
ILCs—Crucial Players in Enteric Infectious Diseases


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Different ILC Subsets
3. ILC Plasticity
4. ILC Subsests in Intestinal Homeostasis and Inflammation
5. ILCs in Immune Response to Gut Intracellular Pathogens
5.1. Toxoplasma gondii
5.2. Salmonella Typhimurium
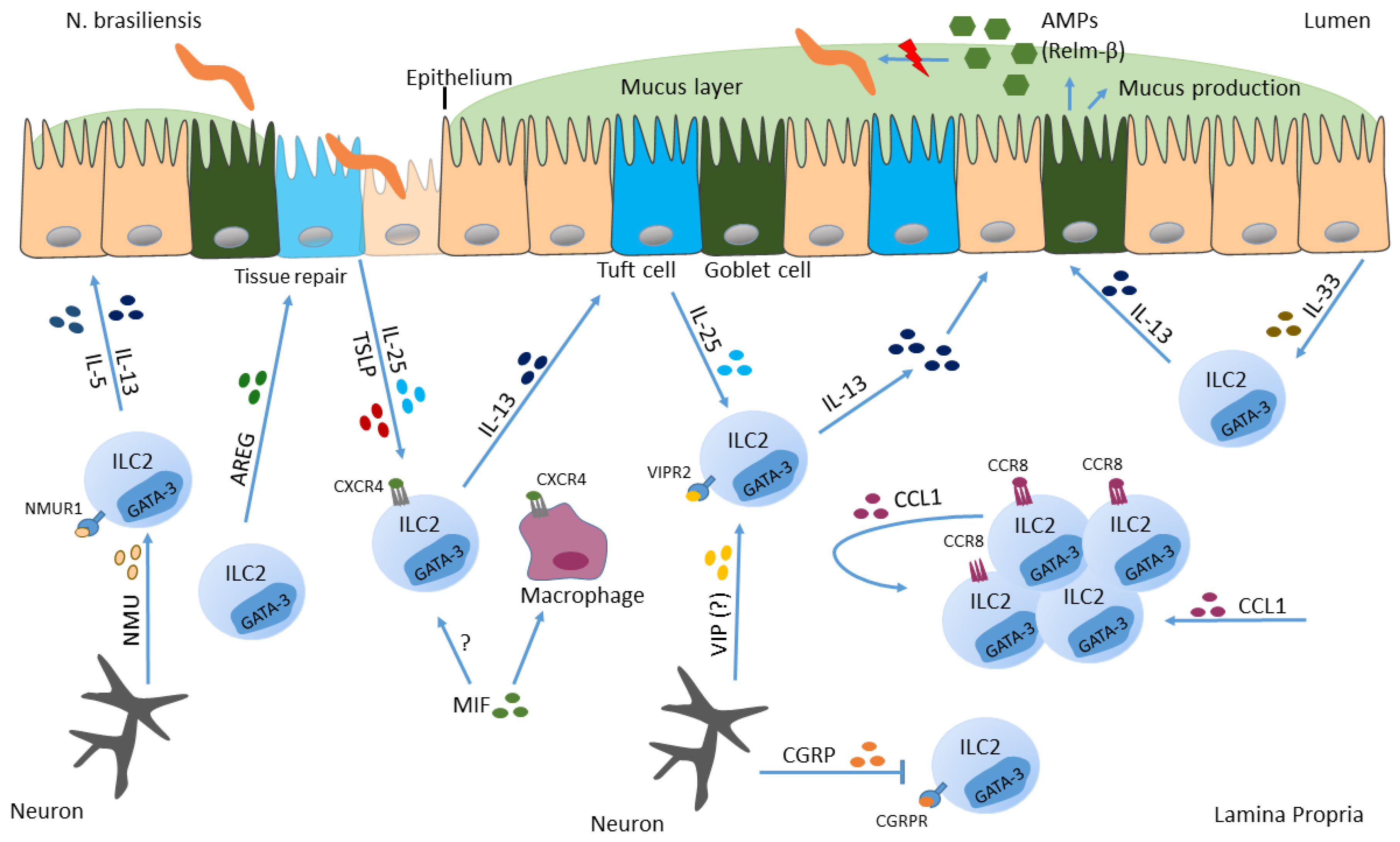
6. ILC2s and Helminths
6.1. Nippostrongylus brasiliensis
6.2. Trichuris muris
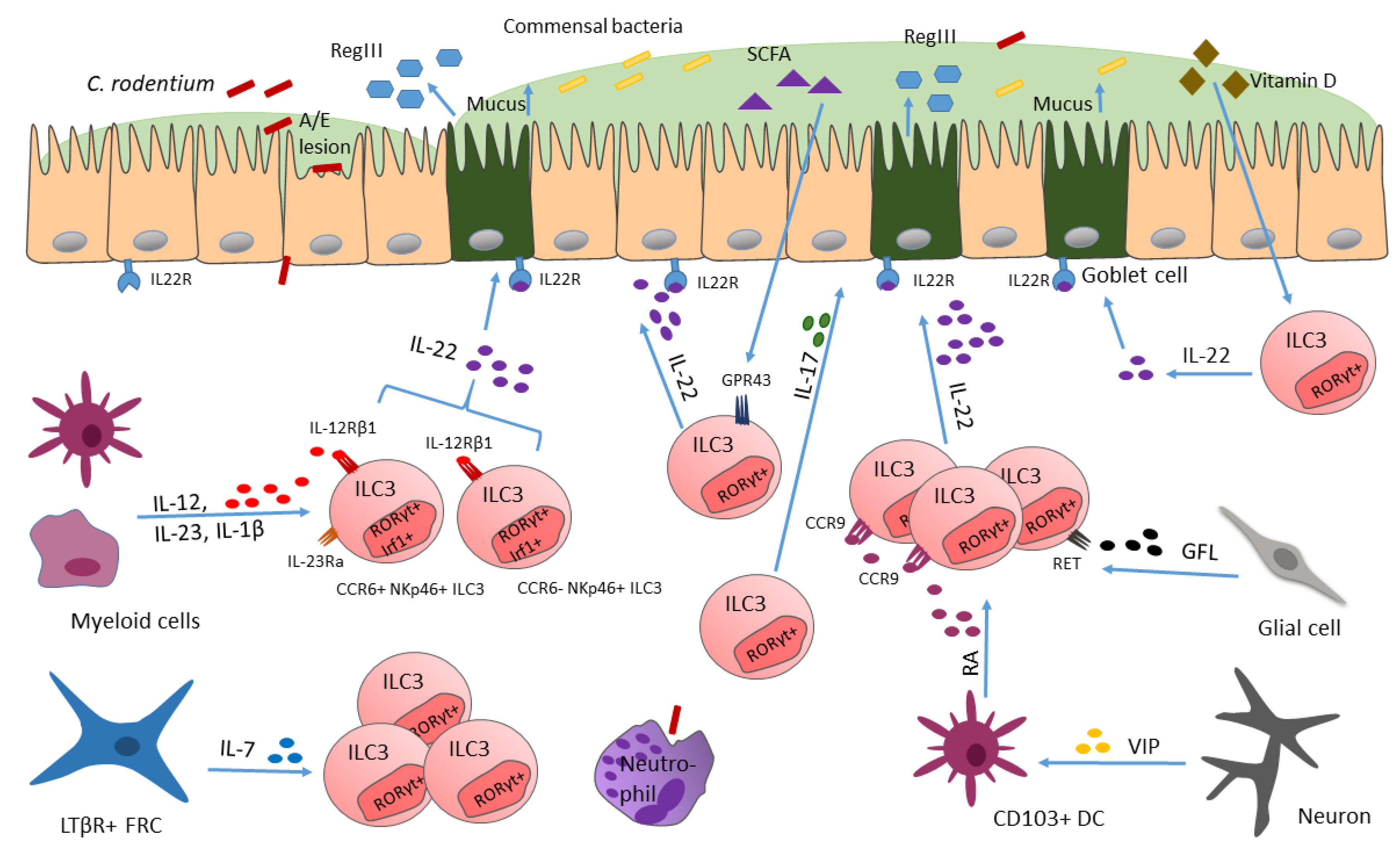
7. ILC3 Respond to (Extracellular) Bacteria
7.1. Citrobacter rodentium
7.2. Helicobacter
8. ILCs and Viruses
9. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Artis, D.; Spits, H. The biology of innate lymphoid cells. Nature 2015, 517, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Mowat, A.M.; Agace, W.W. Regional specialization within the intestinal immune system. Nat. Rev. Immunol. 2014, 14, 667–685. [Google Scholar] [CrossRef] [PubMed]
- Ramos, G.P.; Papadakis, K.A. Mechanisms of Disease: Inflammatory Bowel Diseases. Mayo Clin. Proc. 2019, 94, 155–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alatab, S.; Sepanlou, S.G.; Kuta, K.; Vahe, H.; Bisignano, D.C.; Safiri, S.; Sadeghi, A.; Nixon, M.R.; Abdoli, A.; Abolhassani, H.; et al. Collaborators, The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2020, 5, 17–30. [Google Scholar]
- Kobayashi, T.; Siegmund, B.; Le Berre, C.; Wei, S.C.; Ferrante, M.; Shen, B.; Bernstein, C.N.; Danese, S.; Peyrin-Biroulet, L.; Hibi, T. Ulcerative colitis. Nat. Rev. Dis. Primers 2020, 6, 74. [Google Scholar] [CrossRef]
- Roda, G.; Chien Ng, S.; Kotze, P.G.; Argollo, M.; Panaccione, R.; Spinelli, A.; Kaser, A.; Peyrin-Biroulet, L.; Danese, S. Crohn’s disease. Nat. Rev. Dis. Primers 2020, 6, 22. [Google Scholar] [CrossRef]
- Ananthakrishnan, A.N.; Bernstein, C.N.; Iliopoulos, D.; Macpherson, A.; Neurath, M.F.; Ali, R.A.R.; Vavricka, S.R.; Fiocchi, C. Environmental triggers in IBD: A review of progress and evidence. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 39–49. [Google Scholar] [CrossRef]
- Chang, J.T. Pathophysiology of Inflammatory Bowel Diseases. N. Engl. J. Med. 2020, 383, 2652–2664. [Google Scholar] [CrossRef]
- Nishida, A.; Inoue, R.; Inatomi, O.; Bamba, S.; Naito, Y.; Andoh, A. Gut microbiota in the pathogenesis of inflammatory bowel disease. Clin. J. Gastroenterol. 2018, 11, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Thakur, A.; Mikkelsen, H.; Jungersen, G. Intracellular Pathogens: Host Immunity and Microbial Persistence Strategies. J. Immunol. Res. 2019, 2019, 1356540. [Google Scholar] [CrossRef]
- Klose, C.S.; Artis, D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat. Immunol. 2016, 17, 765–774. [Google Scholar] [CrossRef]
- Constantinides, M.G.; McDonald, B.D.; Verhoef, P.A.; Bendelac, A. A committed precursor to innate lymphoid cells. Nature 2014, 508, 397–401. [Google Scholar] [CrossRef] [Green Version]
- Zheng, M.; Zhu, J. Innate Lymphoid Cells and Intestinal Inflammatory Disorders. Int. J. Mol. Sci. 2022, 23, 1856. [Google Scholar] [CrossRef] [PubMed]
- Hepworth, M.R.; Monticelli, L.A.; Fung, T.C.; Ziegler, C.G.; Grunberg, S.; Sinha, R.; Mantegazza, A.R.; Ma, H.L.; Crawford, A.; Angelosanto, J.M.; et al. Innate lymphoid cells regulate CD4+ T-cell responses to intestinal commensal bacteria. Nature 2013, 498, 113–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neill, D.R.; Wong, S.H.; Bellosi, A.; Flynn, R.J.; Daly, M.; Langford, T.K.; Bucks, C.; Kane, C.M.; Fallon, P.G.; Pannell, R.; et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 2010, 464, 1367–1370. [Google Scholar] [CrossRef] [Green Version]
- Spencer, S.P.; Wilhelm, C.; Yang, Q.; Hall, J.A.; Bouladoux, N.; Boyd, A.; Nutman, T.B.; Urban, J.F., Jr.; Wang, J.; Ramalingam, T.R.; et al. Adaptation of innate lymphoid cells to a micronutrient deficiency promotes type 2 barrier immunity. Science 2014, 343, 432–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Burg, N.; Turchinovich, G.; Finke, D. Maintenance of Immune Homeostasis through ILC/T Cell Interactions. Front. Immunol. 2015, 6, 416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diefenbach, A.; Colonna, M.; Koyasu, S. Development, differentiation, and diversity of innate lymphoid cells. Immunity 2014, 41, 354–365. [Google Scholar] [CrossRef] [Green Version]
- Vivier, E.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate Lymphoid Cells: 10 Years On. Cell 2018, 174, 1054–1066. [Google Scholar] [CrossRef] [Green Version]
