation of the EGFR-p38/ERK-STAT3/CREB-1-EMT pathway promotes the migration/invasion of non-small cell lung cancer cells and is inhibited by podophyllotoxin acetate. Tumour Biol. 2016, 37, 7315–7325. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Guarino, M.; Rubino, B.; Ballabio, G. The role of epithelial-mesenchymal transition in cancer pathology. Pathology 2007, 39, 305–318. [Google Scholar] [CrossRef] [PubMed]
- Wild-Bode, C.; Weller, M.; Rimner, A.; Dichgans, J.; Wick, W. Sublethal irradiation promotes migration and invasiveness of glioma cells: Implications for radiotherapy of human glioblastoma. Cancer Res. 2001, 61, 2744–2750. [Google Scholar] [PubMed]
- Bensimon, J.; Altmeyer-Morel, S.; Benjelloun, H.; Chevillard, S.; Lebeau, J. CD24(-/low) stem-like breast cancer marker defines the radiation-resistant cells involved in memorization and transmission of radiation-induced genomic instability. Oncogene 2013, 32, 251–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.C.; Tsai, J.T.; Chao, T.Y.; Ma, H.I.; Liu, W.H. The STAT3/Slug Axis Enhances Radiation-Induced Tumor Invasion and Cancer Stem-like Properties in Radioresistant Glioblastoma. Cancers 2018, 10, 512. [Google Scholar] [CrossRef] [PubMed]
- Shintani, Y.; Okimura, A.; Sato, K.; Nakagiri, T.; Kadota, Y.; Inoue, M.; Sawabata, N.; Minami, M.; Ikeda, N.; Kawahara, K.; et al. Epithelial to mesenchymal transition is a determinant of sensitivity to chemoradiotherapy in non-small cell lung cancer. Ann. Thorac. Surg. 2011, 92, 1794–1804; discussion 1804. [Google Scholar] [CrossRef]
- Kim, E.; Youn, H.; Kwon, T.; Son, B.; Kang, J.; Yang, H.J.; Seong, K.M.; Kim, W.; Youn, B. PAK1 tyrosine phosphorylation is required to induce epithelial-mesenchymal transition and radioresistance in lung cancer cells. Cancer Res. 2014, 74, 5520–5531. [Google Scholar] [CrossRef] [Green Version]
- Alhaddad, L.; Pustovalova, M.; Blokhina, T.; Chuprov-Netochin, R.; Osipov, A.N.; Leonov, S. IR-Surviving NSCLC Cells Exhibit Different Patterns of Molecular and Cellular Reactions Relating to the Multifraction Irradiation Regimen and p53-Family Proteins Expression. Cancers 2021, 13, 2669. [Google Scholar] [CrossRef]
- Saleh, T.; Carpenter, V.J.; Bloukh, S.; Gewirtz, D.A. Targeting tumor cell senescence and polyploidy as potential therapeutic strategies. Semin. Cancer Biol. 2022, 81, 37–47. [Google Scholar] [CrossRef]
- Zhang, J.; Si, J.; Gan, L.; Di, C.; Xie, Y.; Sun, C.; Li, H.; Guo, M.; Zhang, H. Research progress on therapeutic targeting of quiescent cancer cells. Artif. Cells Nanomed. Biotechnol. 2019, 47, 2810–2820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ewald, J.A.; Desotelle, J.A.; Wilding, G.; Jarrard, D.F. Therapy-induced senescence in cancer. J. Natl. Cancer Inst. 2010, 102, 1536–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguirre-Ghiso, J.A. Models, mechanisms and clinical evidence for cancer dormancy. Nat. Rev. Cancer 2007, 7, 834–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Triana-Martinez, F.; Loza, M.I.; Dominguez, E. Beyond Tumor Suppression: Senescence in Cancer Stemness and Tumor Dormancy. Cells 2020, 9, 346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, S.G.; Jackson, J.G. SASP: Tumor Suppressor or Promoter? Yes! Trends Cancer 2016, 2, 676–687. [Google Scholar] [CrossRef] [Green Version]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef]
- Santos-de-Frutos, K.; Djouder, N. When dormancy fuels tumour relapse. Commun. Biol. 2021, 4, 747. [Google Scholar] [CrossRef]
- Edelstein, J.E. Prosthetic feet. State of the Art. Phys. Ther. 1988, 68, 1874–1881. [Google Scholar] [CrossRef]
- Crea, F.; Nur Saidy, N.R.; Collins, C.C.; Wang, Y. The epigenetic/noncoding origin of tumor dormancy. Trends Mol. Med. 2015, 21, 206–211. [Google Scholar] [CrossRef]
- Meacham, C.E.; Morrison, S.J. Tumour heterogeneity and cancer cell plasticity. Nature 2013, 501, 328–337. [Google Scholar] [CrossRef] [Green Version]
- Mohammad, K.; Dakik, P.; Medkour, Y.; Mitrofanova, D.; Titorenko, V.I. Quiescence Entry, Maintenance, and Exit in Adult Stem Cells. Int. J. Mol. Sci. 2019, 20, 2158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coller, H.A.; Sang, L.; Roberts, J.M. A new description of cellular quiescence. PLoS Biol. 2006, 4, e83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krenning, L.; van den Berg, J.; Medema, R.H. Life or Death after a Break: What Determines the Choice? Mol. Cell 2019, 76, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Shaltiel, I.A.; Krenning, L.; Bruinsma, W.; Medema, R.H. The same, only different—DNA damage checkpoints and their reversal throughout the cell cycle. J. Cell Sci. 2015, 128, 607–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puig, I.; Tenbaum, S.P.; Chicote, I.; Arques, O.; Martinez-Quintanilla, J.; Cuesta-Borras, E.; Ramirez, L.; Gonzalo, P.; Soto, A.; Aguilar, S.; et al. TET2 controls chemoresistant slow-cycling cancer cell survival and tumor recurrence. J. Clin. Investig. 2018, 128, 3887–3905. [Google Scholar] [CrossRef] [Green Version]
