

Helicobacter pylori in the Oral Cavity: Current Evidence and Potential Survival Strategies


Abstract
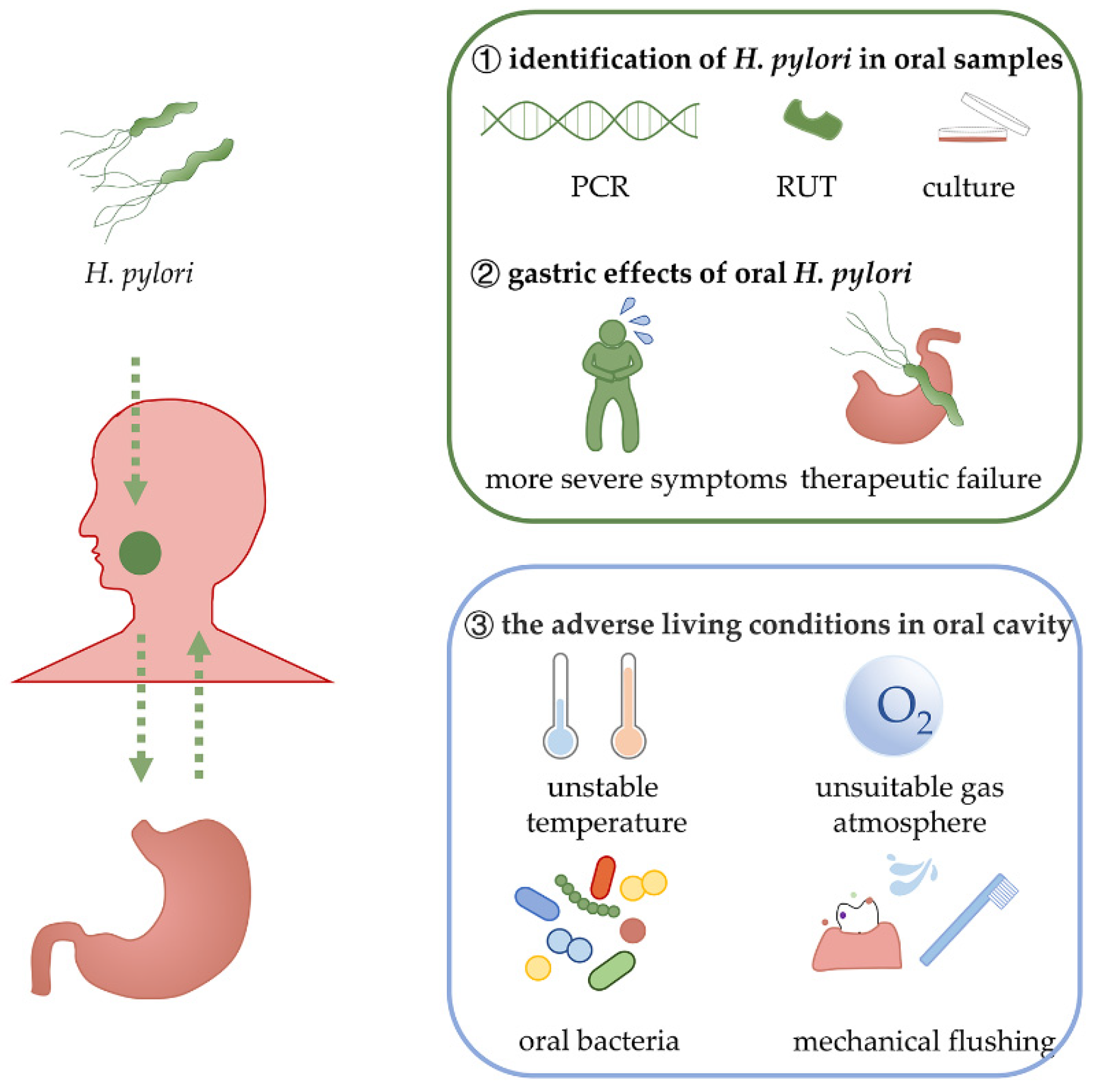
1. Introduction
2. The Evidence for the Persistent Survival of Oral H. pylori
2.1. Various Samples from the Oral Cavity Can Be Detected as H. pylori-Positive
2.2. The Association between Gastric H. pylori Infection and Oral H. pylori Positivity
2.3. H. pylori in the Oral Cavity Is Associated with Oral Diseases and Gastric Infection
2.4. Effects of Oral Hygiene Management on H. pylori Infection
3. Pangenome and Virulence Factors
4. Interactions between H. pylori and Microenvironments in the Oral Cavity
4.1. Oral Microenvironment and H. pylori
4.2. Oral Host Cells and H. pylori
4.3. Human Host Immunity and H. pylori
5. The Synergistic Interactions of Oral H. pylori with Oral Microorganisms
6. Non-Growing State of H. pylori: Viable but Non-Culturable State and Dormant State
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schulz, C.; Kalali, B.; Link, A.; Gerhard, M.; Malfertheiner, P. New Rapid Helicobacter pylori Blood Test Based on Dual Detection of FliD and CagA Antibodies for On-Site Testing. Clin. Gastroenterol. Hepatol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Mladenova, I. Clinical Relevance of Helicobacter pylori Infection. J. Clin. Med. 2021, 10, 3473. [Google Scholar] [CrossRef] [PubMed]
- Ranjbar, R.; Behzadi, P.; Farshad, S. Advances in diagnosis and treatment of Helicobacter pylori infection. Acta Microbiol. Immunol. Hung. 2017, 64, 273–292. [Google Scholar] [CrossRef] [PubMed]
- Momtaz, H.; Souod, N.; Dabiri, H.; Sarshar, M. Study of Helicobacter pylori genotype status in saliva, dental plaques, stool and gastric biopsy samples. World J. Gastroenterol. 2012, 18, 2105–2111. [Google Scholar] [CrossRef]
- Burucoa, C.; Axon, A. Epidemiology of Helicobacter pylori infection. Helicobacter 2017, 22 (Suppl. S1), e12403. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Jakubovics, N.S.; Bachle, M.; Buchalla, W.; Hiller, K.A.; Maisch, T.; Hellwig, E.; Kirschneck, C.; Gessner, A.; Al-Ahmad, A.; et al. Colonization of Helicobacter pylori in the oral cavity—An endless controversy? Crit. Rev. Microbiol. 2021, 47, 612–629. [Google Scholar] [CrossRef]
- Ansari, S.A.; Iqbal, M.U.N.; Khan, T.A.; Kazmi, S.U. Association of oral Helicobacter pylori with gastric complications. Life Sci. 2018, 205, 125–130. [Google Scholar] [CrossRef]
- Miyabayashi, H.; Furihata, K.; Shimizu, T.; Ueno, I.; Akamatsu, T. Influence of oral Helicobacter pylori on the success of eradication therapy against gastric Helicobacter pylori. Helicobacter 2000, 5, 30–37. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, J. Helicobacter pylori recrudescence and its influencing factors. J. Cell Mol. Med. 2019, 23, 7919–7925. [Google Scholar] [CrossRef]
- Gisbert, J.P. The recurrence of Helicobacter pylori infection: Incidence and variables influencing it. A critical review. Am. J. Gastroenterol. 2005, 100, 2083–2099. [Google Scholar] [CrossRef]
- Park, S.A.; Ko, A.; Lee, N.G. Stimulation of growth of the human gastric pathogen Helicobacter pylori by atmospheric level of oxygen under high carbon dioxide tension. BMC Microbiol. 2011, 11, 96. [Google Scholar] [CrossRef] [PubMed]
- Bury-Mone, S.; Kaakoush, N.O.; Asencio, C.; Megraud, F.; Thibonnier, M.; De Reuse, H.; Mendz, G.L. Is Helicobacter pylori a true microaerophile? Helicobacter 2006, 11, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Khosravi, Y.; Dieye, Y.; Loke, M.F.; Goh, K.L.; Vadivelu, J. Streptococcus mitis induces conversion of Helicobacter pylori to coccoid cells during co-culture in vitro. PLoS ONE 2014, 9, e112214. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, K.; Miura, T.; Kimizuka, R.; Ebihara, Y.; Mizuno, Y.; Okuda, K. Oral bacteria inhibit Helicobacter pylori growth. FEMS Microbiol. Lett. 1997, 152, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Mendoza-Cantu, A.; Urrutia-Baca, V.H.; Urbina-Rios, C.S.; De la Garza-Ramos, M.A.; Garcia-Martinez, M.E.; Torre-Martinez, H.H.H. Prevalence of Helicobacter pylori vacA Genotypes and cagA Gene in Dental Plaque of Asymptomatic Mexican Children. Biomed. Res. Int. 2017, 2017, 4923640. [Google Scholar] [CrossRef]
- Ji, Y.; Liang, X.; Lu, H. Analysis of by high-throughput sequencing: Helicobacter pylori infection and salivary microbiome. BMC Oral. Health 2020, 20, 84. [Google Scholar] [CrossRef]
- Seligova, B.; Lukac, L.; Babelova, M.; Vavrova, S.; Sulo, P. Diagnostic reliability of nested PCR depends on the primer design and threshold abundance of Helicobacter pylori in biopsy, stool, and saliva samples. Helicobacter 2020, 25, e12680. [Google Scholar] [CrossRef]
- Zhao, Y.; Gao, X.; Guo, J.; Yu, D.; Xiao, Y.; Wang, H.; Li, Y. Helicobacter pylori infection alters gastric and tongue coating microbial communities. Helicobacter 2019, 24, e12567. [Google Scholar] [CrossRef]
- Ogaya, Y.; Nomura, R.; Watanabe, Y.; Nakano, K. Detection of Helicobacter pylori DNA in inflamed dental pulp specimens from Japanese children and adolescents. J. Med. Microbiol. 2015, 64, 117–123. [Google Scholar] [CrossRef]
- Nomura, R.; Ogaya, Y.; Matayoshi, S.; Morita, Y.; Nakano, K. Molecular and clinical analyses of Helicobacter pylori colonization in inflamed dental pulp. BMC Oral Health 2018, 18, 64. [Google Scholar] [CrossRef]
- Hirsch, C.; Tegtmeyer, N.; Rohde, M.; Rowland, M.; Oyarzabal, O.A.; Backert, S. Live Helicobacter pylori in the root canal of endodontic-infected deciduous teeth. J. Gastroenterol. 2012, 47, 936–940. [Google Scholar] [CrossRef]
- Sruthi, M.A.; Mani, G.; Ramakrishnan, M.; Selvaraj, J. Dental caries as a source of Helicobacter pylori infection in children. Int. J. Paediatr. Dent. 2022. [Google Scholar] [CrossRef]
- Krajden, S.; Fuksa, M.; Anderson, J.; Kempston, J.; Boccia, A.; Petrea, C.; Babida, C.; Karmali, M.; Penner, J.L. Examination of human stomach biopsies, saliva, and dental plaque for Campylobacter pylori. J. Clin. Microbiol. 1989, 27, 1397–1398. [Google Scholar] [CrossRef]
- Navabi, N.; Aramon, M.; Mirzazadeh, A. Does the presence of the Helicobacter pylori in the dental plaque associate with its gastric infection? A meta-analysis and systematic review. Dent. Res. J. 2011, 8, 178–182. [Google Scholar] [CrossRef]
- Roman-Roman, A.; Giono-Cerezo, S.; Camorlinga-Ponce, M.; Martinez-Carrillo, D.N.; Loaiza-Loeza, S.; Fernandez-Tilapa, G. vacA genotypes of Helicobacter pylori in the oral cavity and stomach of patients with chronic gastritis and gastric ulcer. Enferm Infecc. Microbiol. Clin. 2013, 31, 130–135. [Google Scholar] [CrossRef]
- Aksit Bicak, D.; Akyuz, S.; Kiratli, B.; Usta, M.; Urganci, N.; Alev, B.; Yarat, A.; Sahin, F. The investigation of Helicobacter pylori in the dental biofilm and saliva samples of children with dyspeptic complaints. BMC Oral Health 2017, 17, 67. [Google Scholar] [CrossRef]
- Song, Q.; Lange, T.; Spahr, A.; Adler, G.; Bode, G. Characteristic distribution pattern of Helicobacter pylori in dental plaque and saliva detected with nested PCR. J. Med. Microbiol. 2000, 49, 349–353. [Google Scholar] [CrossRef]
- Tongtawee, T.; Wattanawongdon, W.; Simawaranon, T. Effects of periodontal therapy on eradication and recurrence of Helicobacter pylori infection after successful treatment. J. Int. Med. Res. 2019, 47, 875–883. [Google Scholar] [CrossRef]
- Cai, H.; Li, W.; Shu, X.; Peng, K.; Zhang, Y.; Jiang, M. Genetic variation of Helicobacter pylori in the oral cavity and stomach detected using thymine adenine cloning in children with chronic gastritis. Pediatr. Infect. Dis. J. 2014, 33, e1–e6. [Google Scholar] [CrossRef]
- Leszczynska, K.; Namiot, D.B.; Namiot, Z.; Leszczynska, J.K.; Jakoniuk, P.; Kemona, A. Application of immunoassay for detection of Helicobacter pylori antigens in the dental plaque. Adv. Med. Sci. 2009, 54, 194–198. [Google Scholar] [CrossRef]
- Anand, P.S.; Nandakumar, K.; Shenoy, K.T. Are dental plaque, poor oral hygiene, and periodontal disease associated with Helicobacter pylori infection? J. Periodontol. 2006, 77, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Al Asqah, M.; Al Hamoudi, N.; Anil, S.; Al Jebreen, A.; Al-Hamoudi, W.K. Is the presence of Helicobacter pylori in dental plaque of patients with chronic periodontitis a risk factor for gastric infection? Can. J. Gastroenterol. 2009, 23, 177–179. [Google Scholar] [CrossRef]
- Liu, Y.; Yue, H.; Li, A.; Wang, J.; Jiang, B.; Zhang, Y.; Bai, Y. An epidemiologic study on the correlation between oral Helicobacter pylori and gastric H. pylori. Curr. Microbiol. 2009, 58, 449–453. [Google Scholar] [CrossRef]
- Teoman, I.; Ozmeric, N.; Ozcan, G.; Alaaddinoglu, E.; Dumlu, S.; Akyon, Y.; Balos, K. Comparison of different methods to detect Helicobacter pylori in the dental plaque of dyspeptic patients. Clin. Oral. Investig. 2007, 11, 201–205. [Google Scholar] [CrossRef]
- Medina, M.L.; Medina, M.G.; Merino, L.A. Correlation between virulence markers of Helicobacter pylori in the oral cavity and gastric biopsies. Arq. Gastroenterol. 2017, 54, 217–221. [Google Scholar] [CrossRef]
- Rasmussen, L.T.; de Labio, R.W.; Neto, A.C.; Silva, L.C.; Queiroz, V.F.; Smith, M.A.C.; Payao, S.L.M. Detection of Helicobacter pylori in gastric biopsies, saliva and dental plaques of dyspeptic patients from Marilia, Sao Paulo, Brazil: Presence of vacA and cagA genes. J. Venom. Anim. Toxins Incl. Trop. Dis. 2012, 18, 180–187. [Google Scholar] [CrossRef]
- Silva, D.G.; Stevens, R.H.; Macedo, J.M.; Albano, R.M.; Falabella, M.E.; Veerman, E.C.; Tinoco, E.M. Detection of cytotoxin genotypes of Helicobacter pylori in stomach, saliva and dental plaque. Arch. Oral. Biol. 2009, 54, 684–688. [Google Scholar] [CrossRef]
- Bago, I.; Bago, J.; Plecko, V.; Aurer, A.; Majstorovic, K.; Budimir, A. The effectiveness of systemic eradication therapy against oral Helicobacter pylori. J. Oral. Pathol. Med. 2011, 40, 428–432. [Google Scholar] [CrossRef]
- Liu, Y.; Li, R.; Xue, X.; Xu, T.; Luo, Y.; Dong, Q.; Liu, J.; Liu, J.; Pan, Y.; Zhang, D. Periodontal disease and Helicobacter pylori infection in oral cavity: A meta-analysis of 2727 participants mainly based on Asian studies. Clin. Oral. Investig. 2020, 24, 2175–2188. [Google Scholar] [CrossRef]
- Hu, Z.; Zhang, Y.; Li, Z.; Yu, Y.; Kang, W.; Han, Y.; Geng, X.; Ge, S.; Sun, Y. Effect of Helicobacter pylori infection on chronic periodontitis by the change of microecology and inflammation. Oncotarget 2016, 7, 66700–66712. [Google Scholar] [CrossRef]
- Soto, C.; Rojas, V.; Yáñez, L.; Hidalgo, A.; Olivera, M.; Pacheco, M.; Venegas, D.; Salinas, D.; Bravo, D.; Quest, A.F.G. Porphyromonas gingivalis-Helicobacter pylori co-incubation enhances Porphyromonas gingivalis virulence and increases migration of infected human oral keratinocytes. J. Oral. Microbiol. 2022, 14, 2107691. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhang, Y.; Yang, Z.; Li, J.; Li, Y.; Li, H.; Li, W.; Jia, J.; Ge, S.; Sun, Y. Helicobacter pylori infection is correlated with the incidence of erosive oral lichen planus and the alteration of the oral microbiome composition. BMC Microbiol. 2021, 21, 122. [Google Scholar] [CrossRef] [PubMed]
- Kazanowska-Dygdala, M.; Dus, I.; Radwan-Oczko, M. The presence of Helicobacter pylori in oral cavities of patients with leukoplakia and oral lichen planus. J. Appl. Oral. Sci. 2016, 24, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.A.; Kheur, S.; Raj, A.T.; Mahajan, P. Association of Helicobacter pylori with oral potentially malignant disorders and oral squamous cell carcinoma-a systematic review and meta-analysis. Clin. Oral. Investig. 2020, 24, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Wang, Q.; He, C.; Chen, M.; Liu, J.; Liu, W.; Yuan, Y. An inverse association of Helicobacter pylori infection with oral squamous cell carcinoma. J. Oral. Pathol. Med. 2016, 45, 17–22. [Google Scholar] [CrossRef]
- Hulimavu, S.R.; Mohanty, L.; Tondikulam, N.V.; Shenoy, S.; Jamadar, S.; Bhadranna, A. No evidence for Helicobacter pylori in oral lichen planus. J. Oral. Pathol. Med. 2014, 43, 576–578. [Google Scholar] [CrossRef]
- Zou, Q.H.; Li, R.Q. Helicobacter pylori in the oral cavity and gastric mucosa: A meta-analysis. J. Oral. Pathol. Med. 2011, 40, 317–324. [Google Scholar] [CrossRef]
- Gebara, E.C.; Faria, C.M.; Pannuti, C.; Chehter, L.; Mayer, M.P.; Lima, L.A. Persistence of Helicobacter pylori in the oral cavity after systemic eradication therapy. J. Clin. Periodontol. 2006, 33, 329–333. [Google Scholar] [CrossRef]
- Ren, Q.; Yan, X.; Zhou, Y.; Li, W.X. Periodontal therapy as adjunctive treatment for gastric Helicobacter pylori infection. Cochrane Database Syst. Rev. 2016, 2, CD009477. [Google Scholar] [CrossRef]
- Ozturk, A. Periodontal Treatment Is Associated With Improvement in Gastric Helicobacter pylori Eradication: An Updated Meta-analysis of Clinical Trials. Int. Dent. J. 2021, 71, 188–196. [Google Scholar] [CrossRef]
- Yuksel Sert, S.; Ozturk, A.; Bektas, A.; Cengiz, M.I. Periodontal treatment is more effective in gastric Helicobacter pylori eradication in those patients who maintain good oral hygiene. Int. Dent. J. 2019, 69, 392–399. [Google Scholar] [CrossRef]
- Song, H.Y.; Li, Y. Can eradication rate of gastric Helicobacter pylori be improved by killing oral Helicobacter pylori? World J. Gastroenterol. 2013, 19, 6645–6650. [Google Scholar] [CrossRef]
- Zaric, S.; Bojic, B.; Jankovic, L.; Dapcevic, B.; Popovic, B.; Cakic, S.; Milasin, J. Periodontal therapy improves gastric Helicobacter pylori eradication. J. Dent. Res. 2009, 88, 946–950. [Google Scholar] [CrossRef]
- Gao, J.; Li, Y.; Wang, Q.; Qi, C.; Zhu, S. Correlation between distribution of Helicobacter pylori in oral cavity and chronic stomach conditions. J. Huazhong Univ. Sci. Technol. Med. Sci. 2011, 31, 409–412. [Google Scholar] [CrossRef]
- Miller, A.K.; Williams, S.M. Helicobacter pylori infection causes both protective and deleterious effects in human health and disease. Genes Immun. 2021, 22, 218–226. [Google Scholar] [CrossRef]
- Yamaoka, Y. Mechanisms of disease: Helicobacter pylori virulence factors. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 629–641. [Google Scholar] [CrossRef]
- Alexander, S.M.; Retnakumar, R.J.; Chouhan, D.; Devi, T.N.B.; Dharmaseelan, S.; Devadas, K.; Thapa, N.; Tamang, J.P.; Lamtha, S.C.; Chattopadhyay, S. Helicobacter pylori in Human Stomach: The Inconsistencies in Clinical Outcomes and the Probable Causes. Front. Microbiol. 2021, 12, 713955. [Google Scholar] [CrossRef]
- Wen, J.; Lau, H.C.; Peppelenbosch, M.; Yu, J. Gastric Microbiota beyond H. pylori: An Emerging Critical Character in Gastric Carcinogenesis. Biomedicines 2021, 9, 1680. [Google Scholar] [CrossRef]
- Freire, M.; Nelson, K.E.; Edlund, A. The Oral Host-Microbial Interactome: An Ecological Chronometer of Health? Trends Microbiol. 2021, 29, 551–561. [Google Scholar] [CrossRef]
- Bakhti, S.Z.; Latifi-Navid, S. Oral microbiota and Helicobacter pylori in gastric carcinogenesis: What do we know and where next? BMC Microbiol. 2021, 21, 71. [Google Scholar] [CrossRef]
- Maixner, F.; Krause-Kyora, B.; Turaev, D.; Herbig, A.; Hoopmann, M.R.; Hallows, J.L.; Kusebauch, U.; Vigl, E.E.; Malfertheiner, P.; Megraud, F.; et al. The 5300-year-old Helicobacter pylori genome of the Iceman. Science 2016, 351, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.M.; Lu, Q.F.; Li, S.B.; Wang, J.P.; Chen, Y.L.; Huang, Y.Q.; Bi, H.K. Comparative Genomics of H. pylori and Non-Pylori Helicobacter Species to Identify New Regions Associated with Its Pathogenicity and Adaptability. Biomed. Res. Int. 2016, 2016, 6106029. [Google Scholar] [CrossRef] [PubMed]
- Noto, J.M.; Chopra, A.; Loh, J.T.; Romero-Gallo, J.; Piazuelo, M.B.; Watson, M.; Leary, S.; Beckett, A.C.; Wilson, K.T.; Cover, T.L.; et al. Pan-genomic analyses identify key Helicobacter pylori pathogenic loci modified by carcinogenic host microenvironments. Gut 2018, 67, 1793–1804. [Google Scholar] [CrossRef] [PubMed]
- Falush, D.; Wirth, T.; Linz, B.; Pritchard, J.K.; Stephens, M.; Kidd, M.; Blaser, M.J.; Graham, D.Y.; Vacher, S.; Perez-Perez, G.I.; et al. Traces of human migrations in Helicobacter pylori populations. Science 2003, 299, 1582–1585. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Ramirez, Z.Y.; Pascoe, B.; Mendez-Tenorio, A.; Mourkas, E.; Sandoval-Motta, S.; Perez-Perez, G.; Morgan, D.R.; Dominguez, R.L.; Ortiz-Princz, D.; Cavazza, M.E.; et al. A 500-year tale of co-evolution, adaptation, and virulence: Helicobacter pylori in the Americas. ISME J. 2021, 15, 78–92. [Google Scholar] [CrossRef]
- Chang, W.L.; Yeh, Y.C.; Sheu, B.S. The impacts of H. pylori virulence factors on the development of gastroduodenal diseases. J. Biomed. Sci. 2018, 25, 68. [Google Scholar] [CrossRef]
- Kao, C.Y.; Sheu, B.S.; Wu, J.J. Helicobacter pylori infection: An overview of bacterial virulence factors and pathogenesis. Biomed. J. 2016, 39, 14–23. [Google Scholar] [CrossRef]
- Doohan, D.; Rezkitha, Y.A.A.; Waskito, L.A.; Yamaoka, Y.; Miftahussurur, M. Helicobacter pylori BabA-SabA Key Roles in the Adherence Phase: The Synergic Mechanism for Successful Colonization and Disease Development. Toxins 2021, 13, 485. [Google Scholar] [CrossRef]
- Sekar, R.; Murali, P.; Junaid, M. Quantification of Helicobacter pylori and its oncoproteins in the oral cavity: A cross-sectional study. Oral. Dis. 2022. [Google Scholar] [CrossRef]
- Olbermann, P.; Josenhans, C.; Moodley, Y.; Uhr, M.; Stamer, C.; Vauterin, M.; Suerbaum, S.; Achtman, M.; Linz, B. A global overview of the genetic and functional diversity in the Helicobacter pylori cag pathogenicity island. PLoS Genet. 2010, 6, e1001069. [Google Scholar] [CrossRef]
- Plummer, M.; van Doorn, L.J.; Franceschi, S.; Kleter, B.; Canzian, F.; Vivas, J.; Lopez, G.; Colin, D.; Munoz, N.; Kato, I. Helicobacter pylori cytotoxin-associated genotype and gastric precancerous lesions. J. Natl. Cancer Inst. 2007, 99, 1328–1334. [Google Scholar] [CrossRef]
- Higashi, H.; Tsutsumi, R.; Fujita, A.; Yamazaki, S.; Asaka, M.; Azuma, T.; Hatakeyama, M. Biological activity of the Helicobacter pylori virulence factor CagA is determined by variation in the tyrosine phosphorylation sites. Proc. Natl. Acad. Sci. USA 2002, 99, 14428–14433. [Google Scholar] [CrossRef]
- Zeng, J.; Xie, C.; Zhang, L.; Liu, X.; Chan, M.T.V.; Wu, W.K.K.; Chen, H. Host Cell Antimicrobial Responses against Helicobacter pylori Infection: From Biological Aspects to Therapeutic Strategies. Int. J. Mol. Sci. 2022, 23, 941. [Google Scholar] [CrossRef]
- Loh, J.T.; Shaffer, C.L.; Piazuelo, M.B.; Bravo, L.E.; McClain, M.S.; Correa, P.; Cover, T.L. Analysis of cagA in Helicobacter pylori strains from Colombian populations with contrasting gastric cancer risk reveals a biomarker for disease severity. Cancer Epidemiol. Biomark. Prev. 2011, 20, 2237–2249. [Google Scholar] [CrossRef]
- Ferreira, R.M.; Pinto-Ribeiro, I.; Wen, X.; Marcos-Pinto, R.; Dinis-Ribeiro, M.; Carneiro, F.; Figueiredo, C. Helicobacter pylori cagA Promoter Region Sequences Influence CagA Expression and Interleukin 8 Secretion. J. Infect. Dis. 2016, 213, 669–673. [Google Scholar] [CrossRef]
- Forsyth, M.H.; Atherton, J.C.; Blaser, M.J.; Cover, T.L. Heterogeneity in levels of vacuolating cytotoxin gene (vacA) transcription among Helicobacter pylori strains. Infect. Immun. 1998, 66, 3088–3094. [Google Scholar] [CrossRef]
- Mark Welch, J.L.; Rossetti, B.J.; Rieken, C.W.; Dewhirst, F.E.; Borisy, G.G. Biogeography of a human oral microbiome at the micron scale. Proc. Natl. Acad. Sci. USA 2016, 113, E791–E800. [Google Scholar] [CrossRef]
- Zijnge, V.; van Leeuwen, M.B.; Degener, J.E.; Abbas, F.; Thurnheer, T.; Gmur, R.; Harmsen, H.J. Oral biofilm architecture on natural teeth. PLoS ONE 2010, 5, e9321. [Google Scholar] [CrossRef]
- Bowen, W.H.; Burne, R.A.; Wu, H.; Koo, H. Oral Biofilms: Pathogens, Matrix, and Polymicrobial Interactions in Microenvironments. Trends Microbiol. 2018, 26, 229–242. [Google Scholar] [CrossRef]
- Huang, R.; Li, M.; Gregory, R.L. Bacterial interactions in dental biofilm. Virulence 2011, 2, 435–444. [Google Scholar] [CrossRef]
- Donlan, R.M.; Costerton, J.W. Biofilms: Survival mechanisms of clinically relevant microorganisms. Clin. Microbiol. Rev. 2002, 15, 167–193. [Google Scholar] [CrossRef] [PubMed]
- Iwai, K.; Watanabe, I.; Yamamoto, T.; Kuriyama, N.; Matsui, D.; Nomura, R.; Ogaya, Y.; Oseko, F.; Adachi, K.; Takizawa, S.; et al. Association between Helicobacter pylori infection and dental pulp reservoirs in Japanese adults. BMC Oral Health 2019, 19, 267. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Noto, J.M.; Romero-Gallo, J.; Peek, R.M., Jr.; Amieva, M.R. Helicobacter pylori perturbs iron trafficking in the epithelium to grow on the cell surface. PLoS Pathog. 2011, 7, e1002050. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Tompkins, L.S.; Amieva, M.R. Helicobacter pylori usurps cell polarity to turn the cell surface into a replicative niche. PLoS Pathog. 2009, 5, e1000407. [Google Scholar] [CrossRef]
- Koniger, V.; Holsten, L.; Harrison, U.; Busch, B.; Loell, E.; Zhao, Q.; Bonsor, D.A.; Roth, A.; Kengmo-Tchoupa, A.; Smith, S.I.; et al. Helicobacter pylori exploits human CEACAMs via HopQ for adherence and translocation of CagA. Nat. Microbiol. 2016, 2, 16188. [Google Scholar] [CrossRef]
- Bonsor, D.A.; Zhao, Q.; Schmidinger, B.; Weiss, E.; Wang, J.; Deredge, D.; Beadenkopf, R.; Dow, B.; Fischer, W.; Beckett, D.; et al. The Helicobacter pylori adhesin protein HopQ exploits the dimer interface of human CEACAMs to facilitate translocation of the oncoprotein CagA. EMBO J. 2018, 37, e98664. [Google Scholar] [CrossRef]
- Tegtmeyer, N.; Ghete, T.D.; Schmitt, V.; Remmerbach, T.; Cortes, M.C.C.; Bondoc, E.M.; Graf, H.L.; Singer, B.B.; Hirsch, C.; Backert, S. Type IV secretion of Helicobacter pylori CagA into oral epithelial cells is prevented by the absence of CEACAM receptor expression. Gut. Pathog. 2020, 12, 25. [Google Scholar] [CrossRef]
- Mima, J.; Koshino, A.; Oka, K.; Uchida, H.; Hieda, Y.; Nohara, K.; Kogo, M.; Chai, Y.; Sakai, T. Regulation of the epithelial adhesion molecule CEACAM1 is important for palate formation. PLoS ONE 2013, 8, e61653. [Google Scholar] [CrossRef]
- Wang, F.F.; Guan, B.X.; Yang, J.Y.; Wang, H.T.; Zhou, C.J. CEACAM1 is overexpressed in oral tumors and related to tumorigenesis. Med. Mol. Morphol. 2017, 50, 42–51. [Google Scholar] [CrossRef]
- Huynh-Torlakovic, H.; Bjerkan, L.; Schenck, K.; Blix, I.J. Distribution of carcinoembryonic antigen-related cellular adhesion molecules in human gingiva. Eur. J. Oral Sci. 2012, 120, 395–401. [Google Scholar] [CrossRef]
- Liu, G.X.; Xie, Q.; Zhou, C.J.; Zhang, X.Y.; Ma, B.L.; Wang, C.Q.; Wei, F.C.; Qu, X.; Sun, S.Z. The possible roles of OPN-regulated CEACAM1 expression in promoting the survival of activated T cells and the apoptosis of oral keratinocytes in oral lichen planus patients. J. Clin. Immunol. 2011, 31, 827–839. [Google Scholar] [CrossRef]
- Boyle, J.O.; Gumus, Z.H.; Kacker, A.; Choksi, V.L.; Bocker, J.M.; Zhou, X.K.; Yantiss, R.K.; Hughes, D.B.; Du, B.; Judson, B.L.; et al. Effects of cigarette smoke on the human oral mucosal transcriptome. Cancer Prev. Res. 2010, 3, 266–278. [Google Scholar] [CrossRef]
- Gray-Owen, S.D.; Blumberg, R.S. CEACAM1: Contact-dependent control of immunity. Nat. Rev. Immunol. 2006, 6, 433–446. [Google Scholar] [CrossRef]
- Gur, C.; Maalouf, N.; Gerhard, M.; Singer, B.B.; Emgard, J.; Temper, V.; Neuman, T.; Mandelboim, O.; Bachrach, G. The Helicobacter pylori HopQ outermembrane protein inhibits immune cell activities. Oncoimmunology 2019, 8, e1553487. [Google Scholar] [CrossRef]
- Petersen, A.M.; Krogfelt, K.A. Helicobacter pylori: An invading microorganism? A review. FEMS Immunol. Med. Microbiol. 2003, 36, 117–126. [Google Scholar] [CrossRef]
- Dubois, A.; Boren, T. Helicobacter pylori is invasive and it may be a facultative intracellular organism. Cell Microbiol. 2007, 9, 1108–1116. [Google Scholar] [CrossRef]
- Chu, Y.T.; Wang, Y.H.; Wu, J.J.; Lei, H.Y. Invasion and multiplication of Helicobacter pylori in gastric epithelial cells and implications for antibiotic resistance. Infect. Immun. 2010, 78, 4157–4165. [Google Scholar] [CrossRef]
- Li, H.; Liang, D.; Hu, N.; Dai, X.; He, J.; Zhuang, H.; Zhao, W. Helicobacter pylori inhibited cell proliferation in human periodontal ligament fibroblasts through the Cdc25C/CDK1/cyclinB1 signaling cascade. J. Periodontal. Implant. Sci. 2019, 49, 138–147. [Google Scholar] [CrossRef]
- Sit, W.Y.; Chen, Y.A.; Chen, Y.L.; Lai, C.H.; Wang, W.C. Cellular evasion strategies of Helicobacter pylori in regulating its intracellular fate. Semin. Cell. Dev. Biol. 2020, 101, 59–67. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, Q.L.; Cheng, D.D.; Xu, W.T.; Lu, N.H. Adhesion and Invasion of Gastric Mucosa Epithelial Cells by Helicobacter pylori. Front. Cell Infect. Microbiol. 2016, 6, 159. [Google Scholar] [CrossRef]
- Cheok, Y.Y.; Lee, C.Y.Q.; Cheong, H.C.; Vadivelu, J.; Looi, C.Y.; Abdullah, S.; Wong, W.F. An Overview of Helicobacter pylori Survival Tactics in the Hostile Human Stomach Environment. Microorganisms 2021, 9, 2502. [Google Scholar] [CrossRef] [PubMed]
- Cullen, T.W.; Giles, D.K.; Wolf, L.N.; Ecobichon, C.; Boneca, I.G.; Trent, M.S. Helicobacter pylori versus the host: Remodeling of the bacterial outer membrane is required for survival in the gastric mucosa. PLoS Pathog. 2011, 7, e1002454. [Google Scholar] [CrossRef] [PubMed]
- Schmidinger, B.; Petri, K.; Lettl, C.; Li, H.; Namineni, S.; Ishikawa-Ankerhold, H.; Jimenez-Soto, L.F.; Haas, R. Helicobacter pylori binds human Annexins via Lipopolysaccharide to interfere with Toll-like Receptor 4 signaling. PLoS Pathog. 2022, 18, e1010326. [Google Scholar] [CrossRef] [PubMed]
- Behrens, I.K.; Busch, B.; Ishikawa-Ankerhold, H.; Palamides, P.; Shively, J.E.; Stanners, C.; Chan, C.; Leung, N.; Gray-Owen, S.; Haas, R. The HopQ-CEACAM Interaction Controls CagA Translocation, Phosphorylation, and Phagocytosis of Helicobacter pylori in Neutrophils. mBio 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef]
- Cheok, Y.Y.; Tan, G.M.Y.; Lee, C.Y.Q.; Abdullah, S.; Looi, C.Y.; Wong, W.F. Innate Immunity Crosstalk with Helicobacter pylori: Pattern Recognition Receptors and Cellular Responses. Int. J. Mol. Sci. 2022, 23, 7561. [Google Scholar] [CrossRef]
- Go, D.M.; Lee, S.H.; Lee, S.H.; Woo, S.H.; Kim, K.; Kim, K.; Park, K.S.; Park, J.H.; Ha, S.J.; Kim, W.H.; et al. Programmed Death Ligand 1-Expressing Classical Dendritic Cells Mitigate Helicobacter-Induced Gastritis. Cell Mol. Gastroenterol. Hepatol. 2021, 12, 715–739. [Google Scholar] [CrossRef]
- Javed, S.; Skoog, E.C.; Solnick, J.V. Impact of Helicobacter pylori Virulence Factors on the Host Immune Response and Gastric Pathology. Curr. Top. Microbiol. Immunol. 2019, 421, 21–52. [Google Scholar] [CrossRef]
- Liu, Y.; Lin, H.; Bai, Y.; Qin, X.; Zheng, X.; Sun, Y.; Zhang, Y. Study on the relationship between Helicobacter pylori in the dental plaque and the occurrence of dental caries or oral hygiene index. Helicobacter 2008, 13, 256–260. [Google Scholar] [CrossRef]
- Souto, R.; Colombo, A.P. Detection of Helicobacter pylori by polymerase chain reaction in the subgingival biofilm and saliva of non-dyspeptic periodontal patients. J. Periodontol. 2008, 79, 97–103. [Google Scholar] [CrossRef]
- Nomura, R.; Kadota, T.; Ogaya, Y.; Matayoshi, S.; Iwashita, N.; Okawa, R.; Nakano, K. Contribution of Streptococcus mutans to Helicobacter pylori colonisation in oral cavity and gastric tissue. Sci. Rep. 2020, 10, 12540. [Google Scholar] [CrossRef]
- Zhang, W.; Deng, X.; Zhou, X.; Hao, Y.; Li, Y. Influence of Helicobacter pylori culture supernatant on the ecological balance of a dual-species oral biofilm. J. Appl. Oral. Sci. 2018, 26, e20170113. [Google Scholar] [CrossRef]
- Chen, X.; Zhou, X.; Liao, B.; Zhou, Y.; Cheng, L.; Ren, B. The cross-kingdom interaction between Helicobacter pylori and Candida albicans. PLoS Pathog. 2021, 17, e1009515. [Google Scholar] [CrossRef]
- Kadota, T.; Hamada, M.; Nomura, R.; Ogaya, Y.; Okawa, R.; Uzawa, N.; Nakano, K. Distribution of Helicobacter pylori and Periodontopathic Bacterial Species in the Oral Cavity. Biomedicines 2020, 8, 161. [Google Scholar] [CrossRef]
- Andersen, R.N.; Ganeshkumar, N.; Kolenbrander, P.E. Helicobacter pylori adheres selectively to Fusobacterium spp. Oral Microbiol. Immunol. 1998, 13, 51–54. [Google Scholar] [CrossRef]