- Spits, H.; Bernink, J.H.; Lanier, L. NK cells and type 1 innate lymphoid cells: Partners in host defense. Nat. Immunol. 2016, 17, 758–764. [Google Scholar] [CrossRef]
- Bernink, J.H.; Peters, C.P.; Munneke, M.; te Velde, A.A.; Meijer, S.L.; Weijer, K.; Hreggvidsdottir, H.S.; Heinsbroek, S.E.; Legrand, N.; Buskens, C.J.; et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat. Immunol. 2013, 14, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Krabbendam, L.; Bernink, J.H.; Spits, H. Innate lymphoid cells: From helper to killer. Curr. Opin. Immunol. 2021, 68, 28–33. [Google Scholar] [CrossRef]
- Moro, K.; Yamada, T.; Tanabe, M.; Takeuchi, T.; Ikawa, T.; Kawamoto, H.; Furusawa, J.; Ohtani, M.; Fujii, H.; Koyasu, S. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature 2010, 463, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Price, A.E.; Liang, H.E.; Sullivan, B.M.; Reinhardt, R.L.; Eisley, C.J.; Erle, D.J.; Locksley, R.M. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc. Natl. Acad. Sci. USA 2010, 107, 11489–11494. [Google Scholar] [CrossRef] [Green Version]
- Monticelli, L.A.; Osborne, L.C.; Noti, M.; Tran, S.V.; Zaiss, D.M.; Artis, D. IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin-EGFR interactions. Proc. Natl. Acad. Sci. USA 2015, 112, 10762–10767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monticelli, L.A.; Sonnenberg, G.F.; Abt, M.C.; Alenghat, T.; Ziegler, C.G.; Doering, T.A.; Angelosanto, J.M.; Laidlaw, B.J.; Yang, C.Y.; Sathaliyawala, T.; et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat. Immunol. 2011, 12, 1045–1054. [Google Scholar] [CrossRef]
- Ho, J.; Bailey, M.; Zaunders, J.; Mrad, N.; Sacks, R.; Sewell, W.; Harvey, R.J. Group 2 innate lymphoid cells (ILC2s) are increased in chronic rhinosinusitis with nasal polyps or eosinophilia. Clin. Exp. Allergy 2015, 45, 394–403. [Google Scholar] [CrossRef]
- Silver, J.S.; Kearley, J.; Copenhaver, A.M.; Sanden, C.; Mori, M.; Yu, L.; Pritchard, G.H.; Berlin, A.A.; Hunter, C.A.; Bowler, R.; et al. Inflammatory triggers associated with exacerbations of COPD orchestrate plasticity of group 2 innate lymphoid cells in the lungs. Nat. Immunol. 2016, 17, 626–635. [Google Scholar] [CrossRef] [Green Version]
- Wirtz, S.; Schulz-Kuhnt, A.; Neurath, M.F.; Atreya, I. Functional Contribution and Targeted Migration of Group-2 Innate Lymphoid Cells in Inflammatory Lung Diseases: Being at the Right Place at the Right Time. Front. Immunol. 2021, 12, 688879. [Google Scholar] [CrossRef]
- Hoyler, T.; Klose, C.S.; Souabni, A.; Turqueti-Neves, A.; Pfeifer, D.; Rawlins, E.L.; Voehringer, D.; Busslinger, M.; Diefenbach, A. The transcription factor GATA-3 controls cell fate and maintenance of type 2 innate lymphoid cells. Immunity 2012, 37, 634–648. [Google Scholar] [CrossRef] [Green Version]
- Mjosberg, J.; Bernink, J.; Golebski, K.; Karrich, J.J.; Peters, C.P.; Blom, B.; te Velde, A.A.; Fokkens, W.J.; van Drunen, C.M.; Spits, H. The transcription factor GATA3 is essential for the function of human type 2 innate lymphoid cells. Immunity 2012, 37, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.H.; Walker, J.A.; Jolin, H.E.; Drynan, L.F.; Hams, E.; Camelo, A.; Barlow, J.L.; Neill, D.R.; Panova, V.; Koch, U.; et al. Transcription factor RORalpha is critical for nuocyte development. Nat. Immunol. 2012, 13, 229–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngo Thi Phuong, N.; Palmieri, V.; Adamczyk, A.; Klopfleisch, R.; Langhorst, J.; Hansen, W.; Westendorf, A.M.; Pastille, E. IL-33 Drives Expansion of Type 2 Innate Lymphoid Cells and Regulatory T Cells and Protects Mice from Severe, Acute Colitis. Front. Immunol. 2021, 12, 669787. [Google Scholar] [CrossRef] [PubMed]
- You, Y.; Zhang, X.; Wang, X.; Yue, D.; Meng, F.; Zhu, J.; Wang, Y.; Sun, X. ILC2 Proliferated by IL-33 Stimulation Alleviates Acute Colitis in Rag1−/− Mouse through Promoting M2 Macrophage Polarization. J. Immunol. Res. 2020, 2020, 5018975. [Google Scholar] [CrossRef] [PubMed]
- Spits, H.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.; Mebius, R.E.; et al. Innate lymphoid cells—A proposal for uniform nomenclature. Nat. Rev. Immunol. 2013, 13, 145–149. [Google Scholar] [CrossRef]
- Geremia, A.; Arancibia-Carcamo, C.V. Innate Lymphoid Cells in Intestinal Inflammation. Front. Immunol. 2017, 8, 1296. [Google Scholar] [CrossRef] [PubMed]