- Wolter, K.; Zender, L. Therapy-induced senescence—An induced synthetic lethality in liver cancer? Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 135–136. [Google Scholar] [CrossRef]
- Masunaga, S.; Sakurai, Y.; Tanaka, H.; Hirayama, R.; Matsumoto, Y.; Uzawa, A.; Suzuki, M.; Kondo, N.; Narabayashi, M.; Maruhashi, A.; et al. Radiosensitivity of pimonidazole-unlabelled intratumour quiescent cell population to gamma-rays, accelerated carbon ion beams and boron neutron capture reaction. Br. J. Radiol. 2013, 86, 20120302. [Google Scholar] [CrossRef] [Green Version]
- Miller, I.; Min, M.; Yang, C.; Tian, C.; Gookin, S.; Carter, D.; Spencer, S.L. Ki67 is a Graded Rather than a Binary Marker of Proliferation versus Quiescence. Cell Rep. 2018, 24, 1105–1112. [Google Scholar] [CrossRef] [Green Version]
- Zeniou, M.; Feve, M.; Mameri, S.; Dong, J.; Salome, C.; Chen, W.; El-Habr, E.A.; Bousson, F.; Sy, M.; Obszynski, J.; et al. Chemical Library Screening and Structure-Function Relationship Studies Identify Bisacodyl as a Potent and Selective Cytotoxic Agent Towards Quiescent Human Glioblastoma Tumor Stem-Like Cells. PLoS ONE 2015, 10, e0134793. [Google Scholar] [CrossRef]
- Yao, M.; Xie, C.; Kiang, M.Y.; Teng, Y.; Harman, D.; Tiffen, J.; Wang, Q.; Sved, P.; Bao, S.; Witting, P.; et al. Targeting of cytosolic phospholipase A2alpha impedes cell cycle re-entry of quiescent prostate cancer cells. Oncotarget 2015, 6, 34458–34474. [Google Scholar] [CrossRef] [Green Version]
- Kwon, J.S.; Everetts, N.J.; Wang, X.; Wang, W.; Della Croce, K.; Xing, J.; Yao, G. Controlling Depth of Cellular Quiescence by an Rb-E2F Network Switch. Cell Rep. 2017, 20, 3223–3235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, H.; Guo, Z.; Wang, Z.; Dai, Y.; Zheng, L.; Zhu, L.; Zhang, J.; Hu, W.; Nie, J.; Mao, W.; et al. RAC2 promotes abnormal proliferation of quiescent cells by enhanced JUNB expression via the MAL-SRF pathway. Cell Cycle 2018, 17, 1115–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chitikova, Z.V.; Gordeev, S.A.; Bykova, T.V.; Zubova, S.G.; Pospelov, V.A.; Pospelova, T.V. Sustained activation of DNA damage response in irradiated apoptosis-resistant cells induces reversible senescence associated with mTOR downregulation and expression of stem cell markers. Cell Cycle 2014, 13, 1424–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collado, M.; Gil, J.; Efeyan, A.; Guerra, C.; Schuhmacher, A.J.; Barradas, M.; Benguría, A.; Zaballos, A.; Flores, J.M.; Barbacid, M.; et al. Senescence in premalignant tumours. Nature 2005, 436, 642. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Dhar, D.; Nakagawa, H.; Font-Burgada, J.; Ogata, H.; Jiang, Y.; Shalapour, S.; Seki, E.; Yost, S.E.; Jepsen, K.; et al. Identification of Liver Cancer Progenitors Whose Malignant Progression Depends on Autocrine IL-6 Signaling. Cell 2013, 155, 384–396. [Google Scholar] [CrossRef] [Green Version]
- Joselow, A.; Lynn, D.; Terzian, T.; Box, N.F. Senescence-Like Phenotypes in Human Nevi. Methods Mol. Biol. 2017, 1534, 175–184. [Google Scholar]
- Haugstetter, A.M.; Loddenkemper, C.; Lenze, D.; Gröne, J.; Standfuß, C.; Petersen, I.; Dörken, B.; Schmitt, C.A. Cellular senescence predicts treatment outcome in metastasised colorectal cancer. Br. J. Cancer 2010, 103, 505–509. [Google Scholar] [CrossRef]
- Kahlem, P.; Dörken, B.; Schmitt, C.A. Cellular senescence in cancer treatment: Friend or foe? J. Clin. Investig. 2004, 113, 169–174. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, C.A.; Fridman, J.S.; Yang, M.; Lee, S.; Baranov, E.; Hoffman, R.M.; Lowe, S.W. A Senescence Program Controlled by p53 and p16INK4a Contributes to the Outcome of Cancer Therapy. Cell 2002, 109, 335–346. [Google Scholar] [CrossRef] [Green Version]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef] [Green Version]
- Mikula-Pietrasik, J.; Niklas, A.; Uruski, P.; Tykarski, A.; Ksiazek, K. Mechanisms and significance of therapy-induced and spontaneous senescence of cancer cells. Cell Mol. Life Sci. 2020, 77, 213–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razmik Mirzayans, D.M. Role of Therapy-Induced Cellular Senescence in Tumor Cells and its Modification in Radiotherapy: The Good, The Bad and The Ugly. J. Nucl. Med. Radiat. Ther. 2013, s6. [Google Scholar] [CrossRef]