- Sanchez-Alonzo, K.; Silva-Mieres, F.; Arellano-Arriagada, L.; Parra-Sepulveda, C.; Bernasconi, H.; Smith, C.T.; Campos, V.L.; Garcia-Cancino, A. Nutrient Deficiency Promotes the Entry of Helicobacter pylori Cells into Candida Yeast Cells. Biology 2021, 10, 426. [Google Scholar] [CrossRef]
- Palencia, S.L.; Garcia, A.; Palencia, M. Multiple surface interaction mechanisms direct the anchoring, co-aggregation and formation of dual-species biofilm between Candida albicans and Helicobacter pylori. J. Adv. Res. 2022, 35, 169–185. [Google Scholar] [CrossRef]
- Saniee, P.; Siavoshi, F.; Nikbakht Broujeni, G.; Khormali, M.; Sarrafnejad, A.; Malekzadeh, R. Localization of H.pylori within the vacuole of Candida yeast by direct immunofluorescence technique. Arch. Iran. Med. 2013, 16, 705–710. [Google Scholar]
- Saniee, P.; Siavoshi, F. Endocytotic uptake of FITC-labeled anti-H. pylori egg yolk immunoglobulin Y in Candida yeast for detection of intracellular H. pylori. Front. Microbiol. 2015, 6, 113. [Google Scholar] [CrossRef]
- Sanchez-Alonzo, K.; Belmar, L.; Parra-Sepulveda, C.; Bernasconi, H.; Campos, V.L.; Smith, C.T.; Saez, K.; Garcia-Cancino, A. Antibiotics as a Stressing Factor Triggering the Harboring of Helicobacter pylori J99 within Candida albicans ATCC10231. Pathogens 2021, 10, 382. [Google Scholar] [CrossRef]
- Sanchez-Alonzo, K.; Parra-Sepulveda, C.; Vega, S.; Bernasconi, H.; Campos, V.L.; Smith, C.T.; Saez, K.; Garcia-Cancino, A. In Vitro Incorporation of Helicobacter pylori into Candida albicans Caused by Acidic pH Stress. Pathogens 2020, 9, 489. [Google Scholar] [CrossRef] [PubMed]
- Heydari, S.; Siavoshi, F.; Jazayeri, M.H.; Sarrafnejad, A.; Saniee, P. Helicobacter pylori release from yeast as a vesicle-encased or free bacterium. Helicobacter 2020, 25, e12725. [Google Scholar] [CrossRef] [PubMed]
- Okuda, K.; Ishihara, K.; Miura, T.; Katakura, A.; Noma, H.; Ebihara, Y. Helicobacter pylori may have only a transient presence in the oral cavity and on the surface of oral cancer. Microbiol. Immunol. 2000, 44, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Contaldo, M.; Fusco, A.; Stiuso, P.; Lama, S.; Gravina, A.G.; Itro, A.; Federico, A.; Itro, A.; Dipalma, G.; Inchingolo, F.; et al. Oral Microbiota and Salivary Levels of Oral Pathogens in Gastro-Intestinal Diseases: Current Knowledge and Exploratory Study. Microorganisms 2021, 9, 1064. [Google Scholar] [CrossRef] [PubMed]
- Tefiku, U.; Popovska, M.; Cana, A.; Zendeli-Bedxeti, L.; Recica, B.; Spasovska-Gjorgovska, A.; Spasovski, S. Determination of the Role of Fusobacterium Nucleatum in the Pathogenesis in and Out the Mouth. Prilozi 2020, 41, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Lima, B.P.; Shi, W.; Lux, R. Identification and characterization of a novel Fusobacterium nucleatum adhesin involved in physical interaction and biofilm formation with Streptococcus gordonii. Microbiologyopen 2017, 6, e00444. [Google Scholar] [CrossRef]
- Massarrat, S.; Saniee, P.; Siavoshi, F.; Mokhtari, R.; Mansour-Ghanaei, F.; Khalili-Samani, S. The Effect of Helicobacter pylori Infection, Aging, and Consumption of Proton Pump Inhibitor on Fungal Colonization in the Stomach of Dyspeptic Patients. Front. Microbiol. 2016, 7, 801. [Google Scholar] [CrossRef]
- Siavoshi, F.; Taghikhani, A.; Malekzadeh, R.; Sarrafnejad, A.; Kashanian, M.; Jamal, A.S.; Saniee, P.; Sadeghi, S.; Sharifi, A.H. The role of mother’s oral and vaginal yeasts in transmission of Helicobacter pylori to neonates. Arch. Iran. Med. 2013, 16, 288–294. [Google Scholar]
- Fox, E.P.; Cowley, E.S.; Nobile, C.J.; Hartooni, N.; Newman, D.K.; Johnson, A.D. Anaerobic bacteria grow within Candida albicans biofilms and induce biofilm formation in suspension cultures. Curr. Biol. 2014, 24, 2411–2416. [Google Scholar] [CrossRef]
- Siavoshi, F.; Heydari, S.; Shafiee, M.; Ahmadi, S.; Saniee, P.; Sarrafnejad, A.; Kolahdoozan, S. Sequestration inside the yeast vacuole may enhance Helicobacter pylori survival against stressful condition. Infect. Genet. Evol. 2019, 69, 127–133. [Google Scholar] [CrossRef]
- Saniee, P.; Siavoshi, F.; Nikbakht Broujeni, G.; Khormali, M.; Sarrafnejad, A.; Malekzadeh, R. Immunodetection of Helicobacter pylori-specific proteins in oral and gastric Candida yeasts. Arch. Iran. Med. 2013, 16, 624–630. [Google Scholar]
- Siavoshi, F.; Sahraee, M.; Ebrahimi, H.; Sarrafnejad, A.; Saniee, P. Natural fruits, flowers, honey, and honeybees harbor Helicobacter pylori-positive yeasts. Helicobacter 2018, 23, e12471. [Google Scholar] [CrossRef]
- Siavoshi, F.; Salmanian, A.H.; Akbari, F.; Malekzadeh, R.; Massarrat, S. Detection of Helicobacter pylori-specific genes in the oral yeast. Helicobacter 2005, 10, 318–322. [Google Scholar] [CrossRef]
- Sanchez-Alonzo, K.; Matamala-Valdes, L.; Parra-Sepulveda, C.; Bernasconi, H.; Campos, V.L.; Smith, C.T.; Saez, K.; Garcia-Cancino, A. Intracellular Presence of Helicobacter pylori and Its Virulence-Associated Genotypes within the Vaginal Yeast of Term Pregnant Women. Microorganisms 2021, 9, 131. [Google Scholar] [CrossRef]
- Dong, K.; Pan, H.; Yang, D.; Rao, L.; Zhao, L.; Wang, Y.; Liao, X. Induction, detection, formation, and resuscitation of viable but non-culturable state microorganisms. Compr. Rev. Food Sci. Food Saf. 2020, 19, 149–183. [Google Scholar] [CrossRef]
- Hirukawa, S.; Sagara, H.; Kaneto, S.; Kondo, T.; Kiga, K.; Sanada, T.; Kiyono, H.; Mimuro, H. Characterization of morphological conversion of Helicobacter pylori under anaerobic conditions. Microbiol. Immunol. 2018, 62, 221–228. [Google Scholar] [CrossRef]
- Nilsson, H.O.; Blom, J.; Abu-Al-Soud, W.; Ljungh, A.A.; Andersen, L.P.; Wadstrom, T. Effect of cold starvation, acid stress, and nutrients on metabolic activity of Helicobacter pylori. Appl. Environ. Microbiol. 2002, 68, 11–19. [Google Scholar] [CrossRef]
- Mizoguchi, H.; Fujioka, T.; Kishi, K.; Nishizono, A.; Kodama, R.; Nasu, M. Diversity in protein synthesis and viability of Helicobacter pylori coccoid forms in response to various stimuli. Infect. Immun. 1998, 66, 5555–5560. [Google Scholar] [CrossRef]
- Adams, B.L.; Bates, T.C.; Oliver, J.D. Survival of Helicobacter pylori in a natural freshwater environment. Appl. Environ. Microbiol. 2003, 69, 7462–7466. [Google Scholar] [CrossRef]
- Saito, N.; Konishi, K.; Sato, F.; Kato, M.; Takeda, H.; Sugiyama, T.; Asaka, M. Plural transformation-processes from spiral to coccoid Helicobacter pylori and its viability. J. Infect. 2003, 46, 49–55. [Google Scholar] [CrossRef]
- Sisto, F.; Brenciaglia, M.I.; Scaltrito, M.M.; Dubini, F. Helicobacter pylori: ureA, cagA and vacA expression during conversion to the coccoid form. Int. J. Antimicrob. Agents 2000, 15, 277–282. [Google Scholar] [CrossRef]
- Poursina, F.; Fagri, J.; Mirzaei, N.; Safaei, H.G. Overexpression of spoT gene in coccoid forms of clinical Helicobacter pylori isolates. Folia Microbiol. 2018, 63, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Moreno, Y.; Piqueres, P.; Alonso, J.L.; Jimenez, A.; Gonzalez, A.; Ferrus, M.A. Survival and viability of Helicobacter pylori after inoculation into chlorinated drinking water. Water Res. 2007, 41, 3490–3496. [Google Scholar] [CrossRef] [PubMed]
- Poursina, F.; Faghri, J.; Moghim, S.; Zarkesh-Esfahani, H.; Nasr-Esfahani, B.; Fazeli, H.; Hasanzadeh, A.; Safaei, H.G. Assessment of cagE and babA mRNA expression during morphological conversion of Helicobacter pylori from spiral to coccoid. Curr. Microbiol. 2013, 66, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Piqueres, P.; Moreno, Y.; Alonso, J.L.; Ferrus, M.A. A combination of direct viable count and fluorescent in situ hybridization for estimating Helicobacter pylori cell viability. Res. Microbiol. 2006, 157, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Quaglia, N.C.; Storelli, M.M.; Scardocchia, T.; Lattanzi, A.; Celano, G.V.; Monno, R.; Dambrosio, A. Helicobacter pylori: Survival in cultivable and non-cultivable form in artificially contaminated Mytilus galloprovincialis. Int. J. Food Microbiol. 2020, 312, 108363. [Google Scholar] [CrossRef]
- Cellini, L.; Allocati, N.; Angelucci, D.; Iezzi, T.; Di Campli, E.; Marzio, L.; Dainelli, B. Coccoid Helicobacter pylori not culturable in vitro reverts in mice. Microbiol. Immunol. 1994, 38, 843–850. [Google Scholar] [CrossRef]
- Boehnke, K.F.; Eaton, K.A.; Fontaine, C.; Brewster, R.; Wu, J.; Eisenberg, J.N.S.; Valdivieso, M.; Baker, L.H.; Xi, C. Reduced infectivity of waterborne viable but nonculturable Helicobacter pylori strain SS1 in mice. Helicobacter 2017, 22, e12391. [Google Scholar] [CrossRef]
- Wang, X.; Sturegard, E.; Rupar, R.; Nilsson, H.O.; Aleljung, P.A.; Carlen, B.; Willen, R.; Wadstrom, T. Infection of BALB/c A mice by spiral and coccoid forms of Helicobacter pylori. J. Med. Microbiol. 1997, 46, 657–663. [Google Scholar] [CrossRef]
- Kaprelyants, A.S.; Gottschal, J.C.; Kell, D.B. Dormancy in non-sporulating bacteria. FEMS Microbiol. Rev. 1993, 10, 271–285. [Google Scholar] [CrossRef]
- Sachidanandham, R.; Yew-Hoong Gin, K. A dormancy state in nonspore-forming bacteria. Appl. Microbiol. Biotechnol. 2009, 81, 927–941. [Google Scholar] [CrossRef] [PubMed]
- Kell, D.B.; Kaprelyants, A.S.; Weichart, D.H.; Harwood, C.R.; Barer, M.R. Viability and activity in readily culturable bacteria: A review and discussion of the practical issues. Antonie Van Leeuwenhoek 1998, 73, 169–187. [Google Scholar] [CrossRef] [PubMed]
- Mukamolova, G.V.; Kaprelyants, A.S.; Kell, D.B.; Young, M. Adoption of the transiently non-culturable state--a bacterial survival strategy? Adv. Microb. Physiol. 2003, 47, 65–129. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Brea, M.; Alarcon, T.; Domingo, D.; Diaz-Reganon, J. Inhibitory effect of Gram-negative and Gram-positive microorganisms against Helicobacter pylori clinical isolates. J. Antimicrob. Chemother. 2008, 61, 139–142. [Google Scholar] [CrossRef]
- Krzyzek, P.; Grande, R.; Migdal, P.; Paluch, E.; Gosciniak, G. Biofilm Formation as a Complex Result of Virulence and Adaptive Responses of Helicobacter pylori. Pathogens 2020, 9, 1062. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Oral Sample | Sample Size | H. pylori Detection Method in Oral Samples | Coinfection Rate | Ref. |
|---|---|---|---|---|
| Saliva | 689 H. pylori-associated gastritis patients | PCR-16S rRNA and ureA gene | 79.7% | [28] |
| Saliva | 162 patients with gastric disease | PCR | 24%; 51.1% agreement in vacA genotype in saliva and biopsy from the same patients | [25] |
| Saliva | 300 patients with gastric disease | PCR-ureC, cagA, and vacA gene | 10.72%; high homology (58%) in vacA genotype in saliva and gastric samples from the same patients | [4] |
| Dental plaque | 235 patients with chronic gastritis | PCR-16S rRNA | 56.52% | [29] |
| Dental plaque | 164 dyspeptic patients | Enzyme Immunoassay | 82.1% | [30] |
| Dental plaque | 65 patients with gastric H. pylori infection among 134 dyspeptic patients | RUT | 89.2% among gastric H. pylori-positive patients | [31] |
| Subgingival plaque | 101 dyspeptic patients | RUT | 66% | [32] |
| Subgingival plaque | 443 dyspeptic patients | Nested PCR- 860bp fragment | 71.86% | [33] |
| Subgingival plaque | 67 dyspeptic patients | PCR-ureA gene | 25.4% | [34] |
| Dental plaque and saliva | 70 children with dyspepsia | PCR-16S rRNA and 23S rRNA | Dental plaque (77.6%); saliva (75.9%) | [26] |
| Dental plaque and saliva | 61 patients with dyspepsia | PCR-ureA gene | 50.8%; 38.7% genotype concordance between oral and gastric samples from the same patients | [35] |
| Dental plaque and saliva | 62 patients with dyspepsia | PCR-16S-rRNA | All oral samples (68%) | [36] |
| Dental plaque and saliva | 30 patients with gastric disease | PCR-cagA gene | 60%; 98% agreement between gastric DNA H. pylori sequence and their corresponding saliva or dental plaque DNA | [37] |
| Supragingival plaque, subgingival plaque, and saliva | 56 gastric H. pylori-positive patients with periodontitis | PCR-16S rDNA | All oral samples (41.1%); supragingival plaque (26.8%); subgingival plaque (30.4%); saliva (21.4%); | [38] |
| Oral Hygiene Management | Eradication Therapy | Effects on Gastric H. pylori Infection | Ref. |
|---|---|---|---|
| Scaling and/or combined with root planing and oral hygiene instructions on brushing with the modified Bass technique | 10-day course of triple therapy consisted of a PPI combined with amoxicillin (2 × 1 g daily) and clarithromycin (2 × 500 mg daily) | The eradication rate in the combined therapy group was higher than that in the triple therapy only group (64.71% vs. 51.06%, respectively, p = 0.17). | [51] |
| Scaling and root planing; oral hygiene instruction | 1-week triple therapy (esomeprazole 20 mg twice per day, clarithromycin 500 mg twice per day, or metronidazole 400 mg three times per day (if clarithromycin-resistant), as well as amoxicillin 1000 mg twice per day) | The recurrence rate of gastric H. pylori in the combined therapy group was lower than that in the triple therapy only group (2.04% vs. 15.27%, respectively; OR 0.69; 95% CI 0.52 to 0.99; p = 0.001). | [28] |
| Mouth rinse (0.02% tinidazole and 0.12% chlorhexidine) with 20 mL held in the mouth for 5 min for 10 d; ultrasonic periodontal scaling twice a month | Triple therapy consisted of amoxicillin (1.0 g) and esomeprazole (20 mg) twice a day and levofloxacin (0.5 g) once a day for 10 d | The eradication rate in the combined therapy group was higher than that in the triple therapy only group (94.7% vs. 78.4%, respectively, p = 0.012). | [52] |
| Basic periodontal therapy during triple therapy | 7-day course of triple therapy consisted of amoxicillin 2 g/day (g/d), clarithromycin 1 g/d, and pantoprazole 80 mg/d | The eradication rate in the combined therapy group was higher than that in the triple therapy only group (77.3% vs. 47.6%, respectively, p = 0.044). | [53] |
| Oral hygiene education, dental cleaning, and scaling | 14-day PPI or triple therapy | The eradication rate in the combined therapy group was higher than that in triple therapy only group (62.8% vs. 32.4%, respectively, p < 0. 05). | [54] |
| Interaction Type | Oral Microorganisms | Interaction between H. pylori and Oral Microorganisms | Ref. |
|---|---|---|---|
| Mutualistic relationship | S. mutans | H. pylori can penetrate the biofilm formed by S. mutans. | [111] |
| S. mutans contributes to the formation of “hedgehog” in the dental plaque, which could generate a CO2-rich environment. | [77] | ||
| F. nucleatum | H. pylori can adhere to F. nucleatum and might help to colonize the dental plaque. | [15,115] | |
| P. gingivalis | H. pylori has the ability to coaggregate with P. gingivalis. | [14] | |
| P. gingivalis with a specific filamentous appendage (fimA) genotype may be involved in the colonization by H. pylori. | [114] | ||
| C. albicans | H. pylori can adhere to yeast pseudohyphae. | [116] | |
| H. pylori can anchor on C. albicans and form polymicrobial biofilms. | [117] | ||
| H. pylori can invade yeast cells. | [118,119] | ||
| Nutrient deprivation, acidic pH, and amoxicillin may stimulate the entry of H. pylori into Candida. | [116,120,121] | ||
| H. pylori entering yeast cells can propagate vertically to the vacuoles of progeny yeast cells. | [118] | ||
| C. albicans releases H. pylori as a vesicle-encased or free bacterium, which may facilitate H. pylori invasion of new yeast cells. | [122] | ||
| Antagonistic relationship | S. mitis | The diffusible factors released by S. mitis can inhibit the growth and induce the coccoid conversion of H. pylori during coculture in vitro. | [13] |
| S. mutans, etc. | Bacteriocin-like inhibitory proteins against H. pylori could be produced by oral bacteria. | [14] | |
| S. mutans and Prevotella intermedia | Culture supernatants of these bacteria showed growth inhibitory activity against H. pylori and caused the formation of the coccoid form of H. pylori in vitro. | [123] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Chen, X.; Ren, B.; Zhou, X.; Cheng, L. Helicobacter pylori in the Oral Cavity: Current Evidence and Potential Survival Strategies. Int. J. Mol. Sci. 2022, 23, 13646. https://doi.org/10.3390/ijms232113646
Zhang L, Chen X, Ren B, Zhou X, Cheng L. Helicobacter pylori in the Oral Cavity: Current Evidence and Potential Survival Strategies. International Journal of Molecular Sciences. 2022; 23(21):13646. https://doi.org/10.3390/ijms232113646
Chicago/Turabian StyleZhang, Lin, Xi Chen, Biao Ren, Xuedong Zhou, and Lei Cheng. 2022. "Helicobacter pylori in the Oral Cavity: Current Evidence and Potential Survival Strategies" International Journal of Molecular Sciences 23, no. 21: 13646. https://doi.org/10.3390/ijms232113646
APA StyleZhang, L., Chen, X., Ren, B., Zhou, X., & Cheng, L. (2022). Helicobacter pylori in the Oral Cavity: Current Evidence and Potential Survival Strategies. International Journal of Molecular Sciences, 23(21), 13646. https://doi.org/10.3390/ijms232113646