- Croxatto, D.; Micheletti, A.; Montaldo, E.; Orecchia, P.; Loiacono, F.; Canegallo, F.; Calzetti, F.; Fulcheri, E.; Munari, E.; Zamo, A.; et al. Group 3 innate lymphoid cells regulate neutrophil migration and function in human decidua. Mucosal. Immunol. 2016, 9, 1372–1383. [Google Scholar] [CrossRef] [Green Version]
- Adachi, S.; Yoshida, H.; Kataoka, H.; Nishikawa, S. Three distinctive steps in Peyer’s patch formation of murine embryo. Int. Immunol. 1997, 9, 507–514. [Google Scholar] [CrossRef] [Green Version]
- Mebius, R.E.; Rennert, P.; Weissman, I.L. Developing lymph nodes collect CD4+CD3- LTbeta+ cells that can differentiate to APC, NK cells, and follicular cells but not T or B cells. Immunity 1997, 7, 493–504. [Google Scholar] [CrossRef] [Green Version]
- Shiu, J.; Piazuelo, M.B.; Ding, H.; Czinn, S.J.; Drakes, M.L.; Banerjee, A.; Basappa, N.; Kobayashi, K.S.; Fricke, W.F.; Blanchard, T.G. Gastric LTi cells promote lymphoid follicle formation but are limited by IRAK-M and do not alter microbial growth. Mucosal. Immunol. 2015, 8, 1047–1059. [Google Scholar] [CrossRef] [Green Version]
- Zeng, B.; Shi, S.; Ashworth, G.; Dong, C.; Liu, J.; Xing, F. ILC3 function as a double-edged sword in inflammatory bowel diseases. Cell Death Dis. 2019, 10, 315. [Google Scholar] [CrossRef]
- Klose, C.S.; Kiss, E.A.; Schwierzeck, V.; Ebert, K.; Hoyler, T.; d’Hargues, Y.; Goppert, N.; Croxford, A.L.; Waisman, A.; Tanriver, Y.; et al. A T-bet gradient controls the fate and function of CCR6-RORgammat+ innate lymphoid cells. Nature 2013, 494, 261–265. [Google Scholar] [CrossRef]
- McKenzie, A.N.J.; Spits, H.; Eberl, G. Innate lymphoid cells in inflammation and immunity. Immunity 2014, 41, 366–374. [Google Scholar] [CrossRef] [Green Version]
- Hazenberg, M.D.; Spits, H. Human innate lymphoid cells. Blood 2014, 124, 700–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DuPage, M.; Bluestone, J.A. Harnessing the plasticity of CD4(+) T cells to treat immune-mediated disease. Nat. Rev. Immunol. 2016, 16, 149–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bal, S.M.; Golebski, K.; Spits, H. Plasticity of innate lymphoid cell subsets. Nat. Rev. Immunol. 2020, 20, 552–565. [Google Scholar] [CrossRef] [PubMed]
- Locksley, R.M. Nine lives: Plasticity among T helper cell subsets. J. Exp. Med. 2009, 206, 1643–1646. [Google Scholar] [CrossRef] [PubMed]
- Cella, M.; Otero, K.; Colonna, M. Expansion of human NK-22 cells with IL-7, IL-2, and IL-1beta reveals intrinsic functional plasticity. Proc. Natl. Acad. Sci. USA 2010, 107, 10961–10966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crellin, N.K.; Trifari, S.; Kaplan, C.D.; Satoh-Takayama, N.; Di Santo, J.P.; Spits, H. Regulation of cytokine secretion in human CD127(+) LTi-like innate lymphoid cells by Toll-like receptor 2. Immunity 2010, 33, 752–764. [Google Scholar] [CrossRef] [Green Version]
- Vonarbourg, C.; Mortha, A.; Bui, V.L.; Hernandez, P.P.; Kiss, E.A.; Hoyler, T.; Flach, M.; Bengsch, B.; Thimme, R.; Holscher, C.; et al. Regulated expression of nuclear receptor RORgammat confers distinct functional fates to NK cell receptor-expressing RORgammat(+) innate lymphocytes. Immunity 2010, 33, 736–751. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Domingues, R.G.; Fonseca-Pereira, D.; Ferreira, M.; Ribeiro, H.; Lopez-Lastra, S.; Motomura, Y.; Moreira-Santos, L.; Bihl, F.; Braud, V.; et al. NFIL3 orchestrates the emergence of common helper innate lymphoid cell precursors. Cell Rep. 2015, 10, 2043–2054. [Google Scholar] [CrossRef]
- Sciume, G.; Hirahara, K.; Takahashi, H.; Laurence, A.; Villarino, A.V.; Singleton, K.L.; Spencer, S.P.; Wilhelm, C.; Poholek, A.C.; Vahedi, G.; et al. Distinct requirements for T-bet in gut innate lymphoid cells. J. Exp. Med. 2012, 209, 2331–2338. [Google Scholar] [CrossRef] [Green Version]
- Bernink, J.H.; Krabbendam, L.; Germar, K.; de Jong, E.; Gronke, K.; Kofoed-Nielsen, M.; Munneke, J.M.; Hazenberg, M.D.; Villaudy, J.; Buskens, C.J.; et al. Interleukin-12 and -23 Control Plasticity of CD127(+) Group 1 and Group 3 Innate Lymphoid Cells in the Intestinal Lamina Propria. Immunity 2015, 43, 146–160. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, A.; Vermi, W.; Lee, J.S.; Lonardi, S.; Gilfillan, S.; Newberry, R.D.; Cella, M.; Colonna, M. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-gamma-producing cells. Immunity 2013, 38, 769–781. [Google Scholar] [CrossRef] [Green Version]