- Lee, B.Y.; Han, J.A.; Im, J.S.; Morrone, A.; Johung, K.; Goodwin, E.C.; Kleijer, W.J.; DiMaio, D.; Hwang, E.S. Senescence-associated β-galactosidase is lysosomal β-galactosidase. Aging Cell 2006, 5, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Ogrodnik, M.; Miwa, S.; Tchkonia, T.; Tiniakos, D.; Wilson, C.L.; Lahat, A.; Day, C.P.; Burt, A.; Palmer, A.; Anstee, Q.M.; et al. Cellular senescence drives age-dependent hepatic steatosis. Nat. Commun. 2017, 8, 15691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaushik, S.; Cuervo, A.M. Proteostasis and aging. Nat. Med. 2015, 21, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Segura, A.; de Jong, T.V.; Melov, S.; Guryev, V.; Campisi, J.; Demaria, M. Unmasking Transcriptional Heterogeneity in Senescent Cells. Curr. Biol. 2017, 27, 2652–2660.e2654. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, M.; Boothman, D.A. Stress-induced Premature Senescence (SIPS). J. Radiat. Res. 2008, 49, 105–112. [Google Scholar] [CrossRef]
- Saleh, T.; Tyutynuk-Massey, L.; Cudjoe, E.K.; Idowu, M.O.; Landry, J.W.; Gewirtz, D.A. Non-Cell Autonomous Effects of the Senescence-Associated Secretory Phenotype in Cancer Therapy. Front. Oncol. 2018, 8, 164. [Google Scholar] [CrossRef] [Green Version]
- Antonangeli, F.; Soriani, A.; Ricci, B.; Ponzetta, A.; Benigni, G.; Morrone, S.; Bernardini, G.; Santoni, A. Natural killer cell recognition of in vivo drug-induced senescent multiple myeloma cells. OncoImmunology 2016, 5, e1218105. [Google Scholar] [CrossRef] [Green Version]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Hansel, C.; Jendrossek, V.; Klein, D. Cellular Senescence in the Lung: The Central Role of Senescent Epithelial Cells. Int. J. Mol. Sci. 2020, 21, 3279. [Google Scholar] [CrossRef] [PubMed]
- Coppe, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Munoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef] [PubMed]
- Passos, J.F.; Nelson, G.; Wang, C.; Richter, T.; Simillion, C.; Proctor, C.J.; Miwa, S.; Olijslagers, S.; Hallinan, J.; Wipat, A.; et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 2010, 6, 347. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef] [Green Version]
- Chang, B.D.; Broude, E.V.; Dokmanovic, M.; Zhu, H.; Ruth, A.; Xuan, Y.; Kandel, E.S.; Lausch, E.; Christov, K.; Roninson, I.B. A senescence-like phenotype distinguishes tumor cells that undergo terminal proliferation arrest after exposure to anticancer agents. Cancer Res. 1999, 59, 3761–3767. [Google Scholar]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [Green Version]
- Campisi, J. Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol. 2001, 11, S27–S31. [Google Scholar] [CrossRef] [Green Version]
- Chien, Y.; Scuoppo, C.; Wang, X.; Fang, X.; Balgley, B.; Bolden, J.E.; Premsrirut, P.; Luo, W.; Chicas, A.; Lee, C.S.; et al. Control of the senescence-associated secretory phenotype by NF-kappaB promotes senescence and enhances chemosensitivity. Genes Dev. 2011, 25, 2125–2136. [Google Scholar] [CrossRef]
- Aird, K.M.; Zhang, R. Detection of senescence-associated heterochromatin foci (SAHF). Methods Mol. Biol. 2013, 965, 185–196. [Google Scholar] [CrossRef] [Green Version]
- Ksiazek, K.; Korybalska, K.; Jorres, A.; Witowski, J. Accelerated senescence of human peritoneal mesothelial cells exposed to high glucose: The role of TGF-beta1. Lab. Investig. 2007, 87, 345–356. [Google Scholar] [CrossRef] [Green Version]
- Krouwer, V.J.; Hekking, L.H.; Langelaar-Makkinje, M.; Regan-Klapisz, E.; Post, J.A. Endothelial cell senescence is associated with disrupted cell-cell junctions and increased monolayer permeability. Vasc. Cell 2012, 4, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Statuto, M.; Bianchi, C.; Perego, R.; Del Monte, U. Drop of connexin 43 in replicative senescence of human fibroblasts HEL-299 as a possible biomarker of senescence. Exp. Gerontol. 2002, 37, 1113–1120. [Google Scholar] [CrossRef]
- Wang, Q.; Wu, P.C.; Dong, D.Z.; Ivanova, I.; Chu, E.; Zeliadt, S.; Vesselle, H.; Wu, D.Y. Polyploidy road to therapy-induced cellular senescence and escape. Int. J. Cancer 2013, 132, 1505–1515. [Google Scholar] [CrossRef]
- Saleh, T.; Tyutyunyk-Massey, L.; Murray, G.F.; Alotaibi, M.R.; Kawale, A.S.; Elsayed, Z.; Henderson, S.C.; Yakovlev, V.; Elmore, L.W.; Toor, A.; et al. Tumor cell escape from therapy-induced senescence. Biochem. Pharm. 2019, 162, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Fang, J.; Chen, J. Tumor cell senescence response produces aggressive variants. Cell Death Discov. 2017, 3, 17049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghorai, A.; Mahaddalkar, T.; Thorat, R.; Dutt, S. Sustained inhibition of PARP-1 activity delays glioblastoma recurrence by enhancing radiation-induced senescence. Cancer Lett. 2020, 490, 44–53. [Google Scholar] [CrossRef]
- Cahu, J.; Bustany, S.; Sola, B. Senescence-associated secretory phenotype favors the emergence of cancer stem-like cells. Cell Death Dis. 2012, 3, e446. [Google Scholar] [CrossRef] [Green Version]
- Ye, C.; Zhang, X.; Wan, J.; Chang, L.; Hu, W.; Bing, Z.; Zhang, S.; Li, J.; He, J.; Wang, J.; et al. Radiation-induced cellular senescence results from a slippage of long-term G2 arrested cells into G1 phase. Cell Cycle 2013, 12, 1424–1432. [Google Scholar] [CrossRef]
