Abstract
Ovarian cancer (OC) is the fifth leading cause of women’s death from cancers. The high mortality rate is attributed to the late presence of the disease and the lack of modern diagnostic tools, including molecular biomarkers. Moreover, OC is a highly heterogeneous disease, which contributes to early treatment failure. Thus, exploring OC molecular mechanisms could significantly enhance our understanding of the disease and provide new treatment options. Chromatin remodeling complexes (CRCs) are ATP-dependent molecular machines responsible for chromatin reorganization and involved in many DNA-related processes, including transcriptional regulation, replication, and reparation. Dysregulation of chromatin remodeling machinery may be related to cancer development and chemoresistance in OC. Some forms of OC and other gynecologic diseases have been associated with mutations in specific CRC genes. Most notably, ARID1A in endometriosis-related OC, SMARCA4, and SMARCB1 in hypercalcemic type small cell ovarian carcinoma (SCCOHT), ACTL6A, CHRAC1, RSF1 amplification in high-grade serous OC. Here we review the available literature on CRCs’ involvement in OC to improve our understanding of its development and investigate CRCs as possible biomarkers and treatment targets for OC.
1. Introduction
1.1. Ovarian Cancer Types and Mutations
Ovarian cancer (OC) is the third most common gynecologic malignancy and the second deadliest oncogynecologic disease in the world [1]. It is estimated that just in the United States, more than 12.8 thousand women will die from OC this year [2]. Early-stage OC presents a good diagnosis and a more than 90% 5-year survival rate. However, about 80% of OC patients are diagnosed with stage III-IV disease, where survival, even after therapy, is only 30% [3]. The high OC frequency and mortality rates are attributed to the lack of specific symptoms and insufficiency of the main OC blood biomarker CA125 to reduce mortality. Due to insufficient prognostic power, the CA125 serum biomarker is recommended as a prognostic tool only in high-grade ovarian cancer (HGSOC) patients [4]. Currently, there are no sensitive molecular biomarkers of clinical value for OC screening and patient management. Generally, after diagnosis, OC patients are treated by bilateral salpingo-oophorectomy (surgical removal of ovaries and fallopian tubes) with platinum or taxane therapy. Other therapies are available only after chemoresistance development which almost inevitably occurs in most HGSOC cases [5].
One of the culprits of OC chemoresistance and early therapy failure is the heterogeneity of the disease. Histologically, OCs are subdivided into epithelial and non-epithelial tumors. The non-epithelial OC only comprises 10% of OC cases. These are malignant germ cell tumors (e.g., small cell carcinoma of the ovary) that are usually present in preadolescent women, and sex cord-stromal tumors (granulosa cell tumors, fibroma, Sertoli-Leydig cell tumors, and others) that are more common and typically affect postmenopausal women [6]. Due to its rarity, non-epithelial tumors are rarely studied, and most research focuses on epithelial OC.
Epithelial type of OC is further subdivided into five major types according to histologic differences: the endometrioid, clear cell, mucinous, low-grade, and high-grade serous OCs [7]. According to the dualistic ovarian cancer model, epithelial OCs are grouped into two major types based on the site of origin of the tumor [8]. Type II tumors comprise mainly HGSOC but can also include carcinosarcomas and undifferentiated carcinomas. Unique for its high frequency of TP53 mutations and chromosomal instability, these tumors arise from the intraepithelial carcinoma in the fallopian tube. However, other origins, such as solid pseudoendometrioid transition tumors or borderline or low-grade serous OC, are possible. All other epithelial tumors (low-grade serous, mucinous, clear cell, borderline, seromucinous, and endometrioid) are considered less of a concern due to their slow-growing nature. Although borderline, mucinous, and seromucinous tumor origin is unclear, the endometroid and clear cell tumors are thought to originate from endometriosis and are similar to endometrial cancers [9]. Albeit highly heterogeneous, mutations in PIK3CA, PTEN, KRAS, BRAF, CTNNB1, and ARID1A genes are frequent amongst type I epithelial OCs and are considered potential OC biomarkers [10].
Regardless of type, a third of OC cases are related to homologous recombination gene insufficiency [5]. About 23% of OC cases are inherited. 10–15% of inherited cases are associated with BRCA1 and BRCA2 germline mutations [11,12]. The rest of the inherited OC cases are caused by TP53 mutations due to patients with the Li-Fraumeni syndrome [13]. The second gene group, most often mutated in OC, are the genes encoding proteins that are involved in chromatin remodeling complexes (CRCs) which are crucial for transcription, DNA repair, and most other cell day-to-day fluctuations in chromatin availability states.
1.2. Chromatin Remodelers and Their Functions in Human Cancer
Inside every human cell’s nucleus fits the genome comprising three billion DNA base pairs, almost two meters in length. This is achieved by winding the DNA around histone protein complexes forming nucleosomes that are the building blocks of the chromatin nucleoprotein structure. Each nucleosome has an octamer core made of H2A, H2B, H3, and H4 histone protein pairs and fits 147 bp of DNA. The nucleosomes are connected with linker DNA of 10–60 bp in length [14] and form the chromatin complex, a functionally dynamic structure. During interphase, chromatin hierarchically organizes and compartmentalizes DNA into active (euchromatin) and silent (heterochromatin) regions in the nucleus [15]. The distinct chromatin states are achieved through epigenetic modifications of the DNA, histone N-terminal tails, and nucleosome management done by ATP-dependent chromatin remodeling protein complexes. CRCs aid in nucleosome assembly and maintain dynamic chromatin activity through sliding, rearranging, evicting, and modifying nucleosomes. CRCs achieve these complex functions using their ATPase domain to translocate DNA in regard to histones [16]. Chromatin remodelers are involved in most essential cell processes, most notably regulating RNA pol II activity, gene silencing, homologous recombination (HR), and DNA repair [17].
There are four conserved families of ATP-dependent chromatin remodelers in mammals: chromodomain helicase DNA binding (CHD), imitation switch (ISWI), inositol requiring 80 (INO80), and switch/sucrose non-fermenting (SWI/SNF) [16]. The multiprotein complexes that remodel chromatin all share a common core ATPase-translocase subunit from the RNA/DNA helicase superfamily 2 (Snf2), but differ in accessory proteins that read epigenetic modifications and regulate the enzymatic activity of the complexes. SWI/SNF complexes have bromodomains capable of reading acetylated lysine on histone tails and function as chromatin accessibility regulators by sliding and evicting nucleosomes. ISWI reader domain (C-terminal HAND-SANT-SLIDE) binds to unmodified H4 tails and promotes chromatin assembly as well as proper nucleosome spacing. CHD remodelers are known to have similar functions to ISWI, although they contain two tandem chromodomains that read H3K4me3 active chromatin marks and are also known to bind to AT-rich DNA motifs. INO80 family remodelers are known to act in recombination and DNA replication as it is capable of binding to specialized DNA structures. In addition to nucleosome spacing, the INO80 family facilitates histone exchange by replacing nucleosome dimers and regulating histone variants [18].
Traditionally, SWI/SNF chromatin remodelers are associated with increasing chromatin accessibility, INO80 mainly acts as a chromatin repressor, and ISWI and CHD are most often described as histone chaperones in chromatin assembly [19]. However, these broad classifications are not fixed, as particular chromatin remodelers can be classified into two groups based on the chromatin regions they occupy. BRG1 (SWI/SNF), SNF2H (ISWI), CHD3, and CHD4 proteins are mainly found in active chromatin regions, while BRM (SWI/SNF), INO80, SNF2L (ISWI), CHD1 are associated with inactive chromatin [20].
1.2.1. SWI/SNF Complex Members and Functions
SWI/SNF was the first CRC discovered and, thus, is the most well-studied. The complex gets its SWI/SNF (mating Type SWItch/Sucrose Non-Fermentable) name from the yeast where it was first discovered [21]. The Brg/Brahma-associated factors (BAF), as they are known in mammals, are ATP-dependent CRCs responsible for nucleosome repositioning by nucleosome sliding and eviction, resulting primarily in increased chromatin availability. Besides nucleosome relocation at promoter and enhancer regions, SWI/SNF also facilitates transcription factor binding, histone-modifying enzyme recruitment, and regulation of chromatin looping to facilitate enhancer and promoter interaction [22].
The final assembled SWI/SNF complexes are ∼1 to 1.5 MDa size, 10–15 protein conglomerates subcategorized into three groups: canonical BRGI/BRM-associated factor (cBAF), polybromo-associated BAF (PBAF), and recently characterized non-canonical, GLTSCR1/L-associated BAFs (ncBAF or GBAF) [23]. Each complex contains a common core and ATPase cap that always includes ether BRG1 (encoded by SMARCA4) or BRM (SMARCA2) catalytic subunits. The different SWI/SNF complexes are distinguished by complex-specific accessory subunits (Table 1). cBAF non-core subunits include ARID1A/ARID1B and DPF1/2/3, PBAF has PBRM1, PHF10, ARID2, and BRD7, and ncBAF contains GLTSCR1/GLTSCR1L and BRD9 complexes. Although some of the SWI/SNF domains have known tasks, e.g., bromodomain-containing PBRM1 and BRD7 subunits that read histone acetylation marks such as active chromatin mark H3K27Ac, functions of other non-catalytic subunits are less understood [24]. Moreover, cBAF, PBAF, and ncBAF complexes are preferably assembled on different chromatin sites. The ncBAFs are uniquely localized on CTCF sites, especially ones co-localized with H3K4me1 primed sites, while PBAFs occupy gene body and active promoters (H3K27ac, H3K4me3), and cBAFs are more often localized on distal sites, such as active enhancers (H3K27ac, H3K4me1) [25].

Table 1.
SWI/SNF main subunits and coding genes.
SWI/SNF complexes are well known for their essential functions in many pathways related to cancerogenesis. Most notably, loss of SWI/SNF or its subunits is known to impair DNA damage repair, cell cycle regulation, critical signaling pathways, such as PI3K/AKT/mTOR, and MYC regulation (described in detail below).