- Takayama, T.; Kamada, N.; Chinen, H.; Okamoto, S.; Kitazume, M.T.; Chang, J.; Matuzaki, Y.; Suzuki, S.; Sugita, A.; Koganei, K.; et al. Imbalance of NKp44(+)NKp46(-) and NKp44(-)NKp46(+) natural killer cells in the intestinal mucosa of patients with Crohn’s disease. Gastroenterology 2010, 139, 882–892, 892e1–892e3. [Google Scholar] [CrossRef] [PubMed]
- Krzywinska, E.; Sobecki, M.; Nagarajan, S.; Zacharjasz, J.; Tambuwala, M.M.; Pelletier, A.; Cummins, E.; Gotthardt, D.; Fandrey, J.; Kerdiles, Y.M.; et al. The transcription factor HIF-1alpha mediates plasticity of NKp46+ innate lymphoid cells in the gut. J. Exp. Med. 2022, 219, e20210909. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forkel, M.; Mjosberg, J. Dysregulation of Group 3 Innate Lymphoid Cells in the Pathogenesis of Inflammatory Bowel Disease. Curr. Allergy Asthma Rep. 2016, 16, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, H.Y.; Sciume, G.; Mikami, Y.; Guo, L.; Sun, H.W.; Brooks, S.R.; Urban, J.F., Jr.; Davis, F.P.; Kanno, Y.; O’Shea, J.J. Developmental Acquisition of Regulomes Underlies Innate Lymphoid Cell Functionality. Cell 2016, 165, 1120–1133. [Google Scholar] [CrossRef] [Green Version]
- Kramer, B.; Goeser, F.; Lutz, P.; Glassner, A.; Boesecke, C.; Schwarze-Zander, C.; Kaczmarek, D.; Nischalke, H.D.; Branchi, V.; Manekeller, S.; et al. Compartment-specific distribution of human intestinal innate lymphoid cells is altered in HIV patients under effective therapy. PLoS Pathog. 2017, 13, e1006373. [Google Scholar] [CrossRef] [Green Version]
- Powell, N.; Walker, A.W.; Stolarczyk, E.; Canavan, J.B.; Gokmen, M.R.; Marks, E.; Jackson, I.; Hashim, A.; Curtis, M.A.; Jenner, R.G.; et al. The transcription factor T-bet regulates intestinal inflammation mediated by interleukin-7 receptor+ innate lymphoid cells. Immunity 2012, 37, 674–684. [Google Scholar] [CrossRef]
- Lopez-Yglesias, A.H.; Burger, E.; Camanzo, E.; Martin, A.T.; Araujo, A.M.; Kwok, S.F.; Yarovinsky, F. T-bet-dependent ILC1- and NK cell-derived IFN-gamma mediates cDC1-dependent host resistance against Toxoplasma gondii. PLoS Pathog. 2021, 17, e1008299. [Google Scholar] [CrossRef]
- Cardoso, V.; Chesne, J.; Ribeiro, H.; Garcia-Cassani, B.; Carvalho, T.; Bouchery, T.; Shah, K.; Barbosa-Morais, N.L.; Harris, N.; Veiga-Fernandes, H. Neuronal regulation of type 2 innate lymphoid cells via neuromedin U. Nature 2017, 549, 277–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klose, C.S.N.; Mahlakoiv, T.; Moeller, J.B.; Rankin, L.C.; Flamar, A.L.; Kabata, H.; Monticelli, L.A.; Moriyama, S.; Putzel, G.G.; Rakhilin, N.; et al. The neuropeptide neuromedin U stimulates innate lymphoid cells and type 2 inflammation. Nature 2017, 549, 282–286. [Google Scholar] [CrossRef]
- Talbot, S.; Abdulnour, R.E.; Burkett, P.R.; Lee, S.; Cronin, S.J.; Pascal, M.A.; Laedermann, C.; Foster, S.L.; Tran, J.V.; Lai, N.; et al. Silencing Nociceptor Neurons Reduces Allergic Airway Inflammation. Neuron 2015, 87, 341–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uvnas-Moberg, K. Release of gastrointestinal peptides in response to vagal activation induced by electrical stimulation, feeding and suckling. J. Auton. Nerv. Syst. 1983, 9, 141–155. [Google Scholar] [CrossRef]
- Bitar, K.N.; Makhlouf, G.M. Relaxation of isolated gastric smooth muscle cells by vasoactive intestinal peptide. Science 1982, 216, 531–533. [Google Scholar] [CrossRef]
- Pascal, M.; Kazakov, A.; Chevalier, G.; Dubrule, L.; Deyrat, J.; Dupin, A.; Saha, S.; Jagot, F.; Sailor, K.; Dulauroy, S.; et al. The neuropeptide VIP potentiates intestinal innate type 2 and type 3 immunity in response to feeding. Mucosal. Immunol. 2022, 15, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Robinette, M.L.; Fuchs, A.; Cortez, V.S.; Lee, J.S.; Wang, Y.; Durum, S.K.; Gilfillan, S.; Colonna, M.; Immunological Genome, C. Transcriptional programs define molecular characteristics of innate lymphoid cell classes and subsets. Nat. Immunol. 2015, 16, 306–317. [Google Scholar] [CrossRef]
- Buonocore, S.; Ahern, P.P.; Uhlig, H.H.; Ivanov, I.I.; Littman, D.R.; Maloy, K.J.; Powrie, F. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature 2010, 464, 1371–1375. [Google Scholar] [CrossRef] [Green Version]
- Cella, M.; Fuchs, A.; Vermi, W.; Facchetti, F.; Otero, K.; Lennerz, J.K.; Doherty, J.M.; Mills, J.C.; Colonna, M. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature 2009, 457, 722–725. [Google Scholar] [CrossRef] [PubMed]