- He, J.; Li, J.; Ye, C.; Zhou, L.; Zhu, J.; Wang, J.; Mizota, A.; Furusawa, Y.; Zhou, G. Cell cycle suspension: A novel process lurking in G(2) arrest. Cell Cycle 2011, 10, 1468–1476. [Google Scholar] [CrossRef] [Green Version]
- Sugrue, M.M.; Shin, D.Y.; Lee, S.W.; Aaronson, S.A. Wild-type p53 triggers a rapid senescence program in human tumor cells lacking functional p53. Proc. Natl. Acad. Sci. USA 1997, 94, 9648–9653. [Google Scholar] [CrossRef] [Green Version]
- Calio, A.; Zamo, A.; Ponzoni, M.; Zanolin, M.E.; Ferreri, A.J.; Pedron, S.; Montagna, L.; Parolini, C.; Fraifeld, V.E.; Wolfson, M.; et al. Cellular Senescence Markers p16INK4a and p21CIP1/WAF Are Predictors of Hodgkin Lymphoma Outcome. Clin. Cancer Res. 2015, 21, 5164–5172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Evangelou, K.; Lougiakis, N.; Rizou, S.V.; Kotsinas, A.; Kletsas, D.; Munoz-Espin, D.; Kastrinakis, N.G.; Pouli, N.; Marakos, P.; Townsend, P.; et al. Robust, universal biomarker assay to detect senescent cells in biological specimens. Aging Cell 2017, 16, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Liao, E.C.; Hsu, Y.T.; Chuah, Q.Y.; Lee, Y.J.; Hu, J.Y.; Huang, T.C.; Yang, P.M.; Chiu, S.J. Radiation induces senescence and a bystander effect through metabolic alterations. Cell Death Dis. 2014, 5, e1255. [Google Scholar] [CrossRef] [Green Version]
- Roninson, I.B. Tumor cell senescence in cancer treatment. Cancer Res. 2003, 63, 2705–2715. [Google Scholar]
- Jeon, H.Y.; Kim, J.K.; Ham, S.W.; Oh, S.Y.; Kim, J.; Park, J.B.; Lee, J.Y.; Kim, S.C.; Kim, H. Irradiation induces glioblastoma cell senescence and senescence-associated secretory phenotype. Tumour Biol. 2016, 37, 5857–5867. [Google Scholar] [CrossRef]
- Quick, Q.A.; Gewirtz, D.A. An accelerated senescence response to radiation in wild-type p53 glioblastoma multiforme cells. J. Neurosurg. 2006, 105, 111–118. [Google Scholar] [CrossRef]
- Luo, H.; Yount, C.; Lang, H.; Yang, A.; Riemer, E.C.; Lyons, K.; Vanek, K.N.; Silvestri, G.A.; Schulte, B.A.; Wang, G.Y. Activation of p53 with Nutlin-3a radiosensitizes lung cancer cells via enhancing radiation-induced premature senescence. Lung Cancer 2013, 81, 167–173. [Google Scholar] [CrossRef] [Green Version]
- Jones, K.R.; Elmore, L.W.; Jackson-Cook, C.; Demasters, G.; Povirk, L.F.; Holt, S.E.; Gewirtz, D.A. p53-Dependent accelerated senescence induced by ionizing radiation in breast tumour cells. Int. J. Radiat. Biol. 2005, 81, 445–458. [Google Scholar] [CrossRef]
- Roberson, R.S.; Kussick, S.J.; Vallieres, E.; Chen, S.Y.; Wu, D.Y. Escape from therapy-induced accelerated cellular senescence in p53-null lung cancer cells and in human lung cancers. Cancer Res. 2005, 65, 2795–2803. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Wu, P.C.; Roberson, R.S.; Luk, B.V.; Ivanova, I.; Chu, E.; Wu, D.Y. Survivin and escaping in therapy-induced cellular senescence. Int. J. Cancer 2011, 128, 1546–1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azad, A.; Bukczynska, P.; Jackson, S.; Haupt, Y.; Cullinane, C.; McArthur, G.A.; Solomon, B. Co-targeting deoxyribonucleic acid-dependent protein kinase and poly(adenosine diphosphate-ribose) polymerase-1 promotes accelerated senescence of irradiated cancer cells. Int. J. Radiat. Oncol. Biol. Phys. 2014, 88, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.R.; Amon, A. Aneuploidy: Cancer’s fatal flaw? Cancer Res. 2009, 69, 5289–5291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sudo, T.; Nitta, M.; Saya, H.; Ueno, N.T. Dependence of paclitaxel sensitivity on a functional spindle assembly checkpoint. Cancer Res. 2004, 64, 2502–2508. [Google Scholar] [CrossRef] [Green Version]
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef]
- Zhang, Z.; Feng, X.; Deng, Z.; Cheng, J.; Wang, Y.; Zhao, M.; Zhao, Y.; He, S.; Huang, Q. Irradiation-induced polyploid giant cancer cells are involved in tumor cell repopulation via neosis. Mol. Oncol. 2021, 15, 2219–2234. [Google Scholar] [CrossRef]
- Rand, C.W.; Courville, C.B. Multinucleation of cortical nerve cells at the margins of traumatic lesions of the human brain. J. Neuropathol. Exp. Neurol. 1947, 6, 1–14. [Google Scholar] [CrossRef]
- Liu, J. The dualistic origin of human tumors. Semin. Cancer Biol. 2018, 53, 1–16. [Google Scholar] [CrossRef]
- Barok, M.; Tanner, M.; Koninki, K.; Isola, J. Trastuzumab-DM1 causes tumour growth inhibition by mitotic catastrophe in trastuzumab-resistant breast cancer cells in vivo. Breast Cancer Res. 2011, 13, R46. [Google Scholar] [CrossRef]
- Lin, K.C.; Torga, G.; Sun, Y.; Axelrod, R.; Pienta, K.J.; Sturm, J.C.; Austin, R.H. The role of heterogeneous environment and docetaxel gradient in the emergence of polyploid, mesenchymal and resistant prostate cancer cells. Clin. Exp. Metastasis 2019, 36, 97–108. [Google Scholar] [CrossRef]