1.2.2. ISWI Complex Members and Functions
ISWI (Imitation switch) is a highly conserved family of chromatin remodelers. ISWI contains SWI2/SNF2 family ATPase domain, a part of a broader superfamily of DEAD/H-helicases, a specific HAND-SANT-SLIDE DNA, and histone-tail binding domain. The human ISWI has either SNF2L (SMARCA1) or SNF2H (SMARCA5) ATPases that, together with regulatory domains, form up to 16 CRCs with various functions: ACF, CHARC, RSF perform nucleosome spacing, WICH is primarily involved in replication by incorporating histones into newly synthesized DNA, NoRC perform transcriptional repression, NURF and CERF are responsible for transcriptional activation [26,27]. Many ISWI family CRCs are involved in DSB either by translocating nucleosomes or by recruitment of DSB machinery through protein interactions. Acf1 has been found to interact directly with Ku70/80 to promote non-homologous end joining (NHEJ) repair. Meanwhile, ACF, RSF, NURF, CERF, NoRC, and WICH are also known to regulate transcription under different circumstances due to their influence on RNA polymerases [28].
1.2.3. CHD Family Remodelers
The chromodomain-helicase-DNA binding (CHD) family consists of nine (CHD1-9) proteins. The CHD family shares a highly conservative helicase-ATPase domain from the SWItch2/SNF2 superfamily of ATPases and two tandem chromatin organization modifier domains (chromodomains), that read histone modifications. In particular, CHD1 binds to the H3H4me mark and CHD5 to H3K27me3 [29]. CHD proteins are subdivided into three subfamilies based on the other accessory domains. Subfamily I (CHD1, CHD2) bind to AT-rich DNA motifs through its C-terminus, subfamily II (CHD3-5) has PHD zinc finger domain in its N-terminal region and binds to methylated or acetylated histone residues, subfamily III (CHD6-9) is the most variable family, binding to non-modified DNA through SAINT and BRK domains. Although CHD’s primary function is nucleosome (dis)assembly, CHD proteins are involved in different stages of transcription: CHD7 and CHD8 are associated with transcription activation while CHD1 are more associated with termination [30].
CHD3 and CHD4 proteins are also included in the nucleosome remodeling and histone deacetylase complex (Mi-2/NuRD) that also contains histone deacetylases (HDAC1 and HDAC2), and other proteins (MBD2, MBD4, MTA1-3, GATAD2A/B, RBBP4/7) that bind to methylated DNA or histone tails [31]. Generally, CHD3 or CHD4 containing NuRD complexes are associated with transcriptional repression. However, new studies have found that NuRD also governs transcriptional activation, replication, and DNA repair, adding to NuRD’s involvement in cancer progression [32]. Although CHD5 is not a part of the NuRD complex, immunoprecipitation analysis found CHD5 to bind with most of NuRD’s components, including MTA1/2, GATAD2A, HDAC1/2, RBBP4/7, and MBD2/3, forming a similar complex to NuRD [33].
The first CHD gene linked with human cancer was CHD5, with the discovery that its locus 1p36.31 is often deleted in glioma and other tumors [34]. Curiously, CHD5 expression is low in most tissues except the nervous system and testis. However, in other tissues, CHD5 expression is induced by DNA damage [29]. CHD5 expression is decreased by lysine demethylase JMJD2A/KDM4A leading to increased cell senescence due to p53 pathway dysregulation [35]. CHD5 regulates INK4A/ARF locus that codes p19(Arf) and other genes that stabilize p53; thus, CHD5 loss causes p53 deficiency [34]. Moreover, low CHD5 expression correlates with the upregulation of DNA damage response (DDR) markers -H2AX and CHK2 Thr68 phosphorylation in nuclear loci of pancreatic cancer cells [36].
1.2.4. INO80 Family
Inositol requiring 80 (INO80) family, named after inositol-responsive gene expression regulation [37], is the newest member of CRCs. Humans consist of INO80, SRCAP, and p400/TIP60 complexes. Like SWI/SNF, INO80 family complexes are large, 14–15 protein conglomerations. The defining feature of the INO80 family is the insertion of the spacer region in its SNF2 helicase that interacts with RuvB-like helicases and actin-related proteins (Arps). As RuvB is a bacterial DNA repair factor, RuvB-like subunits suggest INO80’s role in DNA repair [38]. Although the primary biochemical function of INO80 is nucleosome sliding, during DNA repair, TIP60 acetylates H4 and H2A.Z histones, which stimulates incorporation of H2A.Z:H2B dimers by SRCAP, while INO80 reverses this action [39]. INO80 is recruited to the damage site via association with -H2AX (phosphorylated H2AX) histone mark to help with nucleosome eviction during HR [38].
Just like other CRCs, INO80 also governs transcription. INO80 can activate mRNA transcription by −1 and +1 nucleosome sliding at transcription start sites where it is recruited by RNAPII and transcription elongation complex PAF1, however, at heterochromatin sites INO80 silences transcription by removing the H2A.Z mark, preventing H3K79 methylation and RNAPII eviction and degradation at replication fork–transcription complex collision sites [40].
2. Alterations of Chromatin Remodeler Complexes in Ovarian Cancer
2.1. ARID1A Alterations in Ovarian Cancer
About 25% of human cancers harbor mutations in at least one of 29 genes encoding SWI/SNF proteins [41]. Among them, ARID1A (BAF250a, B120, C1orf4, Osa1) is the most frequently altered. ARID1A mutations are particularly prevalent in gynecologic cancers (found in 10–60% of ovarian and endometrioid carcinoma cases) [42] and pre-malignant gynecological lesions, especially of endometrioid origin [43]. The highest ARID1A mutation rates are found in ovarian clear cell carcinoma (OCCC) and endometroid ovarian carcinoma (Table 2) suggesting its role in type I OC development and the potential for ARID1A’s use as a gynecologic cancer biomarker.
Table 2.
ARID1A mutation frequency in gynecologic diseases.
None of the previous research found any sufficient correlation between ARID1A protein expression loss and clinical features such as age, FIGO grade, and disease survival [60]. In a subset of microsatellite stable (MSS) endometrial cancer cases, ARID1A’s loss has been associated with a better prognosis and indicated as a potential prognostic biomarker [61]. Further investigation is highly needed to determine ARID1A’s potential as a gynecologic cancer biomarker.
Usually, ARID1A alterations are loss-of-function mutations such as large deletions, frameshift, or nonsense mutations that lead to the largest BAF complex subunit loss and inactivation of the SWI/SNF complex. However, ARID1A mutations are not sufficient to promote tumorigenesis alone. Tumors with ARID1A loss often harbor mutations of the PI3K/AKT pathway, such as inactivating PTEN or activating PIK3CA mutations. In mice models, separate ARID1A or PI3K/AKT pathway mutations promoted ovarian hyperplasia, while concomitant inactivation of ARID1A together with PIK3CA activating mutations induced tumors, similar to ovarian clear cell carcinoma (OCCC) [62].
ARID1A downregulation due to inactivating mutations has many downstream effects through epigenetic mark displacement, deregulating gene expression, and reduced protein interactions, primarily affecting DNA damage repair and signaling pathways (Table 3).
Table 3.
Effects of ARID1A loss of function mutations and opportunities for cancer therapy.
SWI/SNF complex regulates active H3K27ac histone mark at enhancer and promoter regions through interaction with p300/CBR histone acetyltransferase [63]. The acetylation of +1 nucleosome may be required for normal RNA polymerase II (RNAPII) pausing, a crucial step in effective transcription initiation [64]. Thus, ARID1A’s downregulation results in dysregulated expression of at least 99 target genes [82]. The impairment of ARID1A’s function due to inactivating mutations is not fully compensated by its homolog ARID1B. Most notably, one of the affected genes is tumor suppressor TP53 [64]. ARID1A and TP53 mutations are mutually exclusive in gynecologic malignancies, however, p53 is indirectly regulated by ARID1A. ARID1A is an HDAC6 deacetylase transcriptional repressor. Thus, the loss of ARID1A leads to HDAC6 reactivation, which causes p53 lysine 120 residue deacetylation and apoptosis inhibition [65].
ARID1A’s deficiency has also been linked with telomere impairment. In ARID1A-deficient cells, topoisomerase II (TOP2A) cannot interact with the SWI/SNF ATPase BRG1 (SMARCA4), which it needs to resolve catenanes that develop during replication and transcription [69]. Moreover, due to ARID1A insufficiency, TOP2A is not recruited to resolve R-loop sites and transcription-replication conflicts, which causes replication stress and increased genomic instability [70]. However, during mitosis, ARID1A deficiency results in cohesion protein STAG1 downregulation, and cancer cells with chromosome aberrations are eliminated during mitosis due to telomere cohesion deficiency [68]. ARID1A also downregulates promoters of telomerase reverse transcriptase; thus, ARID1A silencing increases telomerase activity, adding to the survival of cancer cells [71].
ARID1A’s mutational status in OC cells is also associated with reactive oxygen species (ROS) formation. ARID1A knockdown in OC cell lines increased intracellular ROS levels, as well as increased oxidative stress marker 8-hydrixyguanosine levels in OCCC patients with low ARID1A expression. Moreover, ARID1A-mutated cell lines were sensitive to ROS inductor elesclomol [76]. Another study found that ARID1A deficient cells have low antioxidant glutathione (GSH) levels and lower SLC7A11 (cysteine transporter required by GSH) expression making them vulnerable to GSH and glutamate-cysteine ligase synthetase catalytic subunit (GCLC) inhibition and possible cancer treatment through increased ROS formation [75].
Telomere defects and DNA damage caused by ARID1A insufficiency leads to increased reliance on double-stranded DNA break (DSB) repair mechanisms [68]. Inactivation or loss of ARID1A or SWI/SNF complexes increases cell sensitivity to cisplatin and UV treatment through the impairment of multiple DSB repair mechanisms [74]. Most notably, ARID1A is involved in HR through DNA end processing (RPA and RAD51 loading), ATR activation, and G2-M cell-cycle arrest maintenance [72]. BAF factors, particularly ARID1A/B, are required for KU70/KU80 protein recruitment to DSB sites during NHEJ [73] as well as XPA accumulation at UV damage sites during NER (nucleotide excision repair) [74]. Moreover, ARID1A interacts with miss-match repair protein MSH2 and directs it to the damage site, starting the process of DNA damage repair [61]. The loss of functional ARID1A protein may induce DNA damage and decrease the cell’s ability to repair it.