- Satoh-Takayama, N.; Vosshenrich, C.A.; Lesjean-Pottier, S.; Sawa, S.; Lochner, M.; Rattis, F.; Mention, J.J.; Thiam, K.; Cerf-Bensussan, N.; Mandelboim, O.; et al. Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity 2008, 29, 958–970. [Google Scholar] [CrossRef]
- Sonnenberg, G.F.; Monticelli, L.A.; Elloso, M.M.; Fouser, L.A.; Artis, D. CD4(+) lymphoid tissue-inducer cells promote innate immunity in the gut. Immunity 2011, 34, 122–134. [Google Scholar] [CrossRef] [Green Version]
- Savage, A.K.; Liang, H.E.; Locksley, R.M. The Development of Steady-State Activation Hubs between Adult LTi ILC3s and Primed Macrophages in Small Intestine. J. Immunol. 2017, 199, 1912–1922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolk, K.; Kunz, S.; Witte, E.; Friedrich, M.; Asadullah, K.; Sabat, R. IL-22 increases the innate immunity of tissues. Immunity 2004, 21, 241–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Valdez, P.A.; Danilenko, D.M.; Hu, Y.; Sa, S.M.; Gong, Q.; Abbas, A.R.; Modrusan, Z.; Ghilardi, N.; de Sauvage, F.J.; et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat. Med. 2008, 14, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Cash, H.L.; Whitham, C.V.; Behrendt, C.L.; Hooper, L.V. Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science 2006, 313, 1126–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaishnava, S.; Yamamoto, M.; Severson, K.M.; Ruhn, K.A.; Yu, X.; Koren, O.; Ley, R.; Wakeland, E.K.; Hooper, L.V. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science 2011, 334, 255–258. [Google Scholar] [CrossRef] [Green Version]
- Aujla, S.J.; Chan, Y.R.; Zheng, M.; Fei, M.; Askew, D.J.; Pociask, D.A.; Reinhart, T.A.; McAllister, F.; Edeal, J.; Gaus, K.; et al. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat. Med. 2008, 14, 275–281. [Google Scholar] [CrossRef] [Green Version]
- Chun, E.; Lavoie, S.; Fonseca-Pereira, D.; Bae, S.; Michaud, M.; Hoveyda, H.R.; Fraser, G.L.; Gallini Comeau, C.A.; Glickman, J.N.; Fuller, M.H.; et al. Metabolite-Sensing Receptor Ffar2 Regulates Colonic Group 3 Innate Lymphoid Cells and Gut Immunity. Immunity 2019, 51, 871–884e6. [Google Scholar] [CrossRef]
- Kalliolias, G.D.; Ivashkiv, L.B. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat. Rev. Rheumatol. 2016, 12, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zhou, W.; Joseph, A.M.; Chu, C.; Putzel, G.G.; Fang, B.; Teng, F.; Lyu, M.; Yano, H.; Andreasson, K.I.; et al. Group 3 innate lymphoid cells produce the growth factor HB-EGF to protect the intestine from TNF-mediated inflammation. Nat. Immunol. 2022, 23, 251–261. [Google Scholar] [CrossRef]
- Katsanos, K.H.; Papadakis, K.A. Inflammatory Bowel Disease: Updates on Molecular Targets for Biologics. Gut Liver 2017, 11, 455–463. [Google Scholar] [CrossRef] [Green Version]
- Jiang, R.; Wang, H.; Deng, L.; Hou, J.; Shi, R.; Yao, M.; Gao, Y.; Yao, A.; Wang, X.; Yu, L.; et al. IL-22 is related to development of human colon cancer by activation of STAT3. BMC Cancer 2013, 13, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunay, I.R.; Diefenbach, A. Group 1 innate lymphoid cells in Toxoplasma gondii infection. Parasite Immunol. 2018, 40, e12516. [Google Scholar] [CrossRef]
- Yin, S.; Yu, J.; Hu, B.; Lu, C.; Liu, X.; Gao, X.; Li, W.; Zhou, L.; Wang, J.; Wang, D.; et al. Runx3 Mediates Resistance to Intracellular Bacterial Infection by Promoting IL12 Signaling in Group 1 ILC and NCR+ILC3. Front. Immunol. 2018, 9, 2101. [Google Scholar] [CrossRef]
- Konradt, C.; Ueno, N.; Christian, D.A.; Delong, J.H.; Pritchard, G.H.; Herz, J.; Bzik, D.J.; Koshy, A.A.; McGavern, D.B.; Lodoen, M.B.; et al. Endothelial cells are a replicative niche for entry of Toxoplasma gondii to the central nervous system. Nat. Microbiol. 2016, 1, 16001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubey, J.P. History of the discovery of the life cycle of Toxoplasma gondii. Int. J. Parasitol. 2009, 39, 877–882. [Google Scholar] [CrossRef]
- Snyder, L.M.; Doherty, C.M.; Mercer, H.L.; Denkers, E.Y. Induction of IL-12p40 and type 1 immunity by Toxoplasma gondii in the absence of the TLR-MyD88 signaling cascade. PLoS Pathog. 2021, 17, e1009970. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.B.; Denkers, E.Y. Border maneuvers: Deployment of mucosal immune defenses against Toxoplasma gondii. Mucosal. Immunol. 2014, 7, 744–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speer, C.A.; Dubey, J.P. Ultrastructure of early stages of infections in mice fed Toxoplasma gondii oocysts. Parasitology 1998, 116, 35–42. [Google Scholar] [CrossRef]