- Ogden, A.; Rida, P.C.; Knudsen, B.S.; Kucuk, O.; Aneja, R. Docetaxel-induced polyploidization may underlie chemoresistance and disease relapse. Cancer Lett. 2015, 367, 89–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Zhang, D.; Yang, Z.; Zhang, X. Tumor Budding, Micropapillary Pattern, and Polyploidy Giant Cancer Cells in Colorectal Cancer: Current Status and Future Prospects. Stem Cells Int. 2016, 2016, 4810734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fei, F.; Zhang, M.; Li, B.; Zhao, L.; Wang, H.; Liu, L.; Li, Y.; Ding, P.; Gu, Y.; Zhang, X.; et al. Formation of Polyploid Giant Cancer Cells Involves in the Prognostic Value of Neoadjuvant Chemoradiation in Locally Advanced Rectal Cancer. J. Oncol. 2019, 2019, 2316436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Mercado-Uribe, I.; Xing, Z.; Sun, B.; Kuang, J.; Liu, J. Generation of cancer stem-like cells through the formation of polyploid giant cancer cells. Oncogene 2014, 33, 116–128. [Google Scholar] [CrossRef]
- Fujiwara, T.; Bandi, M.; Nitta, M.; Ivanova, E.V.; Bronson, R.T.; Pellman, D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 2005, 437, 1043–1047. [Google Scholar] [CrossRef]
- Revesz, L.; Norman, U. Chromosome ploidy and radiosensitivity of tumours. Nature 1960, 187, 861–862. [Google Scholar] [CrossRef]
- Castedo, M.; Coquelle, A.; Vitale, I.; Vivet, S.; Mouhamad, S.; Viaud, S.; Zitvogel, L.; Kroemer, G. Selective resistance of tetraploid cancer cells against DNA damage-induced apoptosis. Ann. N. Y. Acad. Sci. 2006, 1090, 35–49. [Google Scholar] [CrossRef]
- Ianzini, F.; Kosmacek, E.A.; Nelson, E.S.; Napoli, E.; Erenpreisa, J.; Kalejs, M.; Mackey, M.A. Activation of meiosis-specific genes is associated with depolyploidization of human tumor cells following radiation-induced mitotic catastrophe. Cancer Res. 2009, 69, 2296–2304. [Google Scholar] [CrossRef] [Green Version]
- Prieur-Carrillo, G.; Chu, K.; Lindqvist, J.; Dewey, W.C. Computerized video time-lapse (CVTL) analysis of the fate of giant cells produced by X-irradiating EJ30 human bladder carcinoma cells. Radiat Res. 2003, 159, 705–712. [Google Scholar] [CrossRef]
- Ianzini, F.; Mackey, M.A. Development of the large scale digital cell analysis system. Radiat. Prot. Dosim. 2002, 99, 289–293. [Google Scholar] [CrossRef]
- Erenpreisa, J.A.; Cragg, M.S.; Fringes, B.; Sharakhov, I.; Illidge, T.M. Release of mitotic descendants by giant cells from irradiated Burkitt’s lymphoma cell line. Cell Biol. Int. 2000, 24, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Horbay, R.; Stoika, R. Giant cell formation: The way to cell death or cell survival? Open Life Sci. 2011, 6, 675–684. [Google Scholar] [CrossRef]
- Sliwinska, M.A.; Mosieniak, G.; Wolanin, K.; Babik, A.; Piwocka, K.; Magalska, A.; Szczepanowska, J.; Fronk, J.; Sikora, E. Induction of senescence with doxorubicin leads to increased genomic instability of HCT116 cells. Mech. Ageing Dev. 2009, 130, 24–32. [Google Scholar] [CrossRef]
- Niu, N.; Mercado-Uribe, I.; Liu, J. Dedifferentiation into blastomere-like cancer stem cells via formation of polyploid giant cancer cells. Oncogene 2017, 36, 4887–4900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erenpreisa, J.; Salmina, K.; Huna, A.; Jackson, T.R.; Vazquez-Martin, A.; Cragg, M.S. The "virgin birth", polyploidy, and the origin of cancer. Oncoscience 2015, 2, 3–14. [Google Scholar] [CrossRef]
- Zhang, D.; Yang, X.; Yang, Z.; Fei, F.; Li, S.; Qu, J.; Zhang, M.; Li, Y.; Zhang, X.; Zhang, S. Daughter Cells and Erythroid Cells Budding from PGCCs and Their Clinicopathological Significances in Colorectal Cancer. J. Cancer 2017, 8, 469–478. [Google Scholar] [CrossRef] [Green Version]
- Hosaka, M.; Hatori, M.; Smith, R.; Kokubun, S. Giant cell formation through fusion of cells derived from a human giant cell tumor of tendon sheath. J. Orthop. Sci. 2004, 9, 581–584. [Google Scholar] [CrossRef]
- Brodbeck, W.G.; Anderson, J.M. Giant cell formation and function. Curr. Opin. Hematol. 2009, 16, 53–57. [Google Scholar] [CrossRef] [Green Version]
- Holland, A.J.; Cleveland, D.W. Boveri revisited: Chromosomal instability, aneuploidy and tumorigenesis. Nat. Rev. Mol. Cell Biol. 2009, 10, 478–487. [Google Scholar] [CrossRef]
- Krajcovic, M.; Overholtzer, M. Mechanisms of ploidy increase in human cancers: A new role for cell cannibalism. Cancer Res. 2012, 72, 1596–1601. [Google Scholar] [CrossRef] [Green Version]
- Erenpreisa, J.; Cragg, M.S. Three steps to the immortality of cancer cells: Senescence, polyploidy and self-renewal. Cancer Cell Int. 2013, 13, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beermann, W. Control of Differentiation at the Chromosomal Level. J. Exp. Zool. 1964, 157, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Erenpreisa, J.; Cragg, M.S.; Anisimov, A.P.; Illidge, T.M. Tumor cell embryonality and the ploidy number 32n: Is it a developmental checkpoint? Cell Cycle 2011, 10, 1873–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vakifahmetoglu, H.; Olsson, M.; Zhivotovsky, B. Death through a tragedy: Mitotic catastrophe. Cell Death Differ. 2008, 15, 1153–1162. [Google Scholar] [CrossRef] [Green Version]
- Kaur, E.; Rajendra, J.; Jadhav, S.; Shridhar, E.; Goda, J.S.; Moiyadi, A.; Dutt, S. Radiation-induced homotypic cell fusions of innately resistant glioblastoma cells mediate their sustained survival and recurrence. Carcinogenesis 2015, 36, 685–695. [Google Scholar] [CrossRef] [Green Version]
- Sundaram, M.; Guernsey, D.L.; Rajaraman, M.M.; Rajaraman, R. Neosis: A novel type of cell division in cancer. Cancer Biol. Ther. 2004, 3, 207–218. [Google Scholar] [CrossRef] [Green Version]
- Erenpreisa, J.; Cragg, M.S. Mitotic death: A mechanism of survival? A review. Cancer Cell Int. 2001, 1, 1. [Google Scholar] [CrossRef]
- Niu, N.; Zhang, J.; Zhang, N.; Mercado-Uribe, I.; Tao, F.; Han, Z.; Pathak, S.; Multani, A.S.; Kuang, J.; Yao, J.; et al. Linking genomic reorganization to tumor initiation via the giant cell cycle. Oncogenesis 2016, 5, e281. [Google Scholar] [CrossRef] [Green Version]
- Erenpreisa, J.; Ivanov, A.; Wheatley, S.P.; Kosmacek, E.A.; Ianzini, F.; Anisimov, A.P.; Mackey, M.; Davis, P.J.; Plakhins, G.; Illidge, T.M. Endopolyploidy in irradiated p53-deficient tumour cell lines: Persistence of cell division activity in giant cells expressing Aurora-B kinase. Cell Biol. Int. 2008, 32, 1044–1056. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Cragg, M.S.; Salmina, K.; Hausmann, M.; Scherthan, H. The role of meiotic cohesin REC8 in chromosome segregation in gamma irradiation-induced endopolyploid tumour cells. Exp. Cell Res. 2009, 315, 2593–2603. [Google Scholar] [CrossRef]
- Vitale, I.; Senovilla, L.; Jemaa, M.; Michaud, M.; Galluzzi, L.; Kepp, O.; Nanty, L.; Criollo, A.; Rello-Varona, S.; Manic, G.; et al. Multipolar mitosis of tetraploid cells: Inhibition by p53 and dependency on Mos. EMBO J. 2010, 29, 1272–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajaraman, R.; Guernsey, D.L.; Rajaraman, M.M.; Rajaraman, S.R. Stem cells, senescence, neosis and self-renewal in cancer. Cancer Cell Int. 2006, 6, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajaraman, R.; Rajaraman, M.M.; Rajaraman, S.R.; Guernsey, D.L. Neosis--a paradigm of self-renewal in cancer. Cell Biol. Int. 2005, 29, 1084–1097. [Google Scholar] [CrossRef] [PubMed]
- White-Gilbertson, S.; Voelkel-Johnson, C. Giants and monsters: Unexpected characters in the story of cancer recurrence. Adv. Cancer Res. 2020, 148, 201–232. [Google Scholar] [CrossRef]
- Diaz-Carballo, D.; Saka, S.; Klein, J.; Rennkamp, T.; Acikelli, A.H.; Malak, S.; Jastrow, H.; Wennemuth, G.; Tempfer, C.; Schmitz, I.; et al. A Distinct Oncogenerative Multinucleated Cancer Cell Serves as a Source of Stemness and Tumor Heterogeneity. Cancer Res. 2018, 78, 2318–2331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puck, T.T.; Marcus, P.I. Action of x-rays on mammalian cells. J. Exp. Med. 1956, 103, 653–666. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.C.; Thornton, A.F., Jr.; Sandler, H.M.; Greenberg, H.S. Malignant astrocytomas: Focal tumor recurrence after focal external beam radiation therapy. J. Neurosurg. 1991, 75, 559–563. [Google Scholar] [CrossRef] [Green Version]
- Sneed, P.K.; Gutin, P.H.; Larson, D.A.; Malec, M.K.; Phillips, T.L.; Prados, M.D.; Scharfen, C.O.; Weaver, K.A.; Wara, W.M. Patterns of recurrence of glioblastoma multiforme after external irradiation followed by implant boost. Int. J. Radiat. Oncol. Biol. Phys. 1994, 29, 719–727. [Google Scholar] [CrossRef]
- Sitarz, R.; Leguit, R.J.; de Leng, W.W.; Morsink, F.H.; Polkowski, W.P.; Maciejewski, R.; Offerhaus, G.J.; Milne, A.N. Cyclooxygenase-2 mediated regulation of E-cadherin occurs in conventional but not early-onset gastric cancer cell lines. Cell Oncol. 2009, 31, 475–485. [Google Scholar] [CrossRef]
- Hoskin, D.W.; Mader, J.S.; Furlong, S.J.; Conrad, D.M.; Blay, J. Inhibition of T cell and natural killer cell function by adenosine and its contribution to immune evasion by tumor cells (Review). Int. J. Oncol. 2008, 32, 527–535. [Google Scholar] [CrossRef] [Green Version]
- Malik, S.T.; Griffin, D.B.; Fiers, W.; Balkwill, F.R. Paradoxical effects of tumour necrosis factor in experimental ovarian cancer. Int. J. Cancer 1989, 44, 918–925. [Google Scholar] [CrossRef] [PubMed]
- Sa, G.; Das, T.; Moon, C.; Hilston, C.M.; Rayman, P.A.; Rini, B.I.; Tannenbaum, C.S.; Finke, J.H. GD3, an overexpressed tumor-derived ganglioside, mediates the apoptosis of activated but not resting T cells. Cancer Res. 2009, 69, 3095–3104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toutirais, O.; Chartier, P.; Dubois, D.; Bouet, F.; Leveque, J.; Catros-Quemener, V.; Genetet, N. Constitutive expression of TGF-beta1, interleukin-6 and interleukin-8 by tumor cells as a major component of immune escape in human ovarian carcinoma. Eur. Cytokine Netw. 2003, 14, 246–255. [Google Scholar] [PubMed]
- Inoue, K.; Slaton, J.W.; Kim, S.J.; Perrotte, P.; Eve, B.Y.; Bar-Eli, M.; Radinsky, R.; Dinney, C.P. Interleukin 8 expression regulates tumorigenicity and metastasis in human bladder cancer. Cancer Res. 2000, 60, 2290–2299. [Google Scholar] [CrossRef] [PubMed]
- Doubrovina, E.S.; Doubrovin, M.M.; Vider, E.; Sisson, R.B.; O’Reilly, R.J.; Dupont, B.; Vyas, Y.M. Evasion from NK cell immunity by MHC class I chain-related molecules expressing colon adenocarcinoma. J. Immunol. 2003, 171, 6891–6899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannino, M.; Chalmers, A.J. Radioresistance of glioma stem cells: Intrinsic characteristic or property of the ’microenvironment-stem cell unit’? Mol. Oncol. 2011, 5, 374–386. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.; Ishida, T.; Oyama, T.; Ran, S.; Kravtsov, V.; Nadaf, S.; Carbone, D.P. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood 1998, 92, 4150–4166. [Google Scholar] [CrossRef]
- Whiteside, T.L.; Vujanovic, N.L.; Herberman, R.B. Natural killer cells and tumor therapy. Curr. Top. Microbiol. Immunol. 1998, 230, 221–244. [Google Scholar] [CrossRef]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Wolchok, J.D. General Principles of Tumor Immunotherapy; Springer: Berlin/Heidelberg, Germany, 2007. [Google Scholar]
- Baay, M.; Brouwer, A.; Pauwels, P.; Peeters, M.; Lardon, F. Tumor cells and tumor-associated macrophages: Secreted proteins as potential targets for therapy. Clin. Dev. Immunol. 2011, 2011, 565187. [Google Scholar] [CrossRef] [Green Version]
- Tong, H.; Ke, J.Q.; Jiang, F.Z.; Wang, X.J.; Wang, F.Y.; Li, Y.R.; Lu, W.; Wan, X.P. Tumor-associated macrophage-derived CXCL8 could induce ERalpha suppression via HOXB13 in endometrial cancer. Cancer Lett. 2016, 376, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Augello, A.; Tasso, R.; Negrini, S.M.; Amateis, A.; Indiveri, F.; Cancedda, R.; Pennesi, G. Bone marrow mesenchymal progenitor cells inhibit lymphocyte proliferation by activation of the programmed death 1 pathway. Eur. J. Immunol. 2005, 35, 1482–1490. [Google Scholar] [CrossRef] [PubMed]
- Di Nicola, M.; Carlo-Stella, C.; Magni, M.; Milanesi, M.; Longoni, P.D.; Matteucci, P.; Grisanti, S.; Gianni, A.M. Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood 2002, 99, 3838–3843. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [Green Version]
- Caires, H.R.; Barros da Silva, P.; Barbosa, M.A.; Almeida, C.R. A co-culture system with three different primary human cell populations reveals that biomaterials and MSC modulate macrophage-driven fibroblast recruitment. J. Tissue Eng. Regen. Med. 2018, 12, e1433–e1440. [Google Scholar] [CrossRef]
- Almand, B.; Clark, J.I.; Nikitina, E.; van Beynen, J.; English, N.R.; Knight, S.C.; Carbone, D.P.; Gabrilovich, D.I. Increased production of immature myeloid cells in cancer patients: A mechanism of immunosuppression in cancer. J. Immunol. 2001, 166, 678–689. [Google Scholar] [CrossRef] [Green Version]
- Leonard, W.; Dufait, I.; Schwarze, J.K.; Law, K.; Engels, B.; Jiang, H.; Van den Berge, D.; Gevaert, T.; Storme, G.; Verovski, V.; et al. Myeloid-derived suppressor cells reveal radioprotective properties through arginase-induced l-arginine depletion. Radiother. Oncol. 2016, 119, 291–299. [Google Scholar] [CrossRef]
- Ochoa, A.C.; Zea, A.H.; Hernandez, C.; Rodriguez, P.C. Arginase, prostaglandins, and myeloid-derived suppressor cells in renal cell carcinoma. Clin. Cancer Res. 2007, 13, 721s–726s. [Google Scholar] [CrossRef] [Green Version]
- Serafini, P.; Borrello, I.; Bronte, V. Myeloid suppressor cells in cancer: Recruitment, phenotype, properties, and mechanisms of immune suppression. Semin. Cancer Biol. 2006, 16, 53–65. [Google Scholar] [CrossRef]
- Munn, D.H.; Mellor, A.L. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J. Clin. Investig. 2007, 117, 1147–1154. [Google Scholar] [CrossRef] [Green Version]