In ovarian and endometrial cancers, ARID1A mutations often co-occur with PI3K/AKT pathway gene KRAS, PIK3CA, and PTEN alterations [77]. Additionally, ARID1A’s loss activates the pathway by ANXA1 (AKT activator) upregulation and PIK3IP1 (PI3K inhibitor) expression downregulation. In particular, PIK3IP1 is regulated by ARID1A, suppressing EZH2 methyltransferase that inhibits PIK3IP1 expression through the H3K27me3 epigenetic mark [83]. Therefore, targeting epigenetic regulators EZH2 (polycomb repressive complex (PRC2)), HDAC2 (PRC2 binding partner), and GSK126 (EZH2 inhibitor) are considered viable treatment options for ARID1A mutated tumors [66].
Besides its role in SWI/SNF, ARID members have also been reported to co-precipitate with members of E3 ubiquitin ligase. Specifically, ARID1B is linked with elongin C (EloC) through BC box motif and with the addition of cullin 2 and ROC1 form E3 ubiquitin ligase that targets H2B histone explicitly at lysine 120 for monoubiquitination. ARID1B is a paralog that shares most of its sequence with ARID1A. Thus, a similar E3 ubiquitin ligase complex may be formed with ARID1A too. Mutation and depletion of the ARID domain result in decreased ubiquitination of H2B, similar to VHL mutations, causing decreased ubiquitination of HIF1 in clear cell renal cell carcinoma (ccRCC) [80]. The two genes are often mutated together in ccRCC. The reduced H2B ubiquitination is responsible for reduced H3 histone lysine 79 di-methylation and decreased gene expression [84].
To sum up, ARID1A is highly involved in many pathways related to cancerogenesis (Figure 1), and its loss is associated with DNA damage repair, cell cycle, and apoptosis regulation through TP53 [65], possible ROS formation [75], telomere maintenance [68], and PI3K/AKT/mTOR pathway deregulation [66], providing ample opportunity for ARID1A-mutated OC treatment.
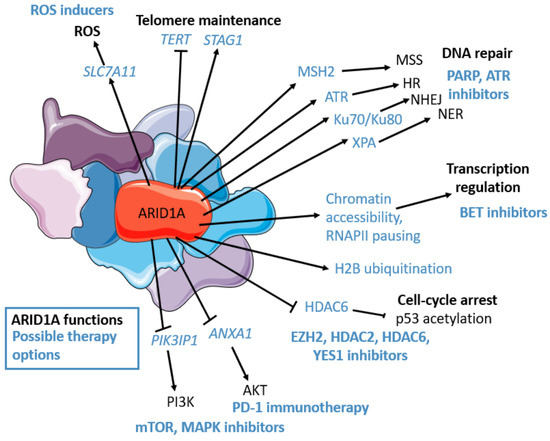
Figure 1.
ARID1A’s functions (in black): transcriptional regulation (chromatin accessibility, epigenetic mark placement, RNAPII pausing regulation), PI3K/AKT/mTOR pathway regulation, cell-cycle regulation (p53 deacetylation through HDAC6), DNA repair: Homologous repair (HR) (RPA and RAD51 loading, ATR interaction, and activation), non-homologous end joining (NHEJ) (Ku70/Ku80 interaction), nucleotide excision repair (NER) (XPA interaction), miss match repair (MSS) (MSH2 interaction), Telomere regulation (STAG1 maintenance, TERT downregulation), oxidative stress repression (though SLC7A11 maintenance). ARID1A’s possible therapeutic options (in blue): cisplatin, PD-L1, PARP, ATR, YES1, BET (BRD2), HDAC6, HDAC2, EZH2, PI3K, AKT inhibitors, mTOR, MAPK inhibitors, ROS inducers. The figure was generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license.
The frequent loss of function ARID1A mutations in cancers arising from endometriosis (endometrial tissue growth outside the uterus) makes SWI/SNF an attractive option for targeted therapy approaches. Although ARID1A has a mutually exclusive homolog ARID1B, and both ARID1A and ARID1B are often co-mutated in human cancers [85], the creation of specific inhibitors is challenging due to the 60% homology between ARID domains [24]. A more powerful therapeutic strategy is to target BRD2, which, together with ARID1A, is a member of BET (bromodomain and extra terminal domain) family proteins. BRD2 inhibitors reduce ARID1B and other SWI/SNF member expression in ARID1A mutant cancers [78].
Due to ARID1A’s involvement in DDR responses, ARID1A mutant cancers may be more susceptible to ATR blockade [86] and PARP inhibition [72]. Clinical trials involving ATR and PARP inhibition in OC patients are underway [87]. Although initially, ARID1A suppression in cancer cell line showed increased cisplatin sensitivity [74], ARID1A deficient ovarian cancer cells showed decreased chemosensitivity to cisplatin [88]. However, ARID1A mutational status may be a good predictor of immunotherapy outcomes: ARID1A deficient OC mouse model showed higher mutational burden, more tumor-infiltrating lymphocytes, and elevated levels of programmed cell death-ligand 1 (PD-1), providing an opportunity for immunotherapy [61]. A study found that patients with ARID1A deficiency that undergone anti-PD-1/PD-L1 immunotherapy had longer progression-free survival than patients with wild-type ARID1A tumors [89]. In another study involving ARID1A deficient OCCC cell lines, dasatinib, a tyrosine kinase inhibitor, was identified as a potential drug target. ARID1A mutated cell lines were sensitive to dasatinib treatment because of impaired cell cycle and YES1 (dasatinib target) deregulation [67].
2.2. Other SWI/SNF Alterations in Ovarian Cancer
Although ARID1A is the most frequently mutated SWI/SNF subunit coding gene, other SWI/SNF members, albeit less often, are also altered in OC. In particular, up to 90% of a rare form of OC, small cell ovarian carcinoma, hypercalcemic type (SCCOHT), a rhabdoid-like tumor has germline or somatic mutations of SWI/SNF ATPase domain BRG1 coding gene SMARCA4 [90]. Just like ARID1A, the SMARCA4 is not only involved in gene expression through the primary SWI/SNF function as chromatin remodeler (SMARCA4 mutations cause hyperactivation of PRC2 repressive complex) [43], but it also directly participates in DNA repair. BRG1 (SMARCA4) is recruited to DNA damage response (DDR) sites by PARP1 and is activated by deacetylation by SIRT1 to open the affected chromatin region in preparation for HR [91]. Moreover, BRG1 has been associated with -H2AX (a histone mark that indicates damaged DNA) formation, thus likely to promote DSB [92].
Meanwhile, other forms of OC, such as undifferentiated uterine or ovarian carcinoma and OCCC, are found to harbor alterations in other SMARC genes [93]. Most notably, other SMARC gene mutations are found in OCCC: SMARCA4 in up to 5%, SMARCA1 at 2%, SMARCA2 at 1%, and SMARCC1 at 2% [94]. Although SMARCA2 mutations are rare, most SCCOHT cases present with dual inactivation of both BGR1 and BRM, though, unlike SMARCA4, SMARCA2 is silenced epigenetically [95]. Similar to other rhabdoid tumors, SCCOHT may also harbor SWI/SNF core subunit SMARCB1 (also known as INI1/SNF5/BAF47) mutations instead of SMARCA4 [90]. Moreover, SMARCB1 mutations are also found in other undifferentiated OC cases and myoepithelium-like tumors of the vulvar region [43]. Interestingly, SMARC coding SWI/SNF together with ARID1A is involved in oncogene MYC regulation: MYC is a transcriptional modulator of SMARCB1. However, ARID1A regulates MYC through binding with the MYC promoter region, and BRG1 (coded by SMARCA4) controls MYC partner MAX in a similar manner [81].
Curiously, in HGSOC, SMARC protein SMARCC1 was shown to be regulated on the protein level through methylation by coactivator-associated arginine methyltransferase 1 (CARM1/PRTM4), which is overexpressed in HGSOC. The methylation causes BAF complex eviction from certain loci, including promoters of c-Myc pathway genes, thus altering gene expression [96].
Although the loss of function mutations in SWI/SNF complex encoding genes occur in more than 20% of human cancers, some amplifications of genes coding for SWI/SNF subunits are also found. Most notably, a scaffolding protein, coded by ACTL6A and ncBAF subunit gene BRD9, are overexpressed in most cancers, including OC. The amplification of these genes co-occurs with notable oncogene TP63 amplification, suggesting an oncogenic mechanism of OC [97]. However, ACTL6A amplification has also been associated with follicle-stimulating hormone regulation through PIK3/AKT pathway. ACTL6A increase leads to enhanced glycolysis due to increased expression of phosphoglycerate kinase 1 (PGK1) [98]. Moreover, ACTL6A is involved in regulating cisplatin resistance through DNA repair enhancement [99].
Overall, SWI/SNF gene alterations, especially inactivating mutations, are frequent events in OC development. Besides ARID1A, SMARC genes coding ATPase and regulatory subunits are frequently altered as well. An investigation is needed on whether these genes could serve as novel biomarkers for OC.
2.3. ISWI Alterations in Ovarian Cancer
Similarly, to SWI/SNF, ISWI ATPase domains are also encoded by SMARC genes that frequent alterations in OC. Most notably, ISWI ATPase SNF2L (SMARCA1) mutations have been linked with OCCC [94]. Although SNF2H is the dominant ISWI ATPase, SNF2L has been described as a regulator of specific gene expression during differentiation. Crucially, SNF2L has been associated with ovarian development and meiotic progression of germ cells during its differentiation [100]. SNF2L interacts with progesterone receptor and steroidogenic acute regulatory protein in ovarian granulosa cells to promote differentiation of the ovary [101]. SMARCA1 deletion has been associated with increased apoptosis through caspase activator Apaf-1 expression upregulation [102] and enhanced proliferation and migration due to WNT signaling regulation in various cancer cells [103].
Depending on which ATPase it binds, ISWI protein remodeling and spacing factor 1 (coded by RSF1, also known as BAZ1A, HBXAP), forms either RSF-1 or RSF-5 CRC [27]. Immunohistochemical analysis showed that ISWI ATPase SNF2H (SMARCA5) is overexpressed in OC tissues together with its binding partner RSF1 which is also often upregulated in various tumors, including OC [104]. RSF1 is an essential interphase centromere protein that maintains chromosome stability and protein homeostasis but also has roles in DSB and transcription regulator functions through its interactions with HDAC1, CENP-A, ATM, SNF2H, cyclin E1, CBP, NF-B, BubR1, and many other cancer-related proteins [105]. The RSF-1 complex protein expression correlates with cancer stage and poor clinical outcomes in OC patients [106]. The upregulation of RSF-1 and SMARCA5 expression leads to increased double-stranded breaks (DSB) and subsequent DDR [107]. However, in HGSOC cells that are almost invariably deficient in TP53, the DNA damage caused by RSF-5 upregulation goes unsolved [108]. Moreover, RSF1 has been co-immunoprecipitated with cyclin E1, which is also overexpressed in OC cells. In HGSOC cells, cyclin activation by RSF1 leads to proliferation and tumor formation via activation of cyclin E1-associated kinase (CDK2) [109]. RSF1 overexpression has also been linked with chemoresistance in OC. RSF1 overexpression in SKOV3 OC cell line activated NF-B transcription factor and subsequent gene expression changes resulting in decreased apoptosis (CFLAR, XIAP, BCL2, BCL2L1) and inflammation (PTGS2) gene expression adding to paclitaxel resistance in ovarian cells [110]. Thus, RSF1 overexpression may serve as a prognostic biomarker for OC patients.
In addition to RSF1, amplification of CHARC1 and RBBP4 has also been identified in HGSOC, along with RSF1 and RBBP4 gene fusions. CHRAC complexes (CHRAC1, RSF1, POLE3) are known oncogenic drivers regulating proliferation and survival in many cancer types [27]. In cisplatin-resistant OC cell lines, CHRAC1 expression was found to be increased due to miR-512-3p downregulation and was related to enhanced proliferation, invasion, migration, and apoptosis [111]. RBBP4 is not the only ISWI family NURF CRC component, but also a member of other transcription regulation complexes, such as MuvB, which is a part of DREAM, MMB, and FoxM1-MuvB regulatory complexes. During different cell cycle phases, MuvB acts either as a transcriptional repressor or activator. DREAM transcriptional complex has been associated with carboplatin chemoresistance, and depletion of DREAM proteins might prove a viable option for OC treatment [112].
2.4. CHD Family Alterations in Ovarian Cancer
CHD5 is the main CHD family protein linked with cancer [34]. Although mutations in the CHD5 gene are detected in OC, CHD5 is also downregulated by increased promoter methylation in OC patient samples [113]. In one study analyzing CHD gene promoter methylation in various cancer cell lines, out of all CHD genes, CHD5 promoter was methylated the most often, although the OC cell line (MDAH2774) showed no increase in methylation [114]. CHD5 polymorphism (rs9434741) has also been associated with endometriosis, an OC precursor [115], however, the clinical significance of this variant is not known. In alignment with CHD5 increased mutation and methylation, CHD5 mRNA expression was found to be downregulated by at least 2-fold in 41% (32/72) of invasive epithelial ovarian carcinomas in comparison with 12 controls and correlated with shorter disease-free survival times [116].
Besides CHD5, CHD CRCs are also found to be dysregulated in ovarian and other cancers and have a role in chemoresistance. CHD8 overexpression has been linked with poor survival in both HGOSC and endometrial cancer cases [117]. CHD8 binds to -catenin and -catenin regulated gene promoters to act as a negative regulator of WNT, a prominent pathway OC [118]. Similarly, CHD4 overexpression also correlated with poor survival and was significantly higher in platinum-resistant HGSOC cases. CHD4 was found to induce chemoresistance through multi-drug transporter MDR1 expression regulation [119]. Other NuRD factors, besides CHD, are also found to confer platinum resistance: methyl-CpG binding domain protein 3 (MBD3) is required for platinum resistance through H3K27ac mark regulation at enhancer regions and altered expression of JAK/STAT and FGF signaling pathway genes [120]. Meanwhile, another NuRD protein, MTA1 overexpression in OC tissues correlates with cancer stage and grade, while in the OVCAR-3 cell culture model MTA1 upregulation reduced estrogen receptor expression and cytokine GRO (CXCL1) upregulation possibly explaining MTA1 role in OC progression and metastasis [121].
2.5. INO80 in Ovarian Cancer
Although several INO80 subunits are overexpressed in melanoma [122], cervical [123], and non-small cell lung cancer [124], according to TCGA data analysis [125,126] the only INO80 gene significantly amplified in OC patients is ACT6LA, shared with SWI/SNF family remodelers. ACT6LA amplification affects multiple CRCs and causes platinum resistance in OC [99]. However, other INO80 family domains are found to be significantly altered in OC cell models. TRRAP (transformation/transcription domain-associated protein)—a TIP60 remodeler/histone acetyltransferase complex adaptor protein is overexpressed in OC A2780 sphere cultures and was found to govern stemness marker NANOG expression and OC cell proliferation [127]. Yin yang 1 (YY1)—a non-conserved human INO80 family subunit [38] in OC cell line models, has been associated with chemoresistance through upregulation of lncRNA PART1, which targets miR-512-3p and causes CHRAC1 (another CRC gene) upregulation and cisplatin-resistant OC cell proliferation and migration [111]. In OC patients and cell lines YY1 itself was found to be suppressed by miR-381, which is downregulated in OC tissues [128]. In another study, higher YY1 protein expression in OC was associated with reduced survival [129].
In 495 Chinese epithelial OC patient study analyzing INO80 module gene RUVBL1, single nucleotide polymorphisms rs1057156 (A>G) and rs149652370 (A>G) were significantly associated with poor overall survival. The rs1057156 (A>G) variant was found to lower miR-4294 binding affinity causing increased RUVBL1 expression in OC cells [130]. In OC patients, RUVBL1/2 has also been found to be crucial for mTOR inhibitor sensitivity as it mitigates cell stress caused by mTORC1-Myc-DNA damage axis [131].
3. Conclusions and Perspectives
Overall, at least 18 different CRC subunits are affected in OC. While SWI/SNF genes are mostly affected by inactivating mutations, amplification and epigenetic changes also affect CRC coding genes (Table 4). These alterations, especially mutations and changes in gene expression, could be helpful as prognostic, predictive, and diagnostic markers for gynecologic malignancies (Figure 2). Although OC is a highly heterogeneous disease, a better understanding of the mutational landscape of individual tumors will help to choose the right therapy for each patient. Unlike mutations in classical cancer-related genes such as TP53, BRCA1/2, MYC or RAS, CRCs and their involvement in cancer pathways are known only for a few decades [132], thus, are much less studied. Arguably, SWI/SNF is the main CRC of interest, it was the first CRC discovered, it’s involved in a wide variety of cellular functions and shows high mutation load in ARID1A and other genes in OC and other cancers, providing ample ground for possible therapeutic options [66]. However, other CRCs are also deregulated in OC, and may also be useful for OC diagnostics and treatment.
Table 4.
Overview of CRC alterations other than ARID1A in ovarian cancer. OC—ovarian cancer, SCCOHT—small cell ovarian carcinoma, hypercalcemic type, OCCC—Ovarian clear cell carcinoma, HGSOC—High-grade serous ovarian cancer.
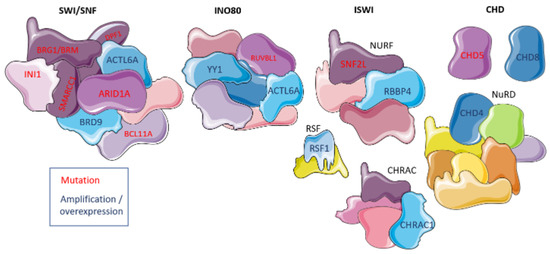
Figure 2.
Chromatin remodeling complexes and their involvement in ovarian cancer. Deregulated domains are shown in either purple with red text denoting mutations in coding genes or blue with dark blue text denoting expression up-regulation. The figure was generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license.
Recently identified numerous mutations and amplifications in chromatin remodeling genes open a possibility for new therapeutic approaches. Of note, FDA approval of EZH2 inhibitor tazemetostat therapy for follicular lymphoma shows a promising step in epigenetic therapy application, and increases hope that the same inhibitor could be used for SWI/SNF deficient HGSOC, OCCC, endometrial OC, or SCCOHT patient treatment [135]. Phase II studies for this and other SWI/SNF deficient cancer therapies are already underway [41]. Most of the current research on CRCs only considers separate CRC protein or gene dysregulation effects on OC cells or small groups of patients, however, a broader picture of how CRCs are affected by these particular mutations or amplifications and how this dysregulation affects OC patients is greatly needed. A better understanding of CRCs will aid in the development of new cancer treatment options guided by biomarker-based OC diagnostics.
Author Contributions
Conceptualization, I.V., R.S. and S.J.; data curation, I.V. and R.S.; writing-original draft preparation, I.V. and R.S.; writing-review and editing, all authors; visualization, I.V.; supervision, J.R.L. and S.J.; All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| CRC | Chromatin remodeling complex |
| DDR | DNA damage response |
| DSB | Double-stranded break |
| HGSOC | High-grade ovarian cancer |
| HR | Homologous recombination |
| NER | Nucleotide excision repair |
| NHEJ | Non-homologous DNA end joining |
| OC | Ovarian cancer |
| OCCC | Ovarian clear cell carcinoma |
| ROS | Reactive oxygen species |
| SCCOHT | Small cell ovarian carcinoma, hypercalcemic type |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Testa, U.; Petrucci, E.; Pasquini, L.; Castelli, G.; Pelosi, E. Ovarian Cancers: Genetic Abnormalities, Tumor Heterogeneity and Progression, Clonal Evolution and Cancer Stem Cells. Medicines 2018, 5, 16. [Google Scholar] [CrossRef]
- Colombo, N.; Preti, E.; Landoni, F.; Carinelli, S.; Colombo, A.; Marini, C.; Sessa, C. Endometrial cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2013, 24, vi33–vi38. [Google Scholar] [CrossRef]
- Matulonis, U.A.; Sood, A.K.; Fallowfield, L.; Howitt, B.E.; Sehouli, J.; Karlan, B.Y. Ovarian cancer. Nat. Rev. Dis. Prim. 2016, 2, 1–22. [Google Scholar] [CrossRef]
- Colombo, N.; Sessa, C.; Bois, A.D.; Ledermann, J.; McCluggage, W.G.; McNeish, I.; Morice, P.; Pignata, S.; Ray-Coquard, I.; Vergote, I.; et al. ESMO-ESGO consensus conference recommendations on ovarian cancer: Pathology and molecular biology, early and advanced stages, borderline tumours and recurrent disease. Ann. Oncol. 2019, 30, 672–705. [Google Scholar] [CrossRef]
- Hollis, R.L.; Gourley, C. Genetic and molecular changes in ovarian cancer. Cancer Biol. Med. 2016, 13, 236–247. [Google Scholar] [CrossRef]
- Kurman, R.J.; Shih, I.M. The dualistic model of ovarian carcinogenesis revisited, revised, and expanded. Am. J. Pathol. 2016, 186, 733–747. [Google Scholar] [CrossRef]
- Salazar, C.; Campbell, I.G.; Gorringe, K.L. When Is “type I” Ovarian Cancer Not “type I”? Indications of an Out-Dated Dichotomy. Front. Oncol. 2018, 8, 654. [Google Scholar] [CrossRef]
- Iijima, M.; Banno, K.; Okawa, R.; Yanokura, M.; Iida, M.; Takeda, T.; Kunitomi-Irie, H.; Adachi, M.; Nakamura, K.; Umene, K.; et al. Genome-wide analysis of gynecologic cancer: The Cancer Genome Atlas in ovarian and endometrial cancer. Oncol. Lett. 2017, 13, 1063–1070. [Google Scholar] [CrossRef]
- Liu, G.; Yang, D.; Sun, Y.; Shmulevich, I.; Xue, F.; Sood, A.K.; Zhang, W. Differing clinical impact of BRCA1 and BRCA2 mutations in serous ovarian cancer. Pharmacogenomics 2012, 13, 1523–1535. [Google Scholar] [CrossRef]
- Zhang, S.; Royer, R.; Li, S.; McLaughlin, J.R.; Rosen, B.; Risch, H.A.; Fan, I.; Bradley, L.; Shaw, P.A.; Narod, S.A. Frequencies of BRCA1 and BRCA2 mutations among 1,342 unselected patients with invasive ovarian cancer. Gynecol. Oncol. 2011, 121, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Toss, A.; Tomasello, C.; Razzaboni, E.; Contu, G.; Grandi, G.; Cagnacci, A.; Schilder, R.J.; Cortesi, L. Hereditary ovarian cancer: Not only BRCA 1 and 2 Genes. BioMed Res. Int. 2015, 2015, 341723. [Google Scholar] [CrossRef]
- Wang, S.; Vogirala, V.K.; Soman, A.; Berezhnoy, N.V.; Liu, Z.B.; Wong, A.S.; Korolev, N.; Su, C.J.; Sandin, S.; Nordenskiöld, L. Linker histone defines structure and self-association behaviour of the 177 bp human chromatosome. Sci. Rep. 2021, 11, 380. [Google Scholar] [CrossRef]
- Cutter, A.R.; Hayes, J.J. A brief review of nucleosome structure. FEBS Lett. 2015, 589, 2914–2922. [Google Scholar] [CrossRef]
- Clapier, C.R.; Iwasa, J.; Cairns, B.R.; Peterson, C.L. Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat. Rev. Mol. Cell Biol. 2017, 18, 407–422. [Google Scholar] [CrossRef]
- Magaña-Acosta, M.; Valadez-Graham, V. Chromatin Remodelers in the 3D Nuclear Compartment. Front. Genet. 2020, 11, 600615. [Google Scholar] [CrossRef]
- Längst, G.; Manelyte, L. Chromatin remodelers: From function to dysfunction. Genes 2015, 6, 299–324. [Google Scholar] [CrossRef]
- Sahu, R.K.; Singh, S.; Tomar, R.S. The mechanisms of action of chromatin remodelers and implications in development and disease. Biochem. Pharmacol. 2020, 180, 114200. [Google Scholar] [CrossRef]
- Giles, K.A.; Gould, C.M.; Du, Q.; Skvortsova, K.; Song, J.Z.; Maddugoda, M.P.; Achinger-Kawecka, J.; Stirzaker, C.; Clark, S.J.; Taberlay, P.C. Integrated epigenomic analysis stratifies chromatin remodellers into distinct functional groups. Epigenetics Chromatin 2019, 12, 12. [Google Scholar] [CrossRef]
- Stern, M.; Jensen, R.; Herskowitz, I. Five SWI genes are required for expression of the HO gene in yeast. J. Mol. Biol. 1984, 178, 853–868. [Google Scholar] [CrossRef]
- Wu, J.N.; Roberts, C.W. ARID1A mutations in cancer: Another epigenetic tumor suppressor? Cancer Discov. 2013, 3, 35–43. [Google Scholar] [CrossRef]
- Alpsoy, A.; Dykhuizen, E.C. Glioma tumor suppressor candidate region gene 1 (GLTSCR1) and its paralog GLTSCR1-like form SWI/SNF chromatin remodeling subcomplexes. J. Biol. Chem. 2018, 293, 3892–3903. [Google Scholar] [CrossRef]
- Centore, R.C.; Sandoval, G.J.; Soares, L.M.M.; Kadoch, C.; Chan, H.M. Mammalian SWI/SNF Chromatin Remodeling Complexes: Emerging Mechanisms and Therapeutic Strategies. Trends Genet. 2020, 36, 936–950. [Google Scholar] [CrossRef]
- Michel, B.C.; D’Avino, A.R.; Cassel, S.H.; Mashtalir, N.; McKenzie, Z.M.; McBride, M.J.; Valencia, A.M.; Zhou, Q.; Bocker, M.; Soares, L.M.; et al. A non-canonical SWI/SNF complex is a synthetic lethal target in cancers driven by BAF complex perturbation. Nat. Cell Biol. 2018, 20, 1410–1420. [Google Scholar] [CrossRef]
- Erdel, F.; Schubert, T.; Marth, C.; Längst, G.; Rippe, K. Human ISWI chromatin-remodeling complexes sample nucleosomes via transient binding reactions and become immobilized at active sites. Proc. Natl. Acad. Sci. USA 2010, 107, 19873–19878. [Google Scholar] [CrossRef]
- Li, Y.; Gong, H.; Wang, P.; Zhu, Y.; Peng, H.; Cui, Y.; Li, H.; Liu, J.; Wang, Z. The emerging role of ISWI chromatin remodeling complexes in cancer. J. Exp. Clin. Cancer Res. 2021, 40, 346. [Google Scholar] [CrossRef]
- Erdel, F.; Rippe, K. Chromatin remodelling in mammalian cells by ISWI-type complexes - Where, when and why? FEBS J. 2011, 278, 3608–3618. [Google Scholar] [CrossRef]
- Li, W.; Mills, A.A. Architects of the genome: CHD dysfunction in cancer, developmental disorders and neurological syndromes. Epigenomics 2014, 6, 381–395. [Google Scholar] [CrossRef]
- Tyagi, M.; Imam, N.; Verma, K.; Patel, A.K. Chromatin remodelers: We are the drivers!! Nucleus 2016, 7, 388–404. [Google Scholar] [CrossRef]
- Lai, A.Y.; Wade, P.A. Cancer biology and NuRD: A multifaceted chromatin remodelling complex. Nat. Rev. Cancer 2011, 11, 588–596. [Google Scholar] [CrossRef]
- Stanley, C.E.; Kulathinal, R.J. Genomic signatures of domestication on neurogenetic genes in Drosophila melanogaster. BMC Evol. Biol. 2016, 16, 6. [Google Scholar] [CrossRef]
- Kolla, V.; Naraparaju, K.; Zhuang, T.; Higashi, M.; Kolla, S.; Blobel, G.A.; Brodeur, G.M. The tumour suppressor CHD5 forms a NuRD-type chromatin remodelling complex. Biochem. J. 2015, 468, 345–352. [Google Scholar] [CrossRef][Green Version]
- Bagchi, A.; Papazoglu, C.; Wu, Y.; Capurso, D.; Brodt, M.; Francis, D.; Bredel, M.; Vogel, H.; Mills, A.A. CHD5 Is a Tumor Suppressor at Human 1p36. Cell 2007, 128, 459–475. [Google Scholar] [CrossRef]
- Mallette, F.A.; Richard, S. JMJD2A Promotes Cellular Transformation by Blocking Cellular Senescence through Transcriptional Repression of the Tumor Suppressor CHD5. Cell Rep. 2012, 2, 1233–1243. [Google Scholar] [CrossRef]
- Hall, W.A.; Petrova, A.V.; Colbert, L.E.; Hardy, C.W.; Fisher, S.B.; Saka, B.; Shelton, J.W.; Warren, M.D.; Pantazides, B.G.; Gandhi, K.; et al. Low CHD5 expression activates the DNA damage response and predicts poor outcome in patients undergoing adjuvant therapy for resected pancreatic cancer. Oncogene 2014, 33, 5450–5456. [Google Scholar] [CrossRef]
- Ebbert, R.; Birkmann, A.; Schüller, H.J. The product of the SNF2/SWI2 paralogue INO80 of Saccharomyces cerevisiae required for efficient expression of various yeast structural genes is part of a high-molecular-weight protein complex. Mol. Microbiol. 1999, 32, 741–751. [Google Scholar] [CrossRef]
- Morrison, A.J.; Shen, X. Chromatin remodelling beyond transcription: The INO80 and SWR1 complexes. Nat. Rev. Mol. Cell Biol. 2009, 10, 373–384. [Google Scholar] [CrossRef]
- Willhoft, O.; Wigley, D.B. INO80 and SWR1 complexes: The non-identical twins of chromatin remodelling. Curr. Opin. Struct. Biol. 2020, 61, 50–58. [Google Scholar] [CrossRef]
- Poli, J.; Gasser, S.M.; Papamichos-Chronakis, M. The INO80 remodeller in transcription, replication and repair. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372. [Google Scholar] [CrossRef]
- Mittal, P.; Roberts, C.W. The SWI/SNF complex in cancer—Biology, Biomarkers and Therapy. Nat. Rev. Clin. Oncol. 2020, 17, 435–448. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Tang, C. The Role of ARID1A in Tumors: Tumor Initiation or Tumor Suppression? Front. Oncol. 2021, 11, 745187. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hoang, L.; Ji, J.X.; Huntsman, D.G. SWI/SNF Complex Mutations in Gynecologic Cancers: Molecular Mechanisms and Models. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 467–492. [Google Scholar] [CrossRef] [PubMed]
- Suda, K.; Nakaoka, H.; Yoshihara, K.; Ishiguro, T.; Tamura, R.; Mori, Y.; Yamawaki, K.; Adachi, S.; Takahashi, T.; Kase, H.; et al. Clonal Expansion and Diversification of Cancer-Associated Mutations in Endometriosis and Normal Endometrium. Cell Rep. 2018, 24, 1777–1789. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Zhou, J.Y.; Guo, J.B.; Wang, L.Q.; Luo, Y.; Zhang, Z.Y.; Liu, F.Y.; Tan, J.; Wang, F.; Huang, O.P. The presence of KRAS, PPP2R1A and ARID1A mutations in 101 Chinese samples with ovarian endometriosis. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2018, 809, 1–5. [Google Scholar] [CrossRef]
- Mcconechy, M.K.; Ding, J.; Cheang, M.C.U.; Wiegand, K.; Tone, A.; Yang, W.; Prentice, L.; Tse, K.; Zeng, T.; Schmidt, A.P.; et al. Use of mutation profiles to refine the classification of endometrial carcinomas. J. Pathol. 2012, 228, 20–30. [Google Scholar] [CrossRef]
- Cherniack, A.D.; Shen, H.; Walter, V.; Stewart, C.; Murray, B.A.; Bowlby, R.; Hu, X.; Ling, S.; Soslow, R.A.; Broaddus, R.R.; et al. Integrated Molecular Characterization of Uterine Carcinosarcoma. Cancer Cell 2017, 31, 411–423. [Google Scholar] [CrossRef]
- Wiegand, K.C.; Shah, S.P.; Al-Agha, O.M.; Zhao, Y.; Al, E. ARID1A Mutations in Endometriosis-Associated Ovarian Carcinomas. N. Engl. J. Med. 2010, 363, 1532–1543. [Google Scholar] [CrossRef]
- Guan, B.; Wang, T.L.; Shih, I.M. ARID1A, a factor that promotes formation of SWI/SNF-mediated chromatin remodeling, is a tumor suppressor in gynecologic cancers. Cancer Res. 2011, 71, 6718–6727. [Google Scholar] [CrossRef]
- Cuevas, D.; Valls, J.; Gatius, S.; Roman-Canal, B.; Estaran, E.; Dorca, E.; Santacana, M.; Vaquero, M.; Eritja, N.; Velasco, A.; et al. Targeted sequencing with a customized panel to assess histological typing in endometrial carcinoma. Virchows Arch. 2019, 474, 585–598. [Google Scholar] [CrossRef]
- Hollis, R.L.; Thomson, J.P.; Stanley, B.; Churchman, M.; Meynert, A.M.; Rye, T.; Bartos, C.; Iida, Y.; Croy, I.; Mackean, M.; et al. Molecular stratification of endometrioid ovarian carcinoma predicts clinical outcome. Nat. Commun. 2020, 11, 4995. [Google Scholar] [CrossRef]
- Kuroda, Y.; Chiyoda, T.; Kawaida, M.; Nakamura, K.; Aimono, E.; Yoshimura, T.; Takahashi, M.; Saotome, K.; Yoshihama, T.; Iwasa, N.; et al. ARID1A mutation/ARID1A loss is associated with a high immunogenic profile in clear cell ovarian cancer. Gynecol. Oncol. 2021, 162, 679–685. [Google Scholar] [CrossRef]
- Teer, J.K.; Yoder, S.; Gjyshi, A.; Nicosia, S.V.; Zhang, C.; Alvaro, N.; Monteiro, A. Mutational heterogeneity in non- serous ovarian cancers. Sci. Rep. 2017, 7, 9728. [Google Scholar] [CrossRef] [PubMed]
- Lapke, N.; Chen, C.H.; Chang, T.C.; Chao, A.; Lu, Y.J.; Lai, C.H.; Tan, K.T.; Chen, H.C.; Lu, H.Y.; Chen, S.J. Genetic alterations and their therapeutic implications in epithelial ovarian cancer. BMC Cancer 2021, 21, 499. [Google Scholar] [CrossRef] [PubMed]
- Eoh, K.J.; Kim, H.M.; Lee, J.Y.; Kim, S.; Kim, S.W.; Kim, Y.T.; Nam, E.J. Mutation landscape of germline and somatic BRCA1/2 in patients with high-grade serous ovarian cancer. BMC Cancer 2020, 20, 204. [Google Scholar] [CrossRef]
- Cheasley, D.; Nigam, A.; Zethoven, M.; Hunter, S.; Etemadmoghadam, D.; Semple, T.; Allan, P.; Carey, M.S.; Fernandez, M.L.; Dawson, A.; et al. Genomic analysis of low-grade serous ovarian carcinoma to identify key drivers and therapeutic vulnerabilities. J. Pathol. 2021, 253, 41–54. [Google Scholar] [CrossRef]
- Yachida, N.; Yoshihara, K.; Suda, K.; Nakaoka, H.; Ueda, H.; Sugino, K.; Yamaguchi, M.; Mori, Y.; Yamawaki, K.; Tamura, R.; et al. ARID1A protein expression is retained in ovarian endometriosis with ARID1A loss-of-function mutations: Implication for the two-hit hypothesis. Sci. Rep. 2020, 10, 14260. [Google Scholar] [CrossRef]
- Jones, S.; Wang, T.L.; Shih, I.M.; Mao, T.L.; Nakayama, K.; Roden, R.; Glas, R.; Slamon, D.; Diaz, L.A.; Vogelstein, B.; et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science 2010, 330, 228–231. [Google Scholar] [CrossRef]
- Wu, R.C.; Chen, S.J.; Chen, H.C.; Tan, K.T.; Jung, S.M.; Lin, C.Y.; Chao, A.S.; Huang, K.G.; Chou, H.H.; Chang, T.C.; et al. Comprehensive genomic profiling reveals ubiquitous KRAS mutations and frequent PIK3CA mutations in ovarian seromucinous borderline tumor. Mod. Pathol. 2020, 33, 2534–2543. [Google Scholar] [CrossRef]
- Heinze, K.; Nazeran, T.M.; Lee, S.; Krämer, P.; Cairns, E.S.; Chiu, D.S.; Leung, S.C.; Kang, E.Y.; Meagher, N.S.; Kennedy, C.J.; et al. Prognostic and Immunological Significance of ARID1A Status in Endometriosis-Associated Ovarian Carcinoma Short title: Significance of ARID1A Status in EAOC Authors. medRxiv 2021. [Google Scholar] [CrossRef]
- Shen, J.; Ju, Z.; Zhao, W.; Wang, L.; Peng, Y.; Ge, Z.; Nagel, Z.D.; Zou, J.; Wang, C.; Kapoor, P.; et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat. Med. 2018, 24, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Guan, B.; Rahmanto, Y.S.; Wu, R.C.; Wang, Y.; Wang, Z.; Wang, T.L.; Shih, I.M. Roles of deletion of Arid1a, a tumor suppressor, in mouse ovarian tumorigenesis. J. Natl. Cancer Inst. 2014, 106, dju146. [Google Scholar] [CrossRef] [PubMed]
- Alver, B.H.; Kim, K.H.; Lu, P.; Wang, X.; Manchester, H.E.; Wang, W.; Haswell, J.R.; Park, P.J.; Roberts, C.W. The SWI/SNF chromatin remodelling complex is required for maintenance of lineage specific enhancers. Nat. Commun. 2017, 8, 14648. [Google Scholar] [CrossRef] [PubMed]
- Trizzino, M.; Barbieri, E.; Petracovici, A.; Wu, S.; Welsh, S.A.; Owens, T.A.; Licciulli, S.; Zhang, R.; Gardini, A. The Tumor Suppressor ARID1A Controls Global Transcription via Pausing of RNA Polymerase II. Cell Rep. 2018, 23, 3933–3945. [Google Scholar] [CrossRef]
- Bitler, B.G.; Wu, S.; Park, P.H.; Hai, Y.; Aird, K.M.; Wang, Y.; Zhai, Y.; Kossenkov, A.V.; Vara-Ailor, A.; Rauscher, F.J.; et al. ARID1A-mutated ovarian cancers depend on HDAC6 activity. Nat. Cell Biol. 2017, 19, 962–973. [Google Scholar] [CrossRef]
- Caumanns, J.J.; Wisman, G.B.A.; Berns, K.; van der Zee, A.G.; de Jong, S. ARID1A mutant ovarian clear cell carcinoma: A clear target for synthetic lethal strategies. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 176–184. [Google Scholar] [CrossRef]
- Miller, R.E.; Brough, R.; Bajrami, I.; Williamson, C.T.; McDade, S.; Campbell, J.; Kigozi, A.; Rafiq, R.; Pemberton, H.; Natrajan, R.; et al. Synthetic lethal targeting of ARID1A-Mutant ovarian clear cell tumors with dasatinib. Mol. Cancer Ther. 2016, 15, 1472–1484. [Google Scholar] [CrossRef]
- Zhao, B.; Lin, J.; Rong, L.; Wu, S.; Deng, Z.; Fatkhutdinov, N.; Zundell, J.; Fukumoto, T.; Liu, Q.; Kossenkov, A.; et al. ARID1A promotes genomic stability through protecting telomere cohesion. Nat. Commun. 2019, 10, 4067. [Google Scholar] [CrossRef]
- Dykhuizen, E.C.; Hargreaves, D.C.; Miller, E.L.; Cui, K.; Korshunov, A.; Kool, M.; Pfister, S.; Cho, Y.J.; Zhao, K.; Crabtree, G.R. BAF complexes facilitate decatenation of DNA by topoisomerase IIα. Nature 2013, 497, 624–627. [Google Scholar] [CrossRef]
- Tsai, S.; Fournier, L.A.; Chang, E.Y.C.; Wells, J.P.; Minaker, S.W.; Zhu, Y.D.; Wang, A.Y.H.; Wang, Y.; Huntsman, D.G.; Stirling, P.C. ARID1A regulates R-loop associated DNA replication stress. PLoS Genet. 2021, 17, e1009238. [Google Scholar] [CrossRef]
- Rahmanto, Y.S.; Jung, J.G.; Wu, R.C.; Kobayashi, Y.; Heaphy, C.M.; Meeker, A.K.; Wang, T.L.; Shih, I.M. Inactivating ARID1A tumor suppressor enhances TERT transcription and maintains telomere length in cancer cells. J. Biol. Chem. 2016, 291, 9690–9699. [Google Scholar] [CrossRef]
- Shen, J.; Peng, Y.; Wei, L.; Zhang, W.; Yang, L.; Lan, L.; Kapoor, P.; Ju, Z.; Mo, Q.; Shih, I.M.; et al. ARID1A Deficiency Impairs the DNA Damage Checkpoint and Sensitizes Cells to PARP Inhibitors. Cancer Discov. 2015, 5, 752–767. [Google Scholar] [CrossRef]
- Watanabe, R.; Ui, A.; Kanno, S.I.; Ogiwara, H.; Nagase, T.; Kohno, T.; Yasui, A. SWI/SNF factors required for cellular resistance to dna damage include arid1a and arid1b and show interdependent protein stability. Cancer Res. 2014, 74, 2465–2475. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, R.; Kanno, S.I.; Roushandeh, A.M.; Ui, A.; Yasui, A. Nucleosome remodelling, DNA repair and transcriptional regulation build negative feedback loops in cancer and cellular ageing. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160473. [Google Scholar] [CrossRef] [PubMed]
- Ogiwara, H.; Takahashi, K.; Sasaki, M.; Kuroda, T.; Yoshida, H.; Watanabe, R.; Maruyama, A.; Makinoshima, H.; Chiwaki, F.; Sasaki, H.; et al. Targeting the Vulnerability of Glutathione Metabolism in ARID1A-Deficient Cancers. Cancer Cell 2019, 35, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Kwan, S.Y.; Cheng, X.; Tsang, Y.T.; Choi, J.S.; Kwan, S.Y.; Izaguirre, D.I.; Kwan, H.S.; Gershenson, D.M.; Wong, K.K. Loss of ARID1A expression leads to sensitivity to ROS-inducing agent elesclomol in gynecologic cancer cells. Oncotarget 2016, 7, 56933–56943. [Google Scholar] [CrossRef] [PubMed]
- De, P.; Dey, N. Mutation-driven signals of ARID1A and PI3K pathways in ovarian carcinomas: Alteration is an opportunity. Int. J. Mol. Sci. 2019, 20, 5732. [Google Scholar] [CrossRef]
- Berns, K.; Caumanns, J.J.; Hijmans, E.M.; Gennissen, A.M.; Severson, T.M.; Evers, B.; Wisman, G.B.A.; Meersma, G.J.; Lieftink, C.; Beijersbergen, R.L.; et al. ARID1A mutation sensitizes most ovarian clear cell carcinomas to BET inhibitors. Oncogene 2018, 37, 4611–4625. [Google Scholar] [CrossRef]
- Fukumoto, T.; Magno, E.; Zhang, R. SWI/SNF complexes in ovarian cancer: Mechanistic insights and therapeutic implications. Mol. Cancer Res. 2018, 16, 1819–1825. [Google Scholar] [CrossRef]
- Li, X.S.; Trojer, P.; Matsumura, T.; Treisman, J.E.; Tanese, N. Mammalian SWI/SNF-A Subunit BAF250/ARID1 Is an E3 Ubiquitin Ligase That Targets Histone H2B. Mol. Cell. Biol. 2010, 30, 1673–1688. [Google Scholar] [CrossRef]
- Srikanth, S.; Ramachandran, S.; Mohan, S. Construction of the gene regulatory network identifies MYC as a transcriptional regulator of SWI/SNF complex. Sci. Rep. 2020, 10, 158. [Google Scholar] [CrossRef] [PubMed]
- Lakshminarasimhan, R.; Andreu-Vieyra, C.; Lawrenson, K.; Duymich, C.E.; Gayther, S.A.; Liang, G.; Jones, P.A. Down-regulation of ARID1A is sufficient to initiate neoplastic transformation along with epigenetic reprogramming in non-tumorigenic endometriotic cells. Cancer Lett. 2017, 401, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Bitler, B.G.; Aird, K.M.; Garipov, A.; Li, H.; Amatangelo, M.; Kossenkov, A.V.; Schultz, D.C.; Liu, Q.; Shih, I.M.; Conejo-Garcia, J.R.; et al. Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nat. Med. 2015, 21, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Shigetomi, H.; Oonogi, A.; Tsunemi, T.; Tanase, Y.; Yamada, Y.; Kajihara, H.; Yoshizawa, Y.; Furukawa, N.; Haruta, S.; Yoshida, S.; et al. The role of components of the chromatin modification machinery in carcinogenesis of clear cell carcinoma of the ovary. Oncol. Lett. 2011, 2, 591–597. [Google Scholar] [CrossRef]
- Helming, K.C.; Wang, X.; Wilson, B.G.; Vazquez, F.; Haswell, J.R.; Manchester, H.E.; Kim, Y.; Kryukov, G.V.; Ghandi, M.; Aguirre, A.J.; et al. ARID1B is a specific vulnerability in ARID1A-mutant cancers. Nat. Med. 2014, 20, 251–254. [Google Scholar] [CrossRef]
- Williamson, C.T.; Miller, R.; Pemberton, H.N.; Jones, S.E.; Campbell, J.; Konde, A.; Badham, N.; Rafiq, R.; Brough, R.; Gulati, A.; et al. ATR inhibitors as a synthetic lethal therapy for tumours deficient in ARID1A. Nat. Commun. 2016, 7, 13837. [Google Scholar] [CrossRef]
- Banerjee, S.; Stewart, J.; Porta, N.; Toms, C.; Leary, A.; Lheureux, S.; Khalique, S.; Tai, J.; Attygalle, A.; Vroobel, K.; et al. ATARI trial: ATR inhibitor in combination with olaparib in gynecological cancers with ARID1A loss or no loss (ENGOT/GYN1/NCRI). Int. J. Gynecol. Cancer Off. J. Int. Gynecol. Cancer Soc. 2021, 31, 1471–1475. [Google Scholar] [CrossRef]
- Lyu, C.; Zhang, Y.; Zhou, X.; Lang, J. ARID1A gene silencing reduces the sensitivity of ovarian clear cell carcinoma to cisplatin. Exp. Ther. Med. 2016, 12, 4067–4071. [Google Scholar] [CrossRef][Green Version]
- Okamura, R.; Kato, S.; Lee, S.; Jimenez, R.E.; Sicklick, J.K.; Kurzrock, R. ARID1A alterations function as a biomarker for longer progression-free survival after anti-PD-1/PD-L1 immunotherapy. J. Immunother. Cancer 2020, 8, 1–6. [Google Scholar] [CrossRef]
- Lu, B.; Shi, H. An In-Depth Look at Small Cell Carcinoma of the Ovary, Hypercalcemic Type (SCCOHT): Clinical Implications from Recent Molecular Findings. J. Cancer 2019, 10, 223–237. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, H.; Xu, Z.; Tang, H.; Geng, A.; Cai, B.; Su, T.; Shi, J.; Jiang, C.; Tian, X.; et al. A PARP1-BRG1-SIRT1 axis promotes HR repair by reducing nucleosome density at DNA damage sites. Nucleic Acids Res. 2019, 47, 8563–8580. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Park, E.J.; Lee, H.S.; Kim, S.J.; Hur, S.K.; Imbalzano, A.N.; Kwon, J. Mammalian SWI/SNF complexes facilitate DNA double-strand break repair by promoting γ-H2AX induction. EMBO J. 2006, 25, 3986–3997. [Google Scholar] [CrossRef]
- Coatham, M.; Li, X.; Karnezis, A.N.; Hoang, L.N.; Tessier-Cloutier, B.; Meng, B.; Soslow, R.A.; Gilks, C.B.; Huntsman, D.G.; Stewart, C.J.; et al. Concurrent ARID1A and ARID1B inactivation in endometrial and ovarian dedifferentiated carcinomas. Mod. Pathol. 2016, 29, 1586–1593. [Google Scholar] [CrossRef] [PubMed]
- Itamochi, H.; Oishi, T.; Oumi, N.; Takeuchi, S.; Yoshihara, K.; Mikami, M.; Yaegashi, N.; Terao, Y.; Takehara, K.; Ushijima, K.; et al. Whole-genome sequencing revealed novel prognostic biomarkers and promising targets for therapy of ovarian clear cell carcinoma. Br. J. Cancer 2017, 117, 717–724. [Google Scholar] [CrossRef]
- Karnezis, A.N.; Wang, Y.; Ramos, P.; Hendricks, W.P.; Oliva, E.; D’Angelo, E.; Prat, J.; Nucci, M.R.; Nielsen, T.O.; Chow, C.; et al. Dual loss of the SWI/SNF complex ATPases SMARCA4/BRG1 and SMARCA2/BRM is highly sensitive and specific for small cell carcinoma of the ovary, hypercalcaemic type. J. Pathol. 2016, 238, 389–400. [Google Scholar] [CrossRef]
- Wang, L.; Zhao, Z.; Meyer, M.B.; Saha, S.; Yu, M.; Guo, A.; Wisinski, K.B.; Huang, W.; Cai, W.; Pike, J.W.; et al. CARM1 methylates chromatin remodeling factor BAF155 to enhance tumor progression and metastasis. Cancer Cell 2014, 25, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Sima, X.; He, J.; Peng, J.; Xu, Y.; Zhang, F.; Deng, L. The genetic alteration spectrum of the SWI/SNF complex: The oncogenic roles of BRD9 and ACTL6A. PLoS ONE 2019, 14, e0222305. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, J.; Wei, Y.; Li, Q.; Wang, Q. ACTL6A regulates follicle-stimulating hormone-driven glycolysis in ovarian cancer cells via PGK1. Cell Death Dis. 2019, 10, 811. [Google Scholar] [CrossRef]
- Xiao, Y.; Lin, F.T.; Lin, W.C. ACTL6A promotes repair of cisplatin-induced DNA damage, a new mechanism of platinum resistance in cancer. Proc. Natl. Acad. Sci. USA 2021, 118, e2015808118. [Google Scholar] [CrossRef]
- Pépin, D.; Vanderhyden, B.C.; Picketts, D.J.; Murphy, B.D. ISWI chromatin remodeling in ovarian somatic and germ cells: Revenge of the NURFs. Trends Endocrinol. Metab. 2007, 18, 215–224. [Google Scholar] [CrossRef]
- Lazzaro, M.A.; Pépin, D.; Pescador, N.; Murphy, B.D.; Vanderhyden, B.C.; Picketts, D.J. The imitation switch protein SNF2L regulates steroidogenic acute regulatory protein expression during terminal differentiation of ovarian granulosa cells. Mol. Endocrinol. 2006, 20, 2406–2417. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Xiao, Y.; Wang, W.; Wang, Q.; Yearsley, K.; Wani, A.A.; Yan, Q.; Gao, J.X.; Shetuni, B.S.; Barsky, S.H. Inhibition of expression of the chromatin remodeling inhibition of expression of the chromatin remodeling gene, SNF2L, selectively leads to DNA damage, growth inhibition, and cancer cell death. Mol. Cancer Res. 2009, 7, 1984–1999. [Google Scholar] [CrossRef] [PubMed]
- Manelyte, L. Chromatin remodelers, their implication in cancer and therapeutic potential. J. Rare Dis. Res. Treat. 2017, 2, 34–40. [Google Scholar] [CrossRef][Green Version]
- Sheu, J.J.C.; Jung, H.C.; Yildiz, I.; Tsai, F.J.; Shaul, Y.; Wang, T.L.; Shih, I.M. The roles of human sucrose nonfermenting protein 2 homologue in the tumor-promoting functions of Rsf-1. Cancer Res. 2008, 68, 4050–4057. [Google Scholar] [CrossRef]
- Cai, G.; Yang, Q.; Sun, W. RSF1 in cancer: Interactions and functions. Cancer Cell Int. 2021, 21, 315. [Google Scholar] [CrossRef] [PubMed]
- Maeda, D.; Chen, X.; Guan, B.; Nakagawa, S.; Yano, T.; Taketani, Y.; Fukayama, M.; Wang, T.L.; Shih, I.M. Rsf-1 (HBXAP) expression is associated with advanced stage and lymph node metastasis in ovarian clear cell carcinoma. Int. J. Gynecol. Pathol. Off. J. Int. Soc. Gynecol. Pathol. 2011, 30, 30–35. [Google Scholar] [CrossRef]
- Sheu, J.J.C.; Guan, B.; Choi, J.H.; Lin, A.; Lee, C.H.; Hsiao, Y.T.; Wang, T.L.; Tsai, F.J.; Shih, I.M. Rsf-1, a chromatin remodeling protein, induces DNA damage and promotes genomic instability. J. Biol. Chem. 2010, 285, 38260–38269. [Google Scholar] [CrossRef]
- Kshirsagar, M.; Jiang, W.; Shih, I.M. DNA damage response is prominent in ovarian high-grade serous carcinomas, especially those with Rsf-1 (HBXAP) overexpression. J. Oncol. 2012, 2012, 621685. [Google Scholar] [CrossRef]
- Sheu, J.J.C.; Choi, J.H.; Guan, B.; Tsai, F.J.; Hua, C.H.; Lai, M.T.; Wang, T.L.; Shih, I.M. Rsf-1, a chromatin remodelling protein, interacts with cyclin E1 and promotes tumour development. J. Pathol. 2013, 229, 559–568. [Google Scholar] [CrossRef]
- Yang, Y.I.; Ahn, J.H.; Lee, K.T.; Shih, I.M.; Choi, J.H. RSF1 is a positive regulator of NF-κB-induced gene expression required for ovarian cancer chemoresistance. Cancer Res. 2014, 74, 2258–2269. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, X.; Zhu, L.; Yang, Y.; Yin, X. YY1-Induced lncRNA PART1 Enhanced Resistance of Ovarian Cancer Cells to Cisplatin by Regulating miR-512-3p/CHRAC1 Axis. DNA Cell Biol. 2021, 40, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Iness, A.N.; Litovchick, L. MuvB: A key to cell cycle control in ovarian cancer. Front. Oncol. 2018, 8, 223. [Google Scholar] [CrossRef] [PubMed]
- Gorringe, K.L.; Choong, D.Y.; Williams, L.H.; Ramakrishna, M.; Sridhar, A.; Qiu, W.; Bearfoot, J.L.; Campbell, I.G. Mutation and methylation analysis of the chromodomain-helicase-DNA binding 5 gene in ovarian cancer. Neoplasia 2008, 10, 1253–1258. [Google Scholar] [CrossRef] [PubMed]
- Mulero-Navarro, S.; Esteller, M. Chromatin remodeling factor CHD5 is silenced by promoter CpG island hypermethylation in human cancer. Epigenetics 2008, 3, 210–215. [Google Scholar] [CrossRef]
- Falconer, H.; Sundqvist, J.; Xu, H.; Vodolazkaia, A.; Fassbender, A.; Kyama, C.; Bokor, A.; D’Hooghe, T.M. Analysis of common variations in tumor-suppressor genes on chr1p36 among Caucasian women with endometriosis. Gynecol. Oncol. 2012, 127, 398–402. [Google Scholar] [CrossRef]
- Wong, R.R.; Chan, L.K.; Tsang, T.P.; Lee, C.W.; Cheung, T.H.; Yim, S.F.; Siu, N.S.; Lee, S.N.; Yu, M.Y.; Chim, S.S.; et al. CHD5 downregulation associated with poor prognosis in epithelial ovarian cancer. Gynecol. Obstet. Investig. 2011, 72, 203–207. [Google Scholar] [CrossRef]
- Jones, D.; Lin, D. Amplification of the NSD3-BRD4-CHD8 pathway in pelvic high-grade serous carcinomas of tubo-ovarian and endometrial origin. Mol. Clin. Oncol. 2017, 7, 301–307. [Google Scholar] [CrossRef]
- Thompson, B.A.; Tremblay, V.; Lin, G.; Bochar, D.A. CHD8 Is an ATP-Dependent Chromatin Remodeling Factor That Regulates β-Catenin Target Genes. Mol. Cell. Biol. 2008, 28, 3894–3904. [Google Scholar] [CrossRef]
- Oyama, Y.; Shigeta, S.; Tokunaga, H.; Tsuji, K.; Ishibashi, M.; Shibuya, Y.; Shimada, M.; Yasuda, J.; Yaegashi, N. CHD4 regulates platinum sensitivity through MDR1 expression in ovarian cancer: A potential role of CHD4 inhibition as a combination therapy with platinum agents. PLoS ONE 2021, 16, e0251079. [Google Scholar] [CrossRef]
- Bauer, T.L.; Collmar, K.; Kaltofen, T.; Loeffler, A.K.; Decker, L.; Mueller, J.; Pinter, S.; Eisler, S.A.; Mahner, S.; Fraungruber, P.; et al. Functional analysis of non-genetic resistance to platinum in epithelial ovarian cancer reveals a role for the mbd3-nurd complex in resistance development. Cancers 2021, 13, 3801. [Google Scholar] [CrossRef]
- Dannenmann, C.; Shabani, N.; Friese, K.; Jeschke, U.; Mylonas, I.; Brüning, A. The metastasis-associated gene MTA1 is upregulated in advanced ovarian cancer, represses ERβ, and enhances expression of oncogenic cytokine GRO. Cancer Biol. Ther. 2008, 7, 1460–1467. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhou, B.; Wang, L.; Zhang, S.; Bennett, B.D.; He, F.; Zhang, Y.; Xiong, C.; Han, L.; Diao, L.; Li, P.; et al. INO80 governs superenhancer-mediated oncogenic transcription and tumor growth in melanoma. Genes Dev. 2016, 30, 1440–1453. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Liu, J.; Chen, A.; Lyu, J.; Ai, G.; Zeng, Q.; Sun, Y.; Chen, C.; Wang, J.; Qiu, J.; et al. Ino80 promotes cervical cancer tumorigenesis by activating Nanog expression. Oncotarget 2016, 7, 72250–72262. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhou, B.; Wang, L.; Li, P.; Bennett, B.D.; Snyder, R.; Garantziotis, S.; Fargo, D.C.; Cox, A.D.; Chen, L.; et al. INO80 is required for oncogenic transcription and tumor growth in non-small cell lung cancer. Oncogene 2017, 36, 1430–1439. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Kang, K.T.; Kwon, Y.W.; Kim, D.K.; Lee, S.I.; Kim, K.H.; Suh, D.S.; Kim, J.H. TRRAP stimulates the tumorigenic potential of ovarian cancer stem cells. BMB Rep. 2018, 51, 514–519. [Google Scholar] [CrossRef]
- Xia, B.; Li, H.; Yang, S.; Liu, T.; Lou, G. MiR-381 inhibits epithelial ovarian cancer malignancy via YY1 suppression. Tumor Biol. 2016, 37, 9157–9167. [Google Scholar] [CrossRef]
- Qian, S.; Wang, W.; Li, M. Transcriptional factor Yin Yang 1 facilitates the stemness of ovarian cancer via suppressing miR-99a activity through enhancing its deacetylation level. Biomed. Pharmacother. 2020, 126, 110085. [Google Scholar] [CrossRef]
- Li, H.; Tong, X.; Xu, Y.; Wang, M.; Dai, H.; Shi, T.; Sun, M.; Chen, K.; Cheng, X.; Wei, Q. Functional genetic variants of RUVBL1 predict overall survival of Chinese patients with epithelial ovarian cancer. Carcinogenesis 2019, 40, 1209–1219. [Google Scholar] [CrossRef]
- Shin, S.H.; Lee, J.S.; Zhang, J.M.; Choi, S.; Boskovic, Z.V.; Zhao, R.; Song, M.; Wang, R.; Tian, J.; Lee, M.H.; et al. Synthetic lethality by targeting the RUVBL1/2-TTT complex in mTORC1-hyperactive cancer cells. Sci. Adv. 2020, 6, eaay9131. [Google Scholar] [CrossRef]
- Becker, P.B.; Workman, J.L. Nucleosome Remodeling and Epigenetics. Cold Spring Harb. Perspect. Biol. 2013, 5, a017905. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.I.; Chudnovsky, Y.; Duggan, B.; Zajchowski, D.; Greenbowe, J.; Ross, J.S.; Gay, L.M.; Ali, S.M.; Elvin, J.A. Comprehensive genomic profiling reveals inactivating SMARCA4 mutations and low tumor mutational burden in small cell carcinoma of the ovary, hypercalcemic-type. Gynecol. Oncol. 2017, 147, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Auguste, A.; Blanc-Durand, F.; Deloger, M.; Formal, A.L.; Bareja, R.; Wilkes, D.C.; Richon, C.; Brunn, B.; Caron, O.; Devouassoux-Shisheboran, M.; et al. Small Cell Carcinoma of the Ovary, Hypercalcemic Type (SCCOHT) beyond SMARCA4 Mutations: A Comprehensive Genomic Analysis. Cells 2020, 9, 1496. [Google Scholar] [CrossRef]
- Coughlan, A.Y.; Testa, G. Exploiting epigenetic dependencies in ovarian cancer therapy. Int. J. Cancer 2021, 149, 1732–1743. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).