- Gazzinelli, R.T.; Wysocka, M.; Hayashi, S.; Denkers, E.Y.; Hieny, S.; Caspar, P.; Trinchieri, G.; Sher, A. Parasite-induced IL-12 stimulates early IFN-gamma synthesis and resistance during acute infection with Toxoplasma gondii. J. Immunol. 1994, 153, 2533–2543. [Google Scholar]
- Mashayekhi, M.; Sandau, M.M.; Dunay, I.R.; Frickel, E.M.; Khan, A.; Goldszmid, R.S.; Sher, A.; Ploegh, H.L.; Murphy, T.L.; Sibley, L.D.; et al. CD8alpha(+) dendritic cells are the critical source of interleukin-12 that controls acute infection by Toxoplasma gondii tachyzoites. Immunity 2011, 35, 249–259. [Google Scholar] [CrossRef] [Green Version]
- Lieberman, L.A.; Cardillo, F.; Owyang, A.M.; Rennick, D.M.; Cua, D.J.; Kastelein, R.A.; Hunter, C.A. IL-23 provides a limited mechanism of resistance to acute toxoplasmosis in the absence of IL-12. J. Immunol. 2004, 173, 1887–1893. [Google Scholar] [CrossRef] [Green Version]
- Scharton-Kersten, T.M.; Wynn, T.A.; Denkers, E.Y.; Bala, S.; Grunvald, E.; Hieny, S.; Gazzinelli, R.T.; Sher, A. In the absence of endogenous IFN-gamma, mice develop unimpaired IL-12 responses to Toxoplasma gondii while failing to control acute infection. J. Immunol. 1996, 157, 4045–4054. [Google Scholar]
- Suzuki, Y.; Orellana, M.A.; Schreiber, R.D.; Remington, J.S. Interferon-gamma: The major mediator of resistance against Toxoplasma gondii. Science 1988, 240, 516–518. [Google Scholar] [CrossRef] [PubMed]
- Denkers, E.Y.; Gazzinelli, R.T.; Martin, D.; Sher, A.; Emergence of NK1. 1+ cells as effectors of IFN-gamma dependent immunity to Toxoplasma gondii in MHC class I-deficient mice. J. Exp. Med. 1993, 178, 1465–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldszmid, R.S.; Caspar, P.; Rivollier, A.; White, S.; Dzutsev, A.; Hieny, S.; Kelsall, B.; Trinchieri, G.; Sher, A. NK cell-derived interferon-gamma orchestrates cellular dynamics and the differentiation of monocytes into dendritic cells at the site of infection. Immunity 2012, 36, 1047–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korchagina, A.A.; Koroleva, E.; Tumanov, A.V. Innate Lymphoid Cells in Response to Intracellular Pathogens: Protection versus Immunopathology. Front. Cell Infect. Microbiol. 2021, 11, 775554. [Google Scholar] [CrossRef]
- Klose, C.S.N.; Flach, M.; Mohle, L.; Rogell, L.; Hoyler, T.; Ebert, K.; Fabiunke, C.; Pfeifer, D.; Sexl, V.; Fonseca-Pereira, D.; et al. Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell 2014, 157, 340–356. [Google Scholar] [CrossRef] [Green Version]
- Clark, J.T.; Christian, D.A.; Gullicksrud, J.A.; Perry, J.A.; Park, J.; Jacquet, M.; Tarrant, J.C.; Radaelli, E.; Silver, J.; Hunter, C.A. IL-33 promotes innate lymphoid cell-dependent IFN-gamma production required for innate immunity to Toxoplasma gondii. Elife 2021, 10, e65614. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Patel, S.; Wang, Q.; Andhey, P.; Zaitsev, K.; Porter, S.; Hershey, M.; Bern, M.; Plougastel-Douglas, B.; Collins, P.; et al. Toxoplasma gondii infection drives conversion of NK cells into ILC1-like cells. Elife 2019, 8, e47605. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Souza-Fonseca-Guimaraes, F.; Bald, T.; Ng, S.S.; Young, A.; Ngiow, S.F.; Rautela, J.; Straube, J.; Waddell, N.; Blake, S.J.; et al. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat. Immunol. 2017, 18, 1004–1015. [Google Scholar] [CrossRef]
- Wagage, S.; Harms Pritchard, G.; Dawson, L.; Buza, E.L.; Sonnenberg, G.F.; Hunter, C.A. The Group 3 Innate Lymphoid Cell Defect in Aryl Hydrocarbon Receptor Deficient Mice Is Associated with T Cell Hyperactivation during Intestinal Infection. PLoS ONE 2015, 10, e0128335. [Google Scholar] [CrossRef]
- Snyder, L.M.; Belmares-Ortega, J.; Doherty, C.M.; Denkers, E.Y. Impact of MyD88, Microbiota, and Location on Type 1 and Type 3 Innate Lymphoid Cells during Toxoplasma gondii Infection. Immunohorizons 2022, 6, 660–670. [Google Scholar] [CrossRef]
- Pham, O.H.; McSorley, S.J. Protective host immune responses to Salmonella infection. Future Microbiol. 2015, 10, 101–110. [Google Scholar] [CrossRef] [Green Version]
- Santos, R.L.; Baumler, A.J. Cell tropism of Salmonella enterica. Int. J. Med. Microbiol. 2004, 294, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Tahoun, A.; Mahajan, S.; Paxton, E.; Malterer, G.; Donaldson, D.S.; Wang, D.; Tan, A.; Gillespie, T.L.; O’Shea, M.; Roe, A.J.; et al. Salmonella transforms follicle-associated epithelial cells into M cells to promote intestinal invasion. Cell Host Microbe 2012, 12, 645–656. [Google Scholar] [CrossRef] [Green Version]
- Songhet, P.; Barthel, M.; Stecher, B.; Muller, A.J.; Kremer, M.; Hansson, G.C.; Hardt, W.D. Stromal IFN-gammaR-signaling modulates goblet cell function during Salmonella Typhimurium infection. PLoS ONE 2011, 6, e22459. [Google Scholar] [CrossRef] [Green Version]
- Goto, Y.; Obata, T.; Kunisawa, J.; Sato, S.; Ivanov, I.I.; Lamichhane, A.; Takeyama, N.; Kamioka, M.; Sakamoto, M.; Matsuki, T.; et al. Innate lymphoid cells regulate intestinal epithelial cell glycosylation. Science 2014, 345, 1254009. [Google Scholar] [CrossRef] [Green Version]
- Kastele, V.; Mayer, J.; Lee, E.S.; Papazian, N.; Cole, J.J.; Cerovic, V.; Belz, G.; Tomura, M.; Eberl, G.; Goodyear, C.; et al. Intestinal-derived ILCs migrating in lymph increase IFNgamma production in response to Salmonella Typhimurium infection. Mucosal Immunol. 2021, 14, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Zarepour, M.; Bhullar, K.; Montero, M.; Ma, C.; Huang, T.; Velcich, A.; Xia, L.; Vallance, B.A. The mucin Muc2 limits pathogen burdens and epithelial barrier dysfunction during Salmonella enterica serovar Typhimurium colitis. Infect. Immun. 2013, 81, 3672–3683. [Google Scholar] [CrossRef] [Green Version]
- Xiong, L.; Wang, S.; Dean, J.W.; Oliff, K.N.; Jobin, C.; Curtiss, R., 3rd; Zhou, L. Group 3 innate lymphoid cell pyroptosis represents a host defence mechanism against Salmonella infection. Nat. Microbiol. 2022, 7, 1087–1099. [Google Scholar] [CrossRef]
- Schistosomiasis and soil-transmitted helminthiases: Number of people treated in 2015. Wkly. Epidemiol. Rec. 2016, 91, 585–595.
- Jourdan, P.M.; Lamberton, P.H.L.; Fenwick, A.; Addiss, D.G. Soil-transmitted helminth infections. Lancet 2018, 391, 252–265. [Google Scholar] [CrossRef] [Green Version]
- Jia, T.W.; Melville, S.; Utzinger, J.; King, C.H.; Zhou, X.N. Soil-transmitted helminth reinfection after drug treatment: A systematic review and meta-analysis. PLoS Negl. Trop. Dis. 2012, 6, e1621. [Google Scholar] [CrossRef] [Green Version]
- Henry, E.K.; Inclan-Rico, J.M.; Siracusa, M.C. Type 2 cytokine responses: Regulating immunity to helminth parasites and allergic inflammation. Curr. Pharmacol. Rep. 2017, 3, 346–359. [Google Scholar] [CrossRef] [PubMed]
- Filbey, K.; Bouchery, T.; Le Gros, G. The role of ILC2 in hookworm infection. Parasite Immunol. 2018, 40, e12429. [Google Scholar] [CrossRef]
- Inclan-Rico, J.M.; Siracusa, M.C. First Responders: Innate Immunity to Helminths. Trends Parasitol. 2018, 34, 861–880. [Google Scholar] [CrossRef]
- Allen, J.E.; Sutherland, T.E. Host protective roles of type 2 immunity: Parasite killing and tissue repair, flip sides of the same coin. Semin. Immunol. 2014, 26, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Grencis, R.K. Immunity to helminths: Resistance, regulation, and susceptibility to gastrointestinal nematodes. Annu. Rev. Immunol. 2015, 33, 201–225. [Google Scholar] [CrossRef] [PubMed]
- Gerbe, F.; Sidot, E.; Smyth, D.J.; Ohmoto, M.; Matsumoto, I.; Dardalhon, V.; Cesses, P.; Garnier, L.; Pouzolles, M.; Brulin, B.; et al. Intestinal epithelial tuft cells initiate type 2 mucosal immunity to helminth parasites. Nature 2016, 529, 226–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howitt, M.R.; Lavoie, S.; Michaud, M.; Blum, A.M.; Tran, S.V.; Weinstock, J.V.; Gallini, C.A.; Redding, K.; Margolskee, R.F.; Osborne, L.C.; et al. Tuft cells, taste-chemosensory cells, orchestrate parasite type 2 immunity in the gut. Science 2016, 351, 1329–1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Moltke, J.; Ji, M.; Liang, H.E.; Locksley, R.M. Tuft-cell-derived IL-25 regulates an intestinal ILC2-epithelial response circuit. Nature 2016, 529, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Bouchery, T.; Le Gros, G.; Harris, N. ILC2s-Trailblazers in the Host Response Against Intestinal Helminths. Front. Immunol. 2019, 10, 623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haber, A.L.; Biton, M.; Rogel, N.; Herbst, R.H.; Shekhar, K.; Smillie, C.; Burgin, G.; Delorey, T.M.; Howitt, M.R.; Katz, Y.; et al. A single-cell survey of the small intestinal epithelium. Nature 2017, 551, 333–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varyani, F.; Loser, S.; Filbey, K.J.; Harcus, Y.; Drurey, C.; Poveda, M.C.; Rasid, O.; White, M.P.J.; Smyth, D.J.; Gerbe, F.; et al. The IL-25-dependent tuft cell circuit driven by intestinal helminths requires macrophage migration inhibitory factor (MIF). Mucosal. Immunol. 2022. [Google Scholar] [CrossRef]
- Waddell, A.; Vallance, J.E.; Hummel, A.; Alenghat, T.; Rosen, M.J. IL-33 Induces Murine Intestinal Goblet Cell Differentiation Indirectly via Innate Lymphoid Cell IL-13 Secretion. J. Immunol. 2019, 202, 598–607. [Google Scholar] [CrossRef] [Green Version]
- Knipfer, L.; Schulz-Kuhnt, A.; Kindermann, M.; Greif, V.; Symowski, C.; Voehringer, D.; Neurath, M.F.; Atreya, I.; Wirtz, S. A CCL1/CCR8-dependent feed-forward mechanism drives ILC2 functions in type 2-mediated inflammation. J. Exp. Med. 2019, 216, 2763–2777. [Google Scholar] [CrossRef] [Green Version]
- Nagashima, H.; Mahlakoiv, T.; Shih, H.Y.; Davis, F.P.; Meylan, F.; Huang, Y.; Harrison, O.J.; Yao, C.; Mikami, Y.; Urban, J.F., Jr.; et al. Neuropeptide CGRP Limits Group 2 Innate Lymphoid Cell Responses and Constrains Type 2 Inflammation. Immunity 2019, 51, 682–695.e6. [Google Scholar] [CrossRef]
- Wallrapp, A.; Riesenfeld, S.J.; Burkett, P.R.; Abdulnour, R.E.; Nyman, J.; Dionne, D.; Hofree, M.; Cuoco, M.S.; Rodman, C.; Farouq, D.; et al. The neuropeptide NMU amplifies ILC2-driven allergic lung inflammation. Nature 2017, 549, 351–356. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Ding, J.; Porter, C.B.M.; Wallrapp, A.; Tabaka, M.; Ma, S.; Fu, S.; Guo, X.; Riesenfeld, S.J.; Su, C.; et al. Transcriptional Atlas of Intestinal Immune Cells Reveals that Neuropeptide alpha-CGRP Modulates Group 2 Innate Lymphoid Cell Responses. Immunity 2019, 51, 696–708.e9. [Google Scholar] [CrossRef] [PubMed]
- Seidl, A.; Panzer, M.; Voehringer, D. Protective immunity against the gastrointestinal nematode Nippostrongylus brasiliensis requires a broad T-cell receptor repertoire. Immunology 2011, 134, 214–223. [Google Scholar] [CrossRef]
- Varela, F.; Symowski, C.; Pollock, J.; Wirtz, S.; Voehringer, D. IL-4/IL-13-producing ILC2s are required for timely control of intestinal helminth infection in mice. Eur. J. Immunol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Hotez, P.J.; Brindley, P.J.; Bethony, J.M.; King, C.H.; Pearce, E.J.; Jacobson, J. Helminth infections: The great neglected tropical diseases. J. Clin. Investig. 2008, 118, 1311–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaiss, D.M.W.; Gause, W.C.; Osborne, L.C.; Artis, D. Emerging functions of amphiregulin in orchestrating immunity, inflammation, and tissue repair. Immunity 2015, 42, 216–226. [Google Scholar] [CrossRef] [Green Version]
- Zaiss, D.M.; Yang, L.; Shah, P.R.; Kobie, J.J.; Urban, J.F.; Mosmann, T.R. Amphiregulin, a TH2 cytokine enhancing resistance to nematodes. Science 2006, 314, 1746. [Google Scholar] [CrossRef]
- Turner, J.E.; Morrison, P.J.; Wilhelm, C.; Wilson, M.; Ahlfors, H.; Renauld, J.C.; Panzer, U.; Helmby, H.; Stockinger, B. IL-9-mediated survival of type 2 innate lymphoid cells promotes damage control in helminth-induced lung inflammation. J. Exp. Med. 2013, 210, 2951–2965. [Google Scholar] [CrossRef] [Green Version]
- Sonnenberg, G.F.; Monticelli, L.A.; Alenghat, T.; Fung, T.C.; Hutnick, N.A.; Kunisawa, J.; Shibata, N.; Grunberg, S.; Sinha, R.; Zahm, A.M.; et al. Innate lymphoid cells promote anatomical containment of lymphoid-resident commensal bacteria. Science 2012, 336, 1321–1325. [Google Scholar] [CrossRef] [Green Version]
- Bishop, A.L.; Wiles, S.; Dougan, G.; Frankel, G. Cell attachment properties and infectivity of host-adapted and environmentally adapted Citrobacter rodentium. Microbes Infect. 2007, 9, 1316–1324. [Google Scholar] [CrossRef]
- Nataro, J.P.; Kaper, J.B. Diarrheagenic Escherichia coli. Clin. Microbiol. Rev. 1998, 11, 142–201. [Google Scholar] [CrossRef] [Green Version]
- Frankel, G.; Phillips, A.D. Attaching effacing Escherichia coli and paradigms of Tir-triggered actin polymerization: Getting off the pedestal. Cell Microbiol. 2008, 10, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Ruano-Gallego, D.; Sanchez-Garrido, J.; Kozik, Z.; Nunez-Berrueco, E.; Cepeda-Molero, M.; Mullineaux-Sanders, C.; Naemi-Baghshomali Clark, