
Defective Thyroglobulin: Cell Biology of Disease


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Role of the Endoplasmic Reticulum in Secretory Protein Synthesis and Intracellular Transport
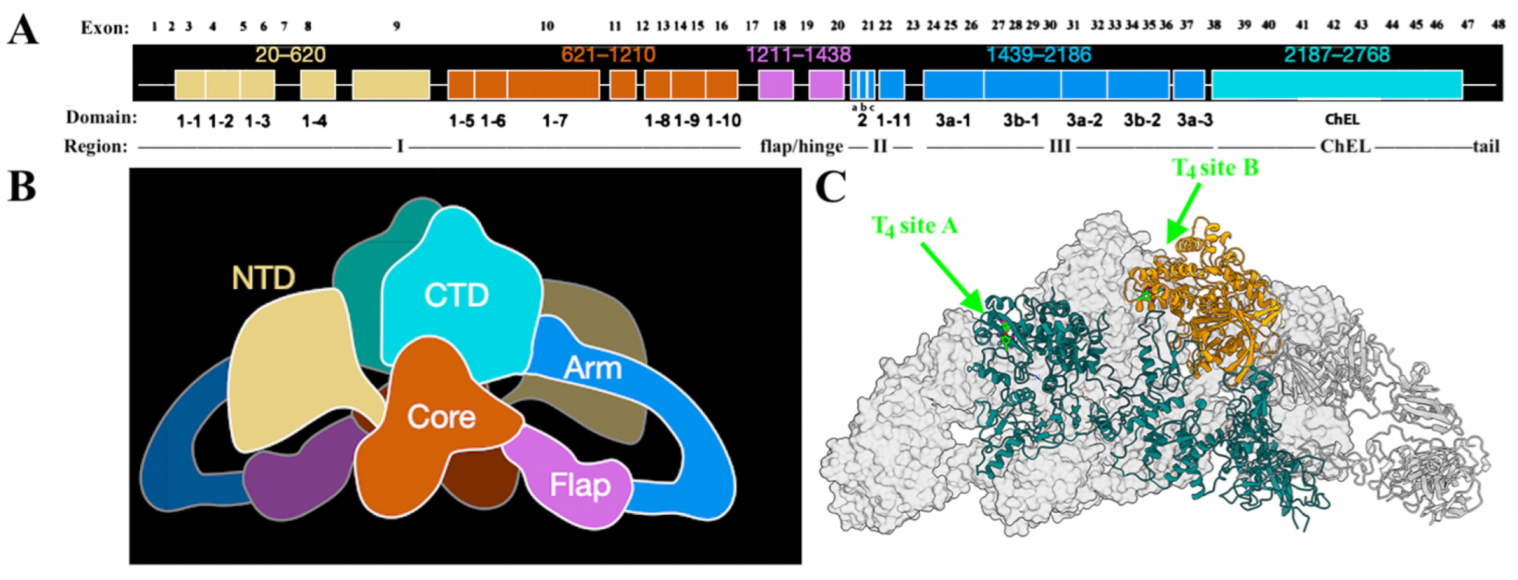
2. Tg Folding and TG Mutations That Trigger Misfolding
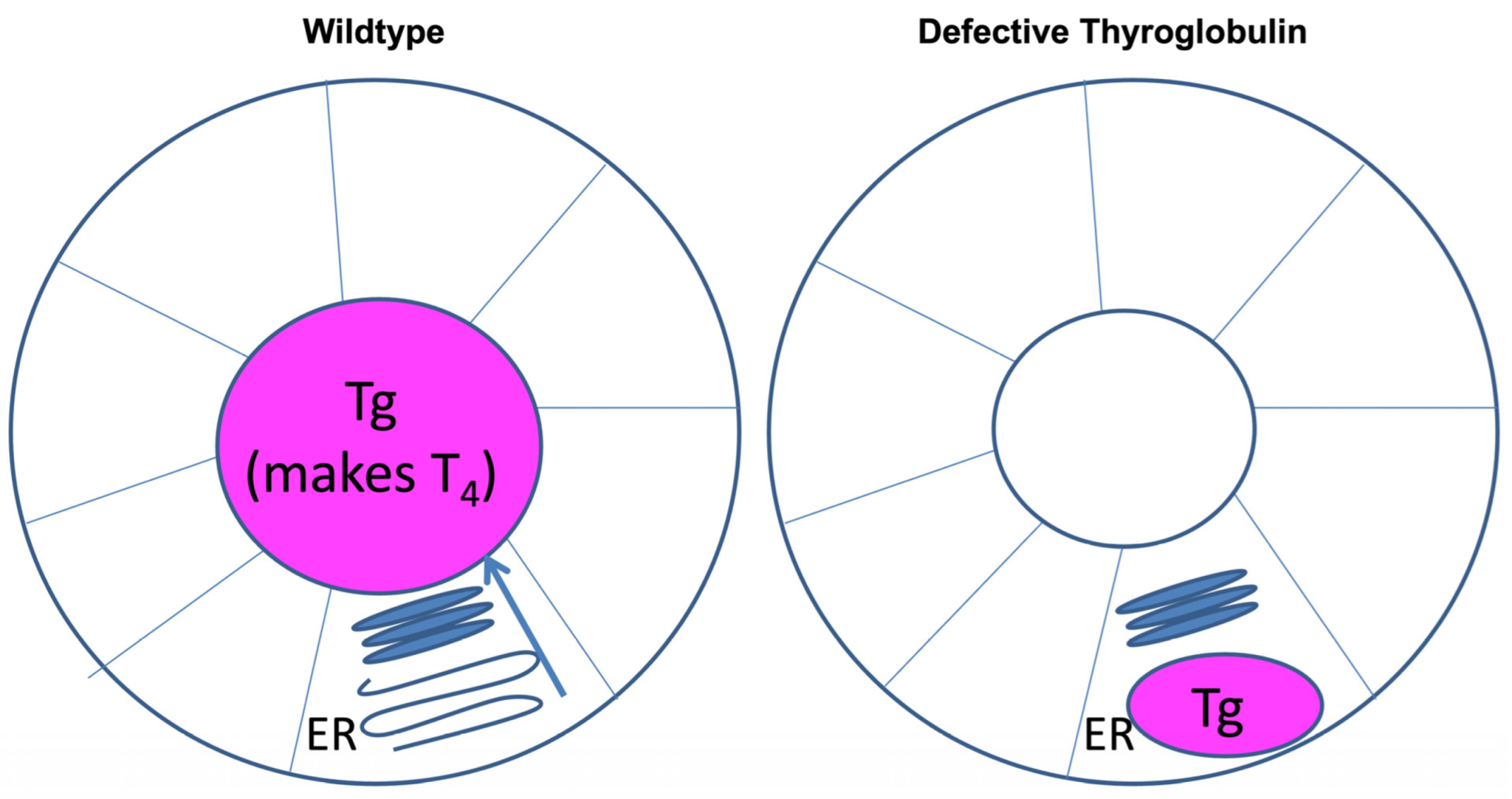
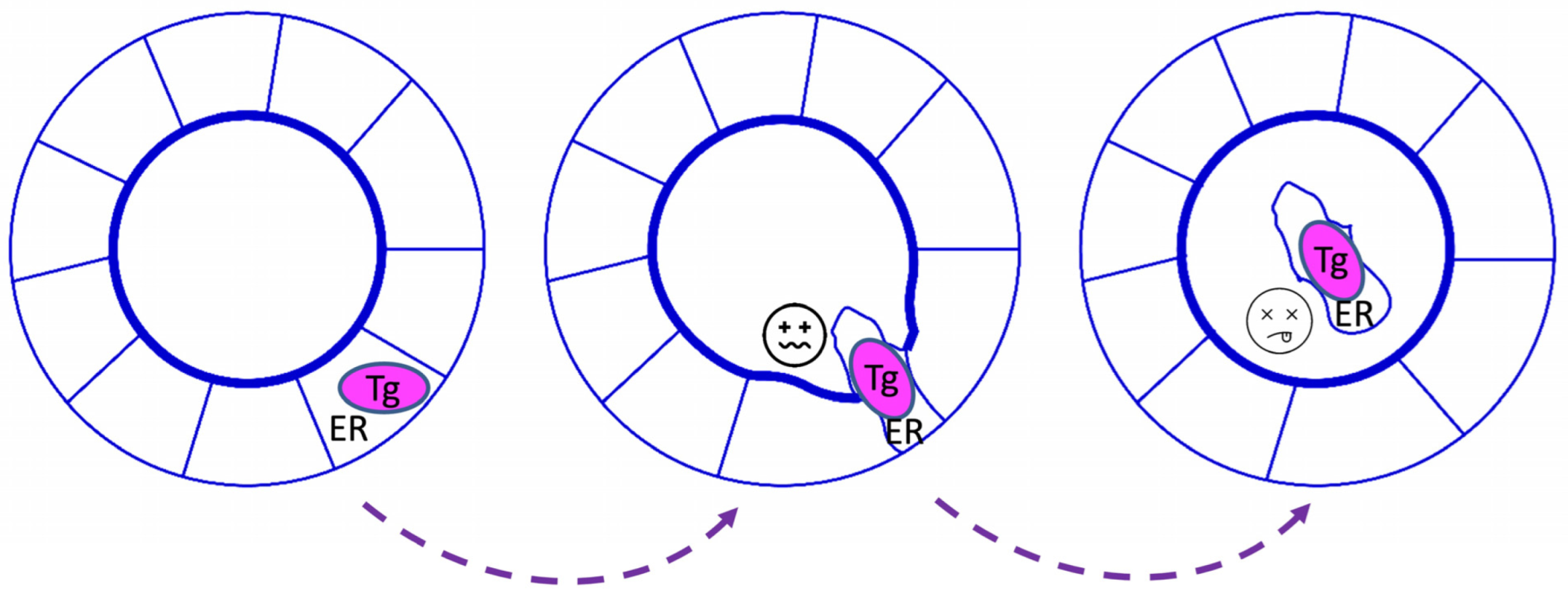
3. Tg Misfolding and Its Consequences in Cell Culture Models
4. Tg Misfolding and Its Consequences for Animal Models and Humans
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Hung, M.C.; Link, W. Protein localization in disease and therapy. J. Cell Sci. 2011, 124 Pt 20, 3381–3392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell. Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef] [Green Version]
- Perkins, H.T.; Allan, V. Intertwined and Finely Balanced: Endoplasmic Reticulum Morphology, Dynamics, Function, and Diseases. Cells 2021, 10, 2341. [Google Scholar] [CrossRef]
- Meras, I.; Maes, J.; Lefrancois, S. Mechanisms regulating the sorting of soluble lysosomal proteins. Biosci. Rep. 2022, 42, BSR20211856. [Google Scholar] [CrossRef] [PubMed]
- Banfield, D.K. Mechanisms of protein retention in the Golgi. Cold Spring Harb. Perspect. Biol. 2011, 3, a005264. [Google Scholar] [CrossRef]
- McRae, M.S.; Wang, B.; Hyson, R.G.; Siddiquee, R.; Duff, A.P.; Ataide, S.F.; Kwan, A.H. Expression and purification of the NG domain from human SRalpha, a key component of the Signal Recognition Particle (SRP) receptor. Protein Expr. Purif. 2022, 198, 106121. [Google Scholar] [CrossRef]
- Song, J.; Becker, T. Fidelity of organellar protein targeting. Curr. Opin. Cell Biol. 2022, 75, 102071. [Google Scholar] [CrossRef] [PubMed]
- Liaci, A.M.; Forster, F. Take Me Home, Protein Roads: Structural Insights into Signal Peptide Interactions during ER Translocation. Int. J. Mol. Sci. 2021, 22, 11871. [Google Scholar] [CrossRef]
- Esmail, S.; Manolson, M.F. Advances in understanding N-glycosylation structure, function, and regulation in health and disease. Eur. J. Cell Biol. 2021, 100, 151186. [Google Scholar] [CrossRef]
- Medinas, D.B.; Rozas, P.; Hetz, C. Critical roles of protein disulfide isomerases in balancing proteostasis in the nervous system. J. Biol. Chem. 2022, 298, 102087. [Google Scholar] [CrossRef]
- Morishita, Y.; Arvan, P. Lessons from animal models of endocrine disorders caused by defects of protein folding in the secretory pathway. Mol. Cell. Endocrinol. 2020, 499, 110613. [Google Scholar] [CrossRef] [PubMed]
- Gidalevitz, T.; Stevens, F.; Argon, Y. Orchestration of secretory protein folding by ER chaperones. Biochim. Biophys. Acta 2013, 1833, 2410–2424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, B.M.; Canniff, N.P.; Guay, K.P.; Hebert, D.N. The Role of Endoplasmic Reticulum Chaperones in Protein Folding and Quality Control. In Progress in Molecular and Subcellular Biology; Springer: Cham, Switzerland, 2021; Volume 59, pp. 27–50. [Google Scholar]
- Phillips, B.P.; Gomez-Navarro, N.; Miller, E.A. Protein quality control in the endoplasmic reticulum. Curr. Opin. Cell Biol. 2020, 65, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Wiseman, R.L.; Mesgarzadeh, J.S.; Hendershot, L.M. Reshaping endoplasmic reticulum quality control through the unfolded protein response. Mol. Cell 2022, 82, 1477–1491. [Google Scholar] [CrossRef]
- Needham, P.G.; Guerriero, C.J.; Brodsky, J.L. Chaperoning Endoplasmic Reticulum-Associated Degradation (ERAD) and Protein Conformational Diseases. Cold Spring Harb. Perspect. Biol. 2019, 11, a033928. [Google Scholar] [CrossRef]
- Mochida, K.; Nakatogawa, H. ER-phagy: Selective autophagy of the endoplasmic reticulum. EMBO Rep. 2022, 23, e55192. [Google Scholar] [CrossRef]
- Graham, K.S.; Le, A.; Sifers, R.N. Accumulation of the insoluble PiZ variant human a1-antitrypsin within the hepatic endoplasmic reticulum does not elevate the steaty-state level of grp78/BiP. J. Biol. Chem. 1990, 265, 20463–20468. [Google Scholar] [CrossRef]
- Di Jeso, B.; Arvan, P. Thyroglobulin from Molecular and Cellular Biology to Clinical Endocrinology. Endocr. Rev. 2016, 37, 2–36. [Google Scholar] [CrossRef] [Green Version]
- Holzer, G.; Morishita, Y.; Fini, J.B.; Lorin, T.; Gillet, B.; Hughes, S.; Tohme, M.; Deleage, G.; Demeneix, B.; Arvan, P.; et al. Thyroglobulin Represents a Novel Molecular Architecture of Vertebrates. J. Biol. Chem. 2016, 291, 16553–16566. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Shimizu, H.; Xiang, Y.-Y.; Sugihara, J.; Lu, W.-Y.; Liao, X.-H.; Cho, H.-R.; Toba, H.; Bai, X.-H.; Asa, S.L.; et al. XB130 Deficiency Causes Congenital Hypothyroidism in Mice due to Disorganized Apical Membrane Structure and Function of Thyrocytes. Thyroid 2021, 31, 1650–1661. [Google Scholar] [CrossRef]
- Wang, Y.; Xiang, Y.-Y.; Sugihara, J.; Lu, W.-Y.; Liao, X.-H.; Arvan, P.; Refetoff, S.; Liu, M. XB130 Plays an Essential Role in Folliculogenesis through Mediating Interactions between Microfilament and Microtubule Systems in Thyrocytes. Thyroid 2022, 32, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Di Jeso, B.; Ulianich, L.; Pacifico, F.; Leonardi, A.; Vito, P.; Consiglio, E.; Formisano, S.; Arvan, P. Folding of thyroglobulin in the calnexin/calreticulin pathway and its alteration by loss of Ca2+ from the endoplasmic reticulum. Biochem. J. 2003, 370 Pt 2, 449–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Herle, A.J.; Vassart, G.; Dumont, J.E. Control of thyroglobulin synthesis and secretion. (First of two parts). N. Engl. J. Med. 1979, 301, 239–249. [Google Scholar] [CrossRef]
- Dunn, J.T.; Dunn, A.D. The importance of thyroglobulin structure for thyroid hormone biosynthesis. Biochimie 1999, 81, 505–509. [Google Scholar] [CrossRef]
- Rivolta, C.M.; Targovnik, H.M. Molecular advances in thyroglobulin disorders. Clin. Chim. Acta 2006, 374, 8–24. [Google Scholar] [CrossRef] [PubMed]
- Kayili, H.M.; Salih, B. Site-specific N-glycosylation analysis of human thyroid thyroglobulin by mass spectrometry-based Glyco-analytical strategies. J. Proteom. 2022, 267, 104700. [Google Scholar] [CrossRef]
- Adaixo, R.; Steiner, E.M.; Righetto, R.D.; Schmidt, A.; Stahlberg, H.; Taylor, N.M.I. Cryo-EM structure of native human thyroglobulin. Nat. Commun. 2022, 13, 61. [Google Scholar] [CrossRef]
- Kim, P.; Bole, D.; Arvan, P. Transient aggregation of nascent thyroglobulin in the endoplasmic reticulum: Relationship to the molecular chaperone, BiP. J. Cell Biol. 1992, 118, 541–549. [Google Scholar] [CrossRef] [Green Version]
- Wright, M.T.; Kouba, L.; Plate, L. Thyroglobulin Interactome Profiling Defines Altered Proteostasis Topology Associated With Thyroid Dyshormonogenesis. Mol. Cell. Proteom. 2021, 20, 100008. [Google Scholar] [CrossRef]
- Di Jeso, B.; Park, Y.-N.; Ulianich, L.; Treglia, A.S.; Urbanas, M.L.; High, S.; Arvan, P. Mixed-disulfide folding intermediates between thyroglobulin and ER resident oxidoreductases ERp57 and PDI. Mol. Cell. Biol. 2005, 25, 9793–9805. [Google Scholar] [CrossRef]
- Di Jeso, B.; Morishita, Y.; Treglia, A.S.; Lofrumento, D.D.; Nicolardi, G.; Beguinot, F.; Kellogg, A.P.; Arvan, P. Transient covalent interactions of newly synthesized thyroglobulin with oxidoreductases of the endoplasmic reticulum. J. Biol. Chem. 2014, 289, 11488–11496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arvan, P.; Kim, P.S.; Kuliawat, R.; Prabakaran, D.; Muresan, Z.; Yoo, S.E.; Hossain, S.A. Intracellular protein transport to the thyrocyte plasma membrane: Potential implications for thyroid physiology. Thyroid 1997, 7, 89–105. [Google Scholar] [CrossRef] [PubMed]
- Coscia, F.; Taler-Vercic, A.; Chang, V.T.; Sinn, L.; O’Reilly, F.J.; Izore, T.; Renko, M.; Berger, I.; Rappsilber, J.; Turk, D.; et al. The structure of human thyroglobulin. Nature 2020, 578, 627–630. [Google Scholar] [CrossRef] [PubMed]
- Tosatto, L.; Coscia, F. A glance at post-translational modifications of human thyroglobulin: Potential impact on function and pathogenesis. Eur. Thyroid J. 2022, 11. [Google Scholar] [CrossRef]
- Citterio, C.E.; Targovnik, H.M.; Arvan, P. The role of thyroglobulin in thyroid hormonogenesis. Nat. Rev. Endocrinol. 2019, 15, 323–338. [Google Scholar] [CrossRef]
- Mendive, F.M.; Rivolta, C.M.; Moya, C.M.; Vassart, G.; Targovnik, H.M. Genomic organization of the human thyroglobulin gene: The complete intron-exon structure. Eur. J. Endocrinol. 2001, 145, 485–496. [Google Scholar] [CrossRef] [Green Version]
- Citterio, C.E.; Rivolta, C.M.; Targovnik, H.M. Structure and genetic variants of thyroglobulin: Pathophysiological implications. Mol. Cell. Endocrinol. 2021, 528, 111227. [Google Scholar] [CrossRef]
- Lee, J.; Di Jeso, B.; Arvan, P. The cholinesterase-like domain of thyrogobulin functions as an intramolecular chaperone. J. Clin. Investig. 2008, 118, 2950–2958. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Kopylov, M.; Bobe, D.; Kelley, K.; Eng, E.T.; Arvan, P.; Clarke, O.B. The structure of natively iodinated bovine thyroglobulin. Acta Crystallogr. D Struct. Biol. 2021, 77 Pt 11, 1451–1459. [Google Scholar] [CrossRef]
- Marechal, N.; Serrano, B.P.; Zhang, X.; Weitz, C.J. Formation of thyroid hormone revealed by a cryo-EM structure of native bovine thyroglobulin. Nat. Commun. 2022, 13, 2380. [Google Scholar] [CrossRef]
- Citterio, C.E.; Morishita, Y.; Dakka, N.; Veluswamy, B.; Arvan, P. Relationship between the dimerization of thyroglobulin and its ability to form triiodothyronine. J. Biol. Chem. 2018, 293, 4860–4869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Lee, J.; Di Jeso, B.; Treglia, A.S.; Comoletti, D.; Dubi, N.; Taylor, P.; Arvan, P. Cis and trans actions of the cholinesterase-like domain within the thyroglobulin dimer. J. Biol. Chem. 2010, 285, 17564–17573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pio, M.G.; Siffo, S.; Scheps, K.G.; Molina, M.F.; Adrover, E.; Abelleyro, M.M.; Rivolta, C.M.; Targovnik, H.M. Curating the gnomAD database: Report of novel variants in the thyrogobulin gene using in silico bioinformatics algorithms. Mol. Cell. Endocrinol. 2021, 534, 111359. [Google Scholar] [CrossRef] [PubMed]
- Park, K.S. Analysis of Worldwide Carrier Frequency and Predicted Genetic Prevalence of Autosomal Recessive Congenital Hypothyroidism Based on a General Population Database. Genes 2021, 12, 863. [Google Scholar] [CrossRef]
- Siffo, S.; Adrover, E.; Citterio, C.E.; Miras, M.B.; Balbi, V.A.; Chiesa, A.; Weill, J.; Sobrero, G.; Gonzalez, V.G.; Papendieck, P.; et al. Molecular analysis of thyroglobulin mutations found in patients with goiter and hypothyroidism. Mol. Cell. Endocrinol. 2018, 473, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Kim, P.S.; Ding, M.; Menon, S.; Jung, C.G.; Cheng, J.M.; Miyamoto, T.; Li, B.; Furudate, S.; Agui, T. A missense mutation G2320R in the thyroglobulin gene causes non-goitrous congenital primary hypothyroidism in the WIC-rdw rat. Mol. Endocrinol. 2000, 14, 1944–1953. [Google Scholar] [CrossRef]
- Wright, J.; Wang, X.; Haataja, L.; Kellogg, A.P.; Lee, J.; Liu, M.; Arvan, P. Dominant protein interactions that influence the pathogenesis of conformational diseases. J. Clin. Investig. 2013, 123, 3124–3134. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Kaufman, R.J. Physiological/pathological ramifications of transcription factors in the unfolded protein response. Genes Dev. 2017, 31, 1417–1438. [Google Scholar] [CrossRef] [Green Version]
- Tsang, K.Y.; Chan, D.; Bateman, J.F.; Cheah, K.S. In Vivo cellular adaptation to ER stress: Survival strategies with double-edged consequences. J. Cell Sci. 2010, 123 Pt 13, 2145–2154. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, R.; Colon-Negron, K.; Papa, F.R. Endoplasmic reticulum stress, degeneration of pancreatic islet β-cells, and therapeutic modulation of the unfolded protein response in diabetes. Mol. Metab. 2019, 27, S60–S68. [Google Scholar] [CrossRef]
- Morishita, Y.; Kabil, O.; Young, K.Z.; Kellogg, A.P.; Chang, A.; Arvan, P. Thyrocyte cell survival and adaptation to chronic endoplasmic reticulum stress due to misfolded thyroglobulin. J. Biol. Chem. 2020, 295, 6876–6887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, P.S.; Arvan, P. Endocrinopathies in the family of endoplasmic reticulum (ER) storage diseases: Disorders of protein trafficking and the role of ER molecular chaperones. Endocr. Rev. 1998, 19, 173–202. [Google Scholar] [PubMed] [Green Version]
- Reiling, J.H.; Clish, C.B.; Carette, J.E.; Varadarajan, M.; Brummelkamp, T.R.; Sabatini, D.M. A haploid genetic screen identifies the major facilitator domain containing 2A (MFSD2A) transporter as a key mediator in the response to tunicamycin. Proc. Natl. Acad. Sci. USA 2011, 108, 11756–11765. [Google Scholar] [CrossRef] [Green Version]
- Kim, P.S.; Kwon, O.-Y.; Arvan, P. An endoplasmic reticulum storage disease causing congenital goiter with hypothyroidism. J. Cell Biol. 1996, 133, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Morishita, Y.; Kellogg, A.P.; Larkin, D.; Chen, W.; Vadrevu, S.; Satin, L.; Liu, M.; Arvan, P. Cell death-associated lipid droplet protein CIDE-A is a noncanonical marker of endoplasmic reticulum stress. JCI Insight 2021, 6, 143980. [Google Scholar] [CrossRef]
- Rohm, M.; Schafer, M.; Laurent, V.; Ustunel, B.E.; Niopek, K.; Algire, C.; Hautzinger, O.; Sijmonsma, T.P.; Zota, A.; Medrikova, D.; et al. An AMP-activated protein kinase-stabilizing peptide ameliorates adipose tissue wasting in cancer cachexia in mice. Nat. Med. 2016, 22, 1120–1130. [Google Scholar] [CrossRef]
- Qi, J.; Gong, J.; Zhao, T.; Zhao, J.; Lam, P.; Ye, J.; Li, J.Z.; Wu, J.; Zhou, H.M.; Li, P. Downregulation of AMP-activated protein kinase by Cidea-mediated ubiquitination and degradation in brown adipose tissue. EMBO J. 2008, 27, 1537–1548. [Google Scholar] [CrossRef] [Green Version]
- Kimura, T.; Van Keymeulen, A.; Golstein, J.; Fusco, A.; Dumont, J.E.; Roger, P.P. Regulation of thyroid cell proliferation by TSH and other factors: A critical evaluation of in vitro models. Endocr. Rev. 2001, 22, 631–656. [Google Scholar] [CrossRef]
- de Vijlder, J.J.; van Voorthuizen, W.F.; van Dijk, J.E.; Rijnberk, A.; Tegelaers, W.H. Hereditary congenital goiter with thyroglobulin deficiency in a breed of goats. Endocrinology 1978, 102, 1214–1222. [Google Scholar] [CrossRef]
- Beamer, W.G.; Maltais, L.J.; DeBaets, M.H.; Eicher, E.M. Inherited congenital goiter in mice. Endocrinology 1987, 120, 838–840. [Google Scholar] [CrossRef]
- Targovnik, H.M.; Vono, J.; Billerbeck, A.E.; Cerrone, G.E.; Varela, V.; Mendive, F.; Wajchenberg, B.L.; Medeiros-Neto, G. A 138-nucleotide deletion in the thyroglobulin ribonucleic acid messenger in a congenital goiter with defective thyroglobulin synthesis. J. Clin. Endocrinol. Metab. 1995, 80, 3356–3360. [Google Scholar] [PubMed]
- Caputo, M.; Rivolta, C.M.; Esperante, S.A.; Gruñeiro-Papendieck, L.; Chiesa, A.; Pellizas, C.G.; González-Sarmiento, R.; Targovnik, H.M. Congenital hypothyroidism with goitre caused by new mutations in the thyroglobulin gene. Clin. Endocrinol. 2007, 67, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Targovnik, H.M.; Rivolta, C.M.; Mendive, F.M.; Moya, C.M.; Vono, J.; Medeiros-Neto, G. Congenital goiter with hypothyroidism caused by a 5′ splice site mutation in the thyroglobulin gene. Thyroid 2001, 11, 685–690. [Google Scholar] [CrossRef]
- Ahlbom, B.E.; Yaqoob, M.; Gustavsson, P.; Abbas, H.G.; Annerén, G.; Larsson, A.; Wadelius, C. Linkage analysis identifies the thyroglobulin gene region as a major locus for familial congenital hypothyroidism. Hum. Genet. 2002, 110, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.S.; Hossain, S.A.; Park, Y.N.; Lee, I.; Yoo, S.E.; Arvan, P. A single amino acid change in the acetylcholinesterase-like domain of thyroglobulin causes congenital goiter with hypothyroidism in the cog/cog mouse: A model of human endoplasmic reticulum storage diseases. Proc. Natl. Acad. Sci. USA 1998, 95, 9909–9913. [Google Scholar] [CrossRef] [Green Version]
- Basche, M.; Beamer, W.G.; Schneider, A.B. Abnormal properties of thyroglobulin in mice with inherited congenital goiter (cog/cog). Endocrinology 1989, 124, 1822–1829. [Google Scholar] [CrossRef]
- Mayerhofer, A.; Amador, A.G.; Beamer, W.G.; Bartke, A. Ultrastructural aspects of the goiter in cog/cog mice. J. Hered. 1988, 79, 200–203. [Google Scholar] [CrossRef]
- Medeiros-Neto, G.; Kim, P.S.; Yoo, S.E.; Vono, J.; Targovnik, H.M.; Camargo, R.; Hossain, S.A.; Arvan, P. Congenital hypothyroid goiter with deficient thyroglobulin. Identification of an endoplasmic reticulum storage disease with induction of molecular chaperones. J. Clin. Investig. 1996, 98, 2838–2844. [Google Scholar] [CrossRef]
- Hishinuma, A.; Furudate, S.; Oh-Ishi, M.; Nagakubo, N.; Namatame, T.; Ieiri, T. A novel missense mutation (G2320R) in thyroglobulin causes hypothyroidism in rdw rats. Endocrinology 2000, 141, 4050–4055. [Google Scholar] [CrossRef]
- Baryshev, M.; Sargsyan, E.; Wallin, G.; Lejnieks, A.; Furudate, S.; Hishinuma, A.; Mkrtchian, S. Unfolded protein response is involved in the pathology of human congenital hypothyroid goiter and rat non-goitrous congenital hypothyroidism. J. Mol. Endocrinol. 2004, 32, 903–920. [Google Scholar] [CrossRef]
- Menon, S.; Lee, J.; Abplanalp, W.A.; Yoo, S.E.; Agui, T.; Furudate, S.; Kim, P.S.; Arvan, P. Oxidoreductase interactions include a role for ERp72 engagement with mutant thyroglobulin from the rdw/rdw rat dwarf. J. Biol. Chem. 2007, 282, 6183–6191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakai, Y.; Yamashina, S.; Furudate, S.I. Missing secretory granules, dilated endoplasmic reticulum, and nuclear dislocation in the thyroid gland of rdw rats with hereditary dwarfism. Anat. Rec. 2000, 259, 60–66. [Google Scholar] [CrossRef]
- Zhang, X.; Malik, B.; Young, C.; Zhang, H.; Larkin, D.; Liao, X.H.; Refetoff, S.; Liu, M.; Arvan, P. Maintaining the thyroid gland in mutant thyroglobulin-induced hypothyroidism requires thyroid cell proliferation that must continue in adulthood. J. Biol. Chem. 2022, 298, 102066. [Google Scholar] [CrossRef] [PubMed]
- Umezu, M.; Kagabu, S.; Jiang, J.; Sato, E. Evaluation and characterization of congenital hypothyroidism in rdw dwarf rats. Lab. Anim. Sci. 1998, 48, 496–501. [Google Scholar]
- Zhang, X.; Kellogg, A.P.; Citterio, C.E.; Zhang, H.; Larkin, D.; Morishita, Y.; Targovnik, H.M.; Balbi, V.A.; Arvan, P. Thyroid hormone synthesis continues despite biallelic thyroglobulin mutation with cell death. JCI Insight 2021, 6, 148496. [Google Scholar] [CrossRef]
- Dumont, J.E.; Ermans, A.M.; Maenhaut, C.; Coppee, F.; Stanbury, J.B. Large goitre as a maladaptation to iodine deficiency. Clin. Endocrinol. 1995, 43, 1–10. [Google Scholar] [CrossRef]
- van de Graaf, S.A.; Ris-Stalpers, C.; Veenboer, G.J.; Cammenga, M.; Santos, C.; Targovnik, H.M.; de Vijlder, J.J.; Medeiros-Neto, G. A premature stopcodon in thyroglobulin messenger RNA results in familial goiter and moderate hypothyroidism. J. Clin. Endocrinol. Metab. 1999, 84, 2537–2542. [Google Scholar] [CrossRef]
- Hermanns, P.; Refetoff, S.; Sriphrapradang, C.; Pohlenz, J.; Okamato, J.; Slyper, L.; Slyper, A.H. A clinically euthyroid child with a large goiter due to a thyroglobulin gene defect: Clinical features and genetic studies. J. Pediatr. Endocrinol. Metab. 2013, 26, 119–123. [Google Scholar] [CrossRef]
- Adkison, L.R.; Taylor, S.; Beamer, W.G. Mutant gene-induced disorders of structure, function and thyroglobulin synthesis in congenital goitre (cog/cog) in mice. J. Endocrinol. 1990, 126, 51–58. [Google Scholar] [CrossRef]
- Hishinuma, A.; Fukata, S.; Kakudo, K.; Murata, Y.; Ieiri, T. High incidence of thyroid cancer in long-standing goiters with thyroglobulin mutations. Thyroid 2005, 15, 1079–1084. [Google Scholar] [CrossRef]
- Raef, H.; Al-Rijjal, R.; Al-Shehri, S.; Zou, M.; Al-Mana, H.; Baitei, E.Y.; Parhar, R.S.; Al-Mohanna, F.A.; Shi, Y. Biallelic p.R2223H mutation in the thyroglobulin gene causes thyroglobulin retention and severe hypothyroidism with subsequent development of thyroid carcinoma. J. Clin. Endocrinol. Metab. 2010, 95, 1000–1006. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; Hong, A.R.; Kim, H.K.; Kang, H.C. Anaplastic Thyroid Cancer Arising from Dyshormonogenetic Goiter: C.3070T>C and Novel c.7070T>C Mutation in the Thyroglobulin Gene. Thyroid 2020, 30, 1676–1680. [Google Scholar] [CrossRef]
- Sato, A.; Abe, K.; Yuzuriha, M.; Fujii, S.; Takahashi, N.; Hojo, H.; Teramoto, S.; Aoyama, H. A novel mutation in the thyroglobulin gene that causes goiter and dwarfism in Wistar Hannover GALAS rats. Mutat. Res. 2014, 762, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Kokoshima, H.; Doi, T.; Yamada, N.; Tsuchitani, M. Proliferative lesions in thyroid follicular cells of dwarfs derived from Wistar Hannover GALAS rats. Toxicol. Pathol. 2014, 42, 565–572. [Google Scholar] [CrossRef] [Green Version]
- van Voorthuizen, W.F.; de Vijlder, J.J.; van Dijk, J.E.; Tegelaers, W.H. Euthyroidism via iodide supplementation in hereditary congenital goiter with thyroglobulin deficiency. Endocrinology 1978, 103, 2105–2111. [Google Scholar] [CrossRef]
- Vono, J.; Lima, N.; Knobel, M.; Medeiros-Neto, G. The effect of oral administration of iodine to patients with goiter and hypothyroidism due to defective synthesis of thyroglobulin. Thyroid 1996, 6, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Arandjelovic, S.; Ravichandran, K.S. Phagocytosis of apoptotic cells in homeostasis. Nat. Immunol. 2015, 16, 907–917. [Google Scholar] [CrossRef] [Green Version]
- Juncadella, I.J.; Kadl, A.; Sharma, A.K.; Shim, Y.M.; Hochreiter-Hufford, A.; Borish, L.; Ravichandran, K.S. Apoptotic cell clearance by bronchial epithelial cells critically influences airway inflammation. Nature 2013, 493, 547–551. [Google Scholar] [CrossRef] [Green Version]
- Pan, H.; Shen, K.; Wang, X.; Meng, H.; Wang, C.; Jin, B. Protective effect of metalloporphyrins against cisplatin-induced kidney injury in mice. PLoS ONE 2014, 9, e86057. [Google Scholar] [CrossRef] [Green Version]
- Munari-Silem, Y.; Mesnil, M.; Selmi, S.; Bernier-Valentin, F.; Rabilloud, R.; Rousset, B. Cell-cell interactions in the process of differentiation of thyroid epithelial cells into follicles: A study by microinjection and fluorescence microscopy on in vitro reconstituted thyroid follicles. J. Cell. Physiol. 1990, 145, 414–427. [Google Scholar] [CrossRef]
- Sastre-Perona, A.; Santisteban, P. Wnt-independent role of β-catenin in thyroid cell proliferation and differentiation. Mol. Endocrinol. 2014, 28, 681–695. [Google Scholar] [CrossRef] [PubMed]
- Dockhorn-Dworniczak, B.; Franke, W.W.; Schröder, S.; Czernobilsky, B.; Gould, V.E.; Böcker, W. Patterns of expression of cytoskeletal proteins in human thyroid gland and thyroid carcinomas. Differentiation 1987, 35, 53–71. [Google Scholar] [CrossRef] [PubMed]
- Lien, W.-H.; Klezovitch, O.; Vasioukhin, V. Cadherin-catenin proteins in vertebrate development. Curr. Opin. Cell Biol. 2006, 18, 499–506. [Google Scholar] [CrossRef]
- Campbell, H.K.; Maiers, J.L.; DeMali, K.A. Interplay between tight junctions & adherens junctions. Exp. Cell Res. 2017, 358, 39–44. [Google Scholar] [PubMed]
- Adil, M.S.; Narayanan, S.P.; Somanath, P.R. Cell-cell junctions: Structure and regulation in physiology and pathology. Tissue Barriers 2021, 9, 1848212. [Google Scholar] [CrossRef]
- Calì, G.; Gentile, F.; Mogavero, S.; Pallante, P.; Nitsch, R.; Ciancia, G.; Ferraro, A.; Fusco, A.; Nitsch, L. CDH16/Ksp-cadherin is expressed in the developing thyroid gland and is strongly down-regulated in thyroid carcinomas. Endocrinology 2011, 153, 522–534. [Google Scholar] [CrossRef] [Green Version]
- Koumarianou, P.; Goméz-López, G.; Santisteban, P. Pax8 controls thyroid follicular polarity through cadherin-16. J. Cell Sci. 2017, 130, 219–231. [Google Scholar] [CrossRef] [Green Version]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Ulianich, L.; Mirra, P.; Garbi, C.; Cali, G.; Conza, D.; Treglia, A.S.; Miraglia, A.; Punzi, D.; Miele, C.; Raciti, G.A.; et al. The Pervasive Effects of ER Stress on a Typical Endocrine Cell: Dedifferentiation, Mesenchymal Shift and Antioxidant Response in the Thyrocyte. Front. Endocrinol. 2020, 11, 588685. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Young, C.; Morishita, Y.; Kim, K.; Kabil, O.O.; Clarke, O.B.; Di Jeso, B.; Arvan, P. Defective Thyroglobulin: Cell Biology of Disease. Int. J. Mol. Sci. 2022, 23, 13605. https://doi.org/10.3390/ijms232113605
Zhang X, Young C, Morishita Y, Kim K, Kabil OO, Clarke OB, Di Jeso B, Arvan P. Defective Thyroglobulin: Cell Biology of Disease. International Journal of Molecular Sciences. 2022; 23(21):13605. https://doi.org/10.3390/ijms232113605
Chicago/Turabian StyleZhang, Xiaohan, Crystal Young, Yoshiaki Morishita, Kookjoo Kim, Omer O. Kabil, Oliver B. Clarke, Bruno Di Jeso, and Peter Arvan. 2022. "Defective Thyroglobulin: Cell Biology of Disease" International Journal of Molecular Sciences 23, no. 21: 13605. https://doi.org/10.3390/ijms232113605
APA StyleZhang, X., Young, C., Morishita, Y., Kim, K., Kabil, O. O., Clarke, O. B., Di Jeso, B., & Arvan, P. (2022). Defective Thyroglobulin: Cell Biology of Disease. International Journal of Molecular Sciences, 23(21), 13605. https://doi.org/10.3390/ijms232113605