- Loukinova, E.; Dong, G.; Enamorado-Ayalya, I.; Thomas, G.R.; Chen, Z.; Schreiber, H.; Van Waes, C. Growth regulated oncogene-alpha expression by murine squamous cell carcinoma promotes tumor growth, metastasis, leukocyte infiltration and angiogenesis by a host CXC receptor-2 dependent mechanism. Oncogene 2000, 19, 3477–3486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakaguchi, S.; Miyara, M.; Costantino, C.M.; Hafler, D.A. FOXP3+ regulatory T cells in the human immune system. Nat. Rev. Immunol. 2010, 10, 490–500. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, C.; Strauss, L.; Zeidler, R.; Lang, S.; Whiteside, T.L. Expansion of human T regulatory type 1 cells in the microenvironment of cyclooxygenase 2 overexpressing head and neck squamous cell carcinoma. Cancer Res. 2007, 67, 8865–8873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, M.P.; Piconese, S. Regulatory-T-cell inhibition versus depletion: The right choice in cancer immunotherapy. Nat. Rev. Cancer 2007, 7, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Arden, K.C. FOXO animal models reveal a variety of diverse roles for FOXO transcription factors. Oncogene 2008, 27, 2345–2350. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Jiang, P.; Wei, S.; Xu, X.; Wang, J. Regulatory T cells in tumor microenvironment: New mechanisms, potential therapeutic strategies and future prospects. Mol. Cancer 2020, 19, 116. [Google Scholar] [CrossRef]
- Kochetkova, I.; Golden, S.; Holderness, K.; Callis, G.; Pascual, D.W. IL-35 stimulation of CD39+ regulatory T cells confers protection against collagen II-induced arthritis via the production of IL-10. J. Immunol. 2010, 184, 7144–7153. [Google Scholar] [CrossRef] [Green Version]
- Kitahata, Y.; Shinkai, M.; Kido, T. Guidance and nursing of a patient with acute myocardial infarction and senile dementia: A case study. Kango Gijutsu 1988, 34, 1052–1056. [Google Scholar]
- Ihara, F.; Sakurai, D.; Takami, M.; Kamata, T.; Kunii, N.; Yamasaki, K.; Iinuma, T.; Nakayama, T.; Motohashi, S.; Okamoto, Y. Regulatory T cells induce CD4(-) NKT cell anergy and suppress NKT cell cytotoxic function. Cancer Immunol. Immunother. 2019, 68, 1935–1947. [Google Scholar] [CrossRef]
- Spolski, R.; Li, P.; Leonard, W.J. Biology and regulation of IL-2: From molecular mechanisms to human therapy. Nat. Rev. Immunol. 2018, 18, 648–659. [Google Scholar] [CrossRef]
- Ohta, A.; Kini, R.; Ohta, A.; Subramanian, M.; Madasu, M.; Sitkovsky, M. The development and immunosuppressive functions of CD4(+) CD25(+) FoxP3(+) regulatory T cells are under influence of the adenosine-A2A adenosine receptor pathway. Front. Immunol. 2012, 3, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raimondi, G.; Turner, M.S.; Thomson, A.W.; Morel, P.A. Naturally occurring regulatory T cells: Recent insights in health and disease. Crit. Rev. Immunol. 2007, 27, 61–95. [Google Scholar] [CrossRef] [PubMed]
- Roncarolo, M.G.; Gregori, S.; Battaglia, M.; Bacchetta, R.; Fleischhauer, K.; Levings, M.K. Interleukin-10-secreting type 1 regulatory T cells in rodents and humans. Immunol. Rev. 2006, 212, 28–50. [Google Scholar] [CrossRef] [PubMed]
- Baratelli, F.; Lin, Y.; Zhu, L.; Yang, S.C.; Heuze-Vourc’h, N.; Zeng, G.; Reckamp, K.; Dohadwala, M.; Sharma, S.; Dubinett, S.M. Prostaglandin E2 induces FOXP3 gene expression and T regulatory cell function in human CD4+ T cells. J. Immunol. 2005, 175, 1483–1490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, N.F.; Carter, T.J.; Ottaviani, D.; Mulholland, P. Harnessing the immune system in glioblastoma. Br. J. Cancer 2018, 119, 1171–1181. [Google Scholar] [CrossRef] [Green Version]
- Abedalthagafi, M.; Barakeh, D.; Foshay, K.M. Immunogenetics of glioblastoma: The future of personalized patient management. NPJ Precis. Oncol. 2018, 2, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, J.; Piao, Y.; Holmes, L.; Fuller, G.N.; Henry, V.; Tiao, N.; de Groot, J.F. Neutrophils promote the malignant glioma phenotype through S100A4. Clin. Cancer Res. 2014, 20, 187–198. [Google Scholar] [CrossRef] [Green Version]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007, 11, 69–82. [Google Scholar] [CrossRef] [Green Version]
- Cui, Y.H.; Suh, Y.; Lee, H.J.; Yoo, K.C.; Uddin, N.; Jeong, Y.J.; Lee, J.S.; Hwang, S.G.; Nam, S.Y.; Kim, M.J.; et al. Radiation promotes invasiveness of non-small-cell lung cancer cells through granulocyte-colony-stimulating factor. Oncogene 2015, 34, 5372–5382. [Google Scholar] [CrossRef]
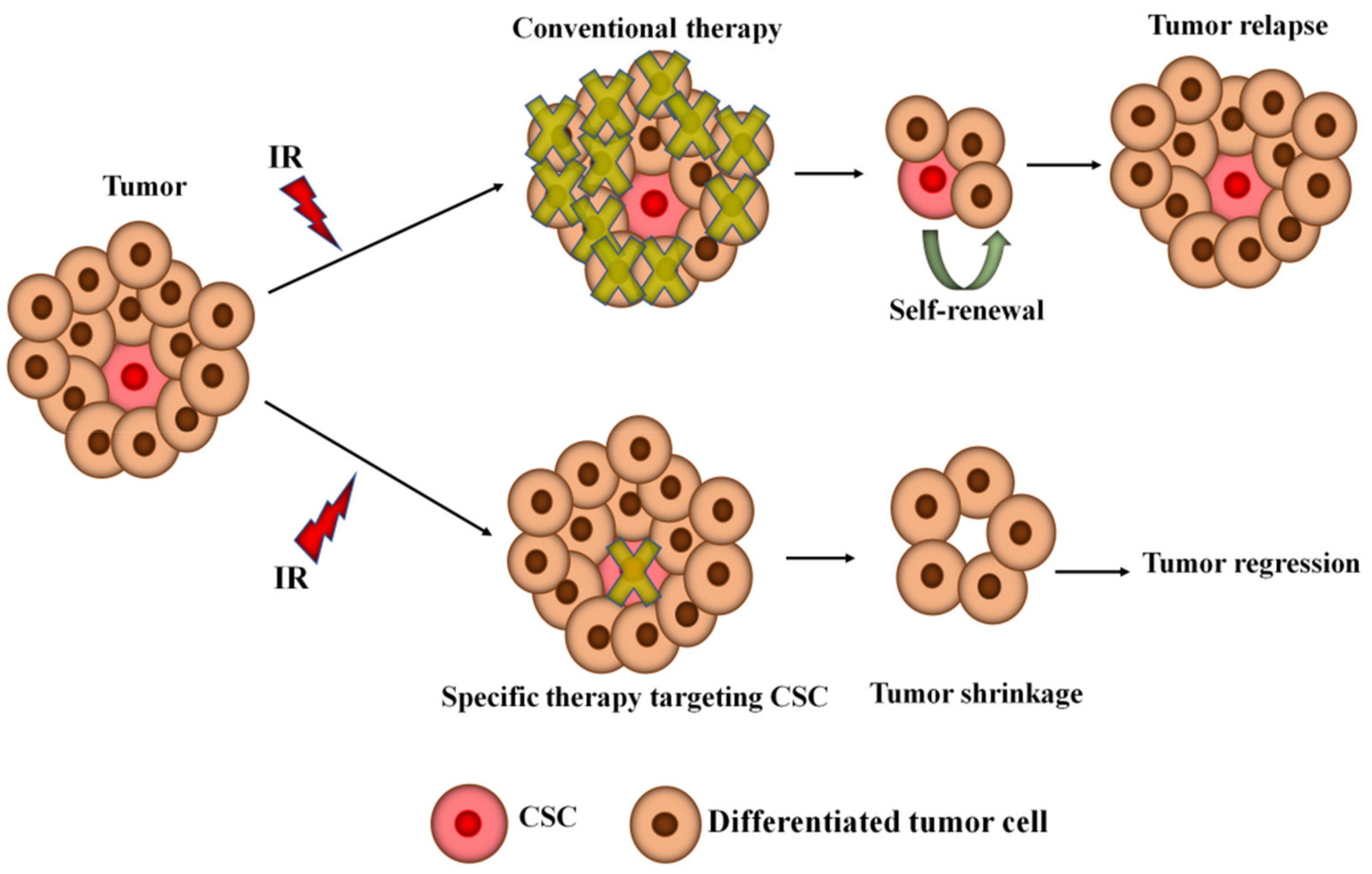
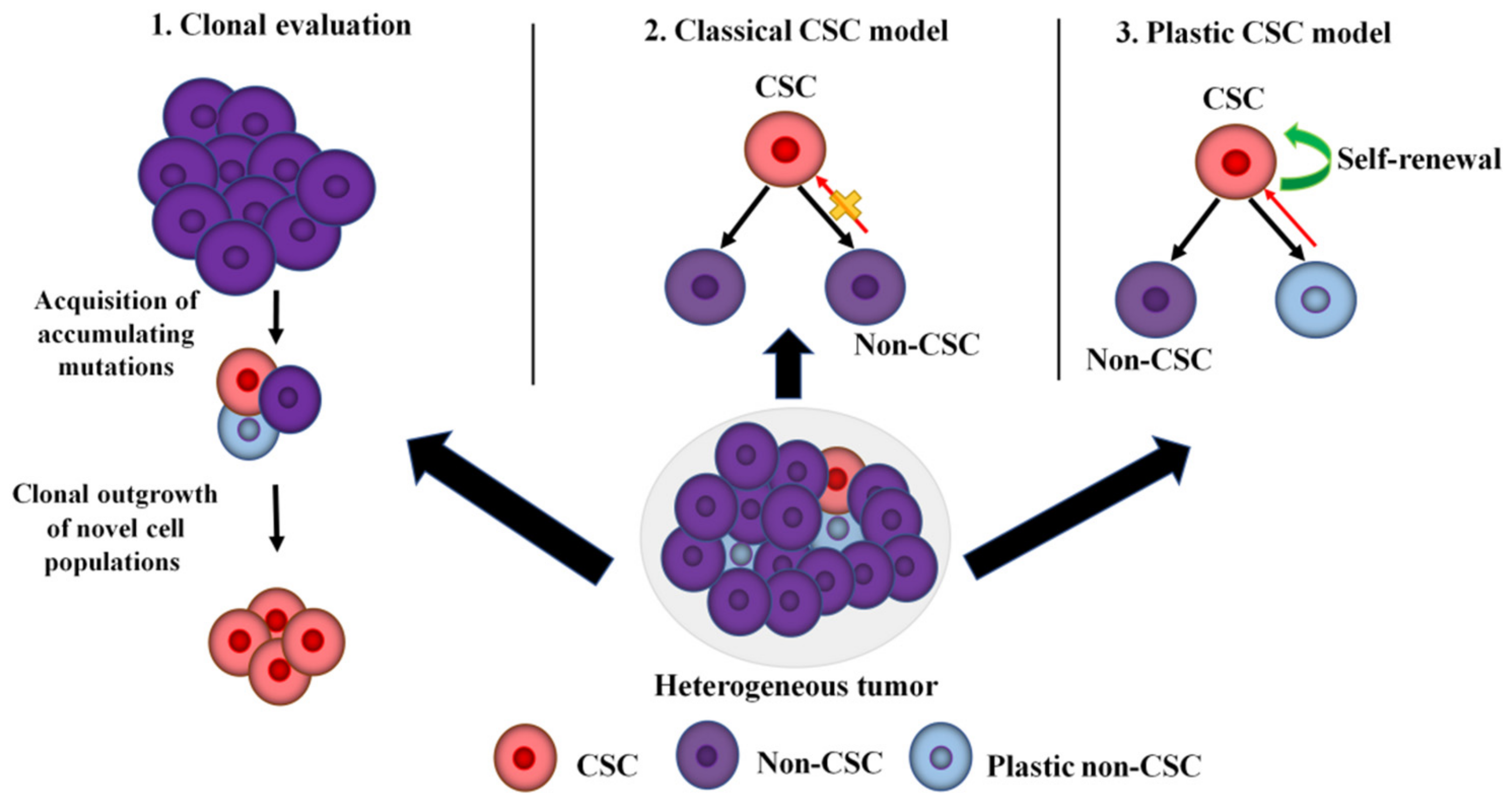
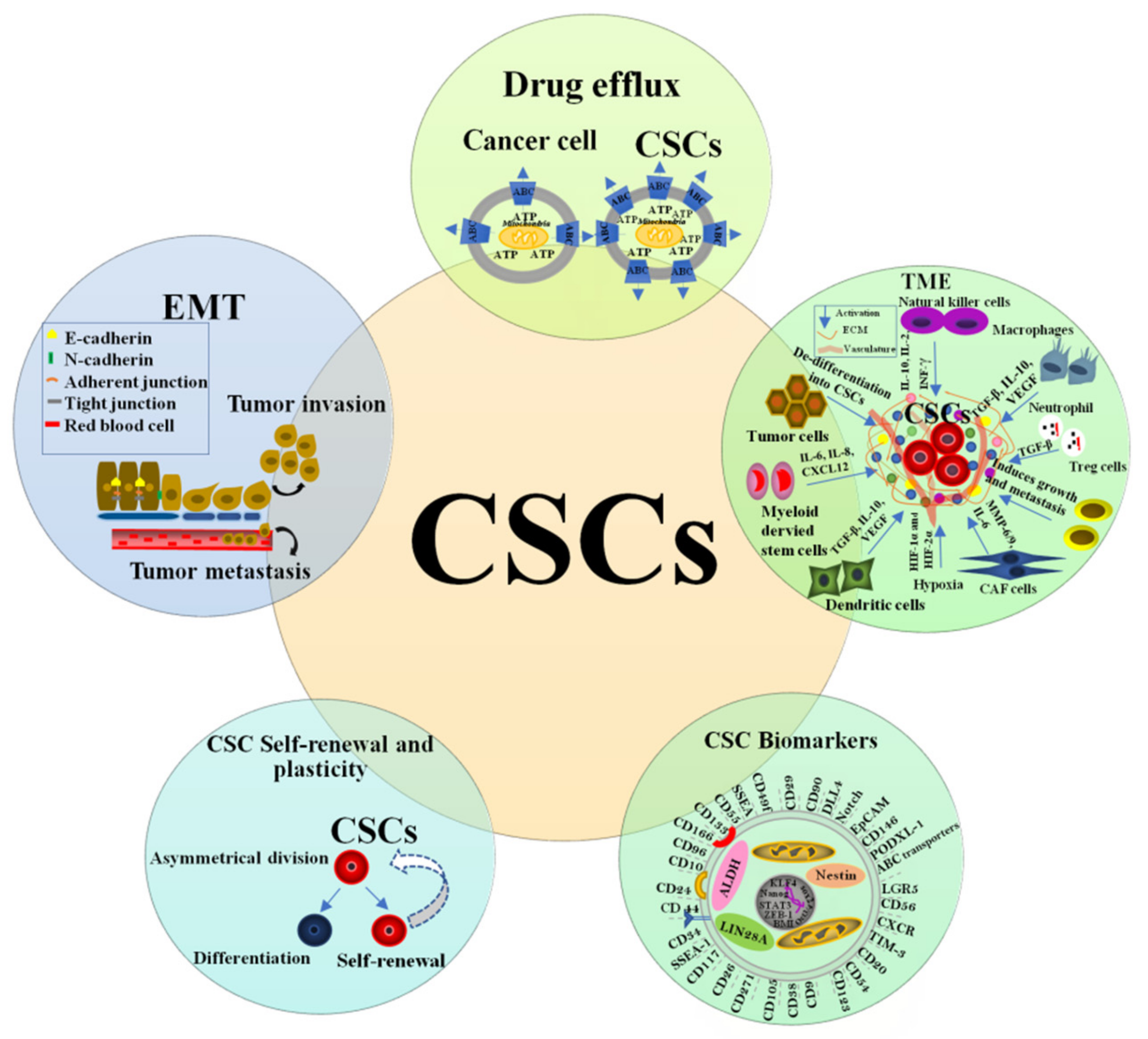
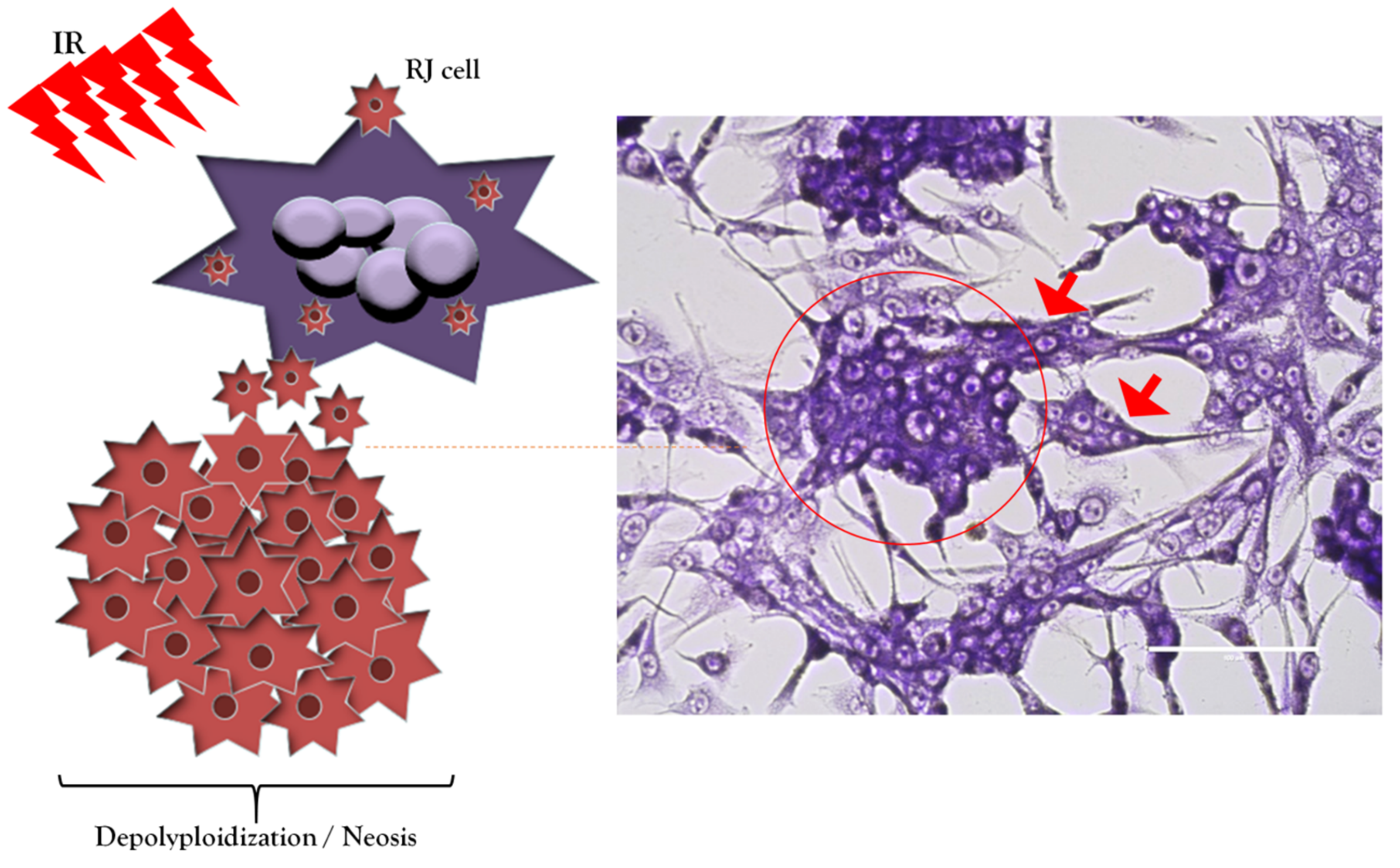
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alhaddad, L.; Osipov, A.N.; Leonov, S. The Molecular and Cellular Strategies of Glioblastoma and Non-Small-Cell Lung Cancer Cells Conferring Radioresistance. Int. J. Mol. Sci. 2022, 23, 13577. https://doi.org/10.3390/ijms232113577
Alhaddad L, Osipov AN, Leonov S. The Molecular and Cellular Strategies of Glioblastoma and Non-Small-Cell Lung Cancer Cells Conferring Radioresistance. International Journal of Molecular Sciences. 2022; 23(21):13577. https://doi.org/10.3390/ijms232113577
Chicago/Turabian StyleAlhaddad, Lina, Andreyan N. Osipov, and Sergey Leonov. 2022. "The Molecular and Cellular Strategies of Glioblastoma and Non-Small-Cell Lung Cancer Cells Conferring Radioresistance" International Journal of Molecular Sciences 23, no. 21: 13577. https://doi.org/10.3390/ijms232113577