

The Role of the Exonic lncRNA PRKDC-210 in Transcription Regulation




Abstract
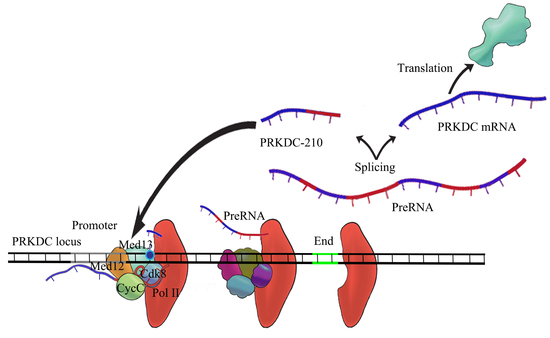
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
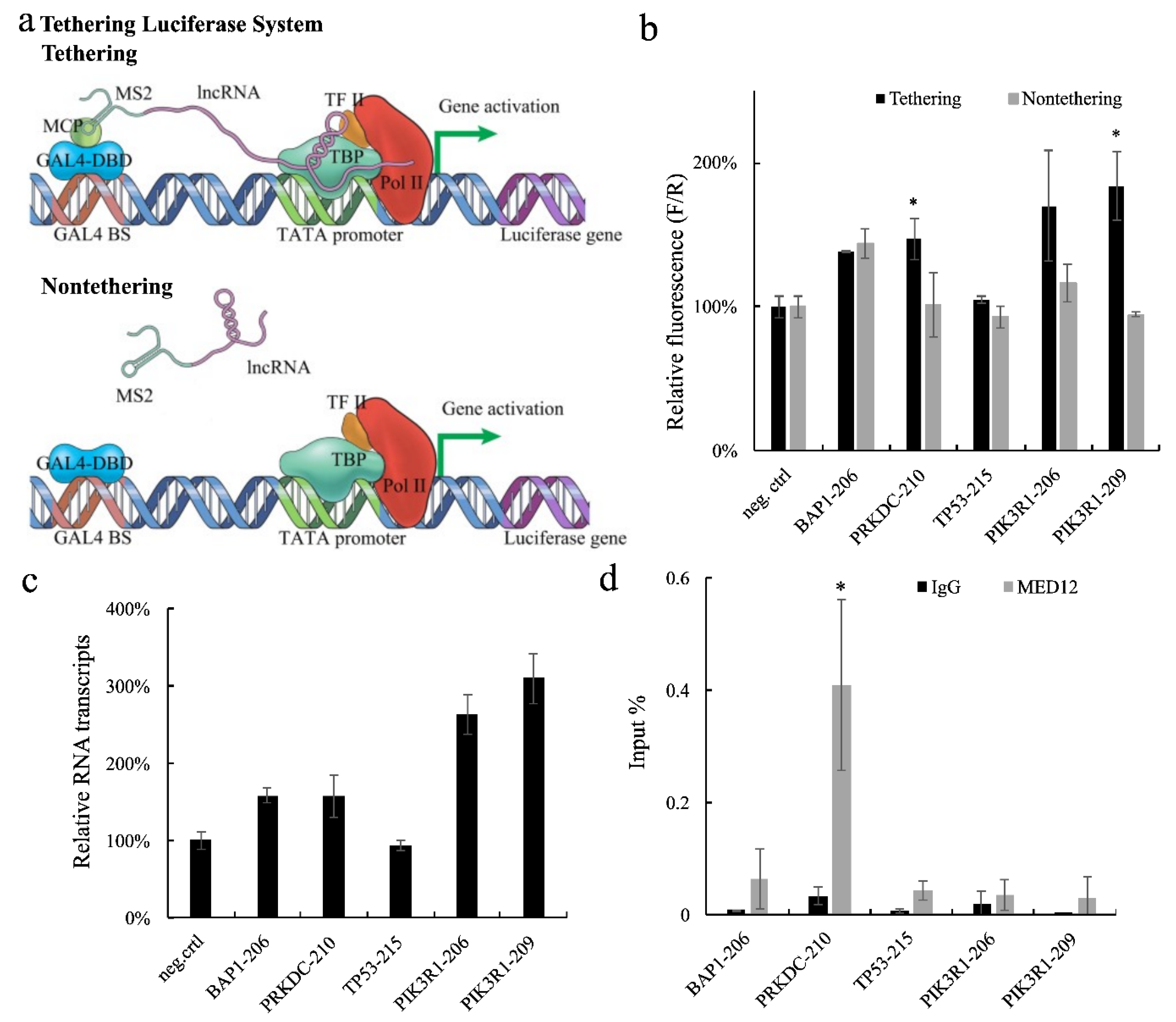
2.1. Tethering Luciferase Assay System
2.2. Identification of Long Exonic Non-Coding RNA with Transcriptional Regulatory Functions
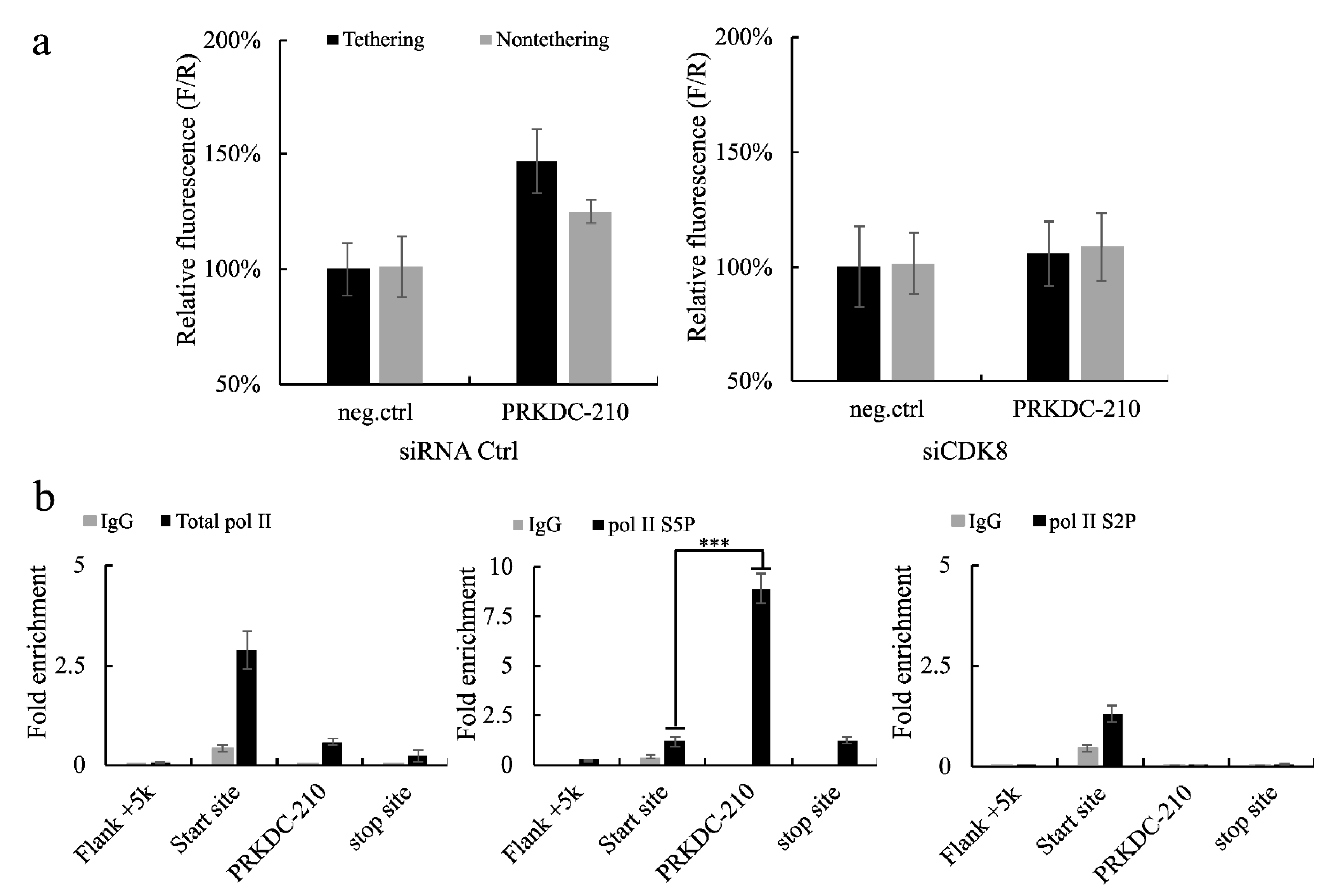
2.3. The long exonic non-coding RNA PRKDC-10 Promotes Activity of the CDK8 Complex to Drive Transcription
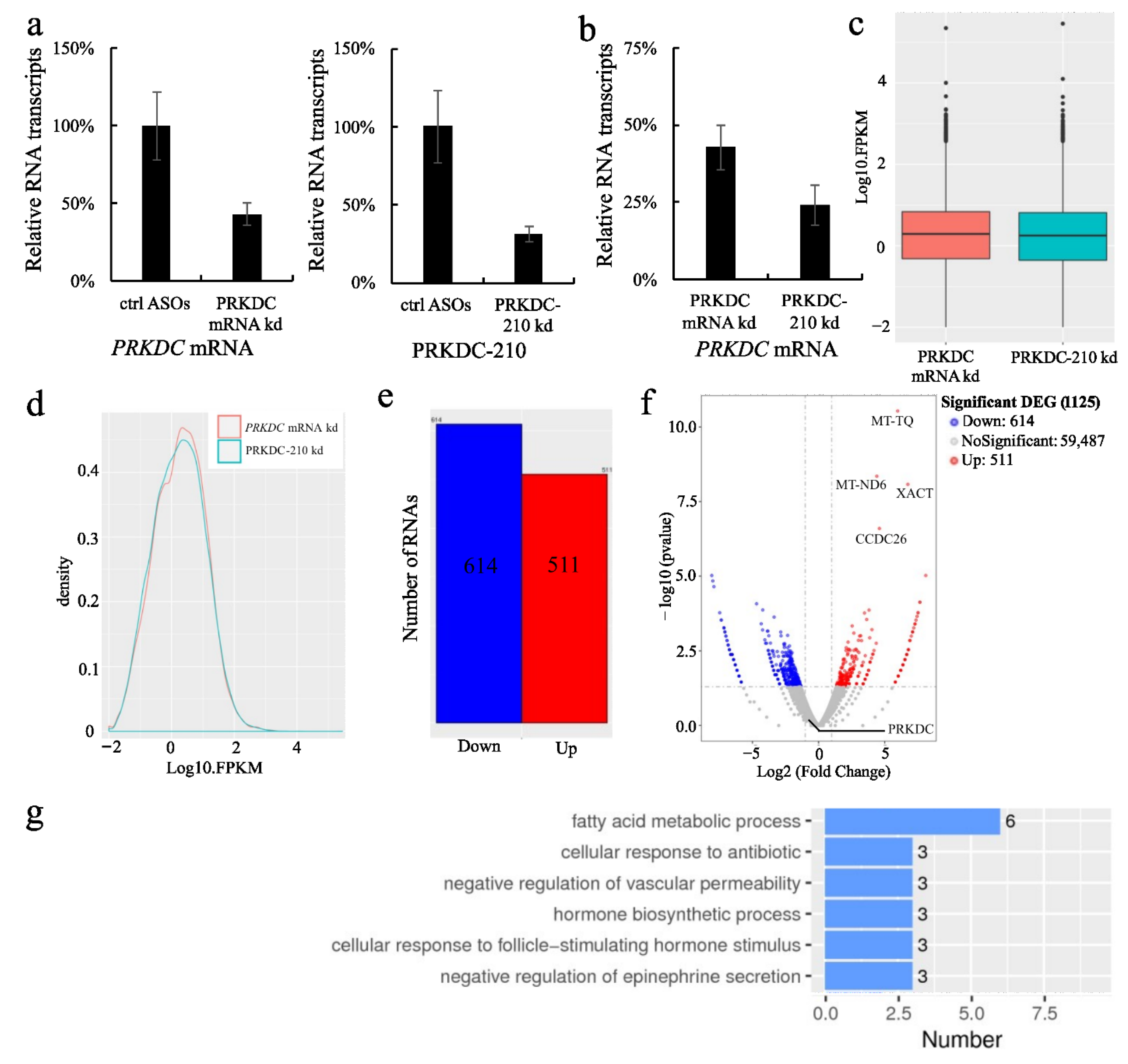
2.4. Functions of PRKDC-210 in MCF7 Cells
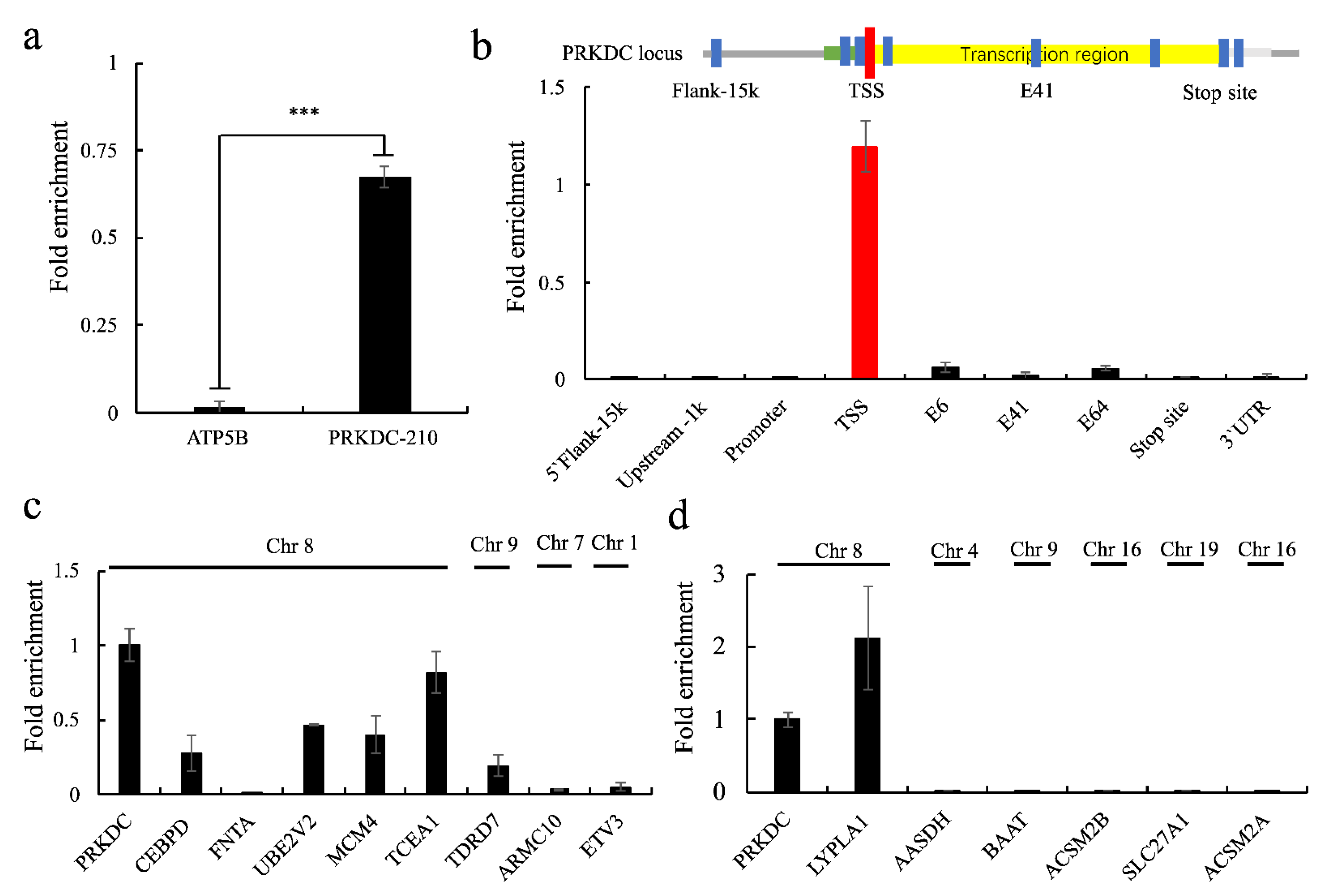
2.5. PRKDC-210 Acts on the Endogenous Genome
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Cell Culture
4.3. Transfection
4.4. Dual-Luciferase Reporter Assay
4.5. RNA Extraction
4.6. RT-qPCR
4.7. RNA Immunoprecipitation
4.8. RNA Sequencing
4.9. Chromatin Isolation by RNA Purification
4.10. Primer
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of Transcription in Human Cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montalbano, A.; Canver, M.C.; Sanjana, N.E. High-Throughput Approaches to Pinpoint Function within the Noncoding Genome. Mol. Cell 2017, 68, 44–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Liang, H.; Yang, H.; Zhou, K.; Xu, L.; Liu, J.; Lai, B.; Song, L.; Luo, H.; Peng, J.; et al. LincRNa-P21: Function and Mechanism in Cancer. Med. Oncol. 2017, 34, 98. [Google Scholar] [CrossRef]
- Atianand, M.K.; Hu, W.; Satpathy, A.T.; Shen, Y.; Ricci, E.P.; Alvarez-Dominguez, J.R.; Bhatta, A.; Schattgen, S.A.; McGowan, J.D.; Blin, J.; et al. A Long Noncoding RNA LincRNA-EPS Acts as a Transcriptional Brake to Restrain Inflammation. Cell 2016, 165, 1672–1685. [Google Scholar] [CrossRef] [Green Version]
- Kuwabara, Y.; Tsuji, S.; Nishiga, M.; Izuhara, M.; Ito, S.; Nagao, K.; Horie, T.; Watanabe, S.; Koyama, S.; Kiryu, H.; et al. Lionheart LincRNA Alleviates Cardiac Systolic Dysfunction under Pressure Overload. Commun. Biol. 2020, 3, 434. [Google Scholar] [CrossRef]
- Sun, Q.; Hao, Q.; Prasanth, K.V. Nuclear Long Noncoding RNAs: Key Regulators of Gene Expression. Trends Genet. 2018, 34, 142–157. [Google Scholar] [CrossRef]
- Kung, J.T.Y.; Colognori, D.; Lee, J.T. Long Noncoding RNAs: Past, Present, and Future. Genetics 2013, 193, 651–669. [Google Scholar] [CrossRef] [Green Version]
- Huarte, M. The Emerging Role of LncRNAs in Cancer. Nat. Med. 2015, 21, 1253–1261. [Google Scholar] [CrossRef]
- Wapinski, O.; Chang, H.Y. Long Noncoding RNAs and Human Disease. Trends Cell Biol. 2011, 21, 354–361. [Google Scholar] [CrossRef]
- Shiraishi, Y.; Kataoka, K.; Chiba, K.; Okada, A.; Kogure, Y.; Tanaka, H.; Ogawa, S.; Miyano, S. A Comprehensive Characterization of Cis-Acting Splicing-Associated Variants in Human Cancer. Genome Res. 2018, 28, 1111–1125. [Google Scholar] [CrossRef]
- Wang, K.C.; Chang, H.Y. Molecular Mechanisms of Long Noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopp, F.; Mendell, J.T. Functional Classification and Experimental Dissection of Long Noncoding RNAs. Cell 2018, 172, 393–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Cho, K.B.; Li, Y.; Tao, G.; Xie, Z.; Guo, B. Long Noncoding RNA (LncRNA)-Mediated Competing Endogenous RNA Networks Provide Novel Potential Biomarkers and Therapeutic Targets for Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 5758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhan, A.; Soleimani, M.; Mandal, S.S. Long Noncoding RNA and Cancer: A New Paradigm. Cancer Res. 2017, 77, 3965–3981. [Google Scholar] [CrossRef] [Green Version]
- Engreitz, J.M.; Haines, J.E.; Perez, E.M.; Munson, G.; Chen, J.; Kane, M.; McDonel, P.E.; Guttman, M.; Lander, E.S. Local Regulation of Gene Expression by LncRNA Promoters, Transcription and Splicing. Nature 2016, 539, 452–455. [Google Scholar] [CrossRef] [Green Version]
- Broude, E.V.; Győrffy, B.; Chumanevich, A.A.; Chen, M.; Mcdermott, M.S.J.; Shtutman, M.; Catroppo, J.F.; Roninson, I.B. Expression of CDK8 and CDK8-Interacting Genes as Potential Biomarkers in Breast Cancer HHS Public Access. Curr. Cancer Drug Targets 2015, 15, 739–749. [Google Scholar] [CrossRef] [Green Version]
- Lai, F.; Orom, U.A.; Cesaroni, M.; Beringer, M.; Taatjes, D.J.; Blobel, G.A.; Shiekhattar, R. Activating RNAs Associate with Mediator to Enhance Chromatin Architecture and Transcription. Nature 2013, 494, 497–501. [Google Scholar] [CrossRef] [Green Version]
- Burley, S.K.; Roeder, R.G. TATA Box Mimicry by TFIID. Cell 1998, 94, 551–553. [Google Scholar] [CrossRef] [Green Version]
- Savinkova, L.K.; Ponomarenko, M.P.; Ponomarenko, P.M.; Drachkova, I.A.; Lysova, M.v.; Arshinova, T.v.; Kolchanov, N.A. TATA Box Polymorphisms in Human Gene Promoters and Associated Hereditary Pathologies. Biochemistry 2009, 74, 117–129. [Google Scholar] [CrossRef]
- Knuesel, M.T.; Meyer, K.D.; Donner, A.J.; Espinosa, J.M.; Taatjes, D.J. The Human CDK8 Subcomplex Is a Histone Kinase That Requires Med12 for Activity and Can Function Independently of Mediator. Mol. Cell Biol. 2009, 29, 650–661. [Google Scholar] [CrossRef]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V. Cellular Fatty Acid Metabolism and Cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, C.; Quinn, J.; Chang, H.Y. Chromatin Isolation by RNA Purification (ChIRP). J. Vis. Exp. 2012, 61, e3912. [Google Scholar] [CrossRef]
- Dixon, J.R.; Gorkin, D.U.; Ren, B. Chromatin Domains: The Unit of Chromosome Organization. Mol. Cell 2016, 62, 668–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Ali, M.; Zhou, Q. Establishment and Evolution of Heterochromatin. Ann. N. Y. Acad. Sci. 2020, 1476, 59–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, K.; Ogawa, S. Splicing Factor Mutations and Cancer. Wiley Interdiscip. Rev. RNA 2014, 5, 445–459. [Google Scholar] [CrossRef] [Green Version]
- Srebrow, A.; Kornblihtt, A.R. The Connection between Splicing and Cancer. J. Cell Sci. 2006, 119, 2635–2641. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.S.; Zhu, T.; Li, L.; Xie, P.; Li, X.; Zhou, H.H.; Liu, Z.Q. LncRNAs and CircRNAs from the Same Gene: Masterpieces of RNA Splicing. Cancer Lett. 2018, 415, 49–57. [Google Scholar] [CrossRef]
- Freshney, R.I. Culture of Animal Cells; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2010; ISBN 9780470649367. [Google Scholar]
- González-Domínguez, I.; Grimaldi, N.; Cervera, L.; Ventosa, N.; Gòdia, F. Impact of Physicochemical Properties of DNA/PEI Complexes on Transient Transfection of Mammalian Cells. N. Biotechnol. 2019, 49, 88–97. [Google Scholar] [CrossRef] [Green Version]
- Yue, Y.; Wu, C. Progress and Perspectives in Developing Polymeric Vectors for in Vitro Gene Delivery. Biomater. Sci. 2013, 1, 152–170. [Google Scholar] [CrossRef]
- Life Technologies. Lipofectamine® RNAiMAX Reagent Lipofectamine® RNAiMAX Transfection Protocol Scaling Up or Down Transfections Limited Product Warranty and Disclaimer Details Package Contents; Protocol Pub. No. MAN0007825 Rev. 1.0; Life Technologies: Carlsbad, CA, USA, 2013. [Google Scholar]
- Life Technologies. Lipofectamine® 2000 Reagent Protocol Outline; Protocol Pub. No. MAN0007824 Rev. 1.0; Life Technologies: Carlsbad, CA, USA, 2013. [Google Scholar]
- Promega Corporation. Dual-Luciferase® Reporter Assay System Instructions for Use of Products E1910 and E1960; Rev 6/15; Promega Corporation: Madison, WI, USA, 2015. [Google Scholar]
- Thermo Fisher Scientific. TRIzol™ Reagent User Guide; Pub. No. MAN0001271 Rev. 8.0; Thermo Fisher Scientific: Waltham, MA, USA, 2016. [Google Scholar]
- Agilent Technologies. Mx3000P & Mx3005P Real-Time QPCR System; Revision# 090530; Agilent Technologies: Santa Clara, CA, USA, 2015. [Google Scholar]
- Gagliardi, M.; Matarazzo, M.R. RIP: RNA Immunoprecipitation. In Polycomb Group Proteins; Humana Press: New York, NY, USA, 2016; pp. 73–86. [Google Scholar]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and Quantifying Mammalian Transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mo, J.; Fan, G.; Tsukahara, T.; Sakari, M. The Role of the Exonic lncRNA PRKDC-210 in Transcription Regulation. Int. J. Mol. Sci. 2022, 23, 13783. https://doi.org/10.3390/ijms232213783
Mo J, Fan G, Tsukahara T, Sakari M. The Role of the Exonic lncRNA PRKDC-210 in Transcription Regulation. International Journal of Molecular Sciences. 2022; 23(22):13783. https://doi.org/10.3390/ijms232213783
Chicago/Turabian StyleMo, Junling, Guangyao Fan, Toshifumi Tsukahara, and Matomo Sakari. 2022. "The Role of the Exonic lncRNA PRKDC-210 in Transcription Regulation" International Journal of Molecular Sciences 23, no. 22: 13783. https://doi.org/10.3390/ijms232213783
APA StyleMo, J., Fan, G., Tsukahara, T., & Sakari, M. (2022). The Role of the Exonic lncRNA PRKDC-210 in Transcription Regulation. International Journal of Molecular Sciences, 23(22), 13783. https://doi.org/10.3390/ijms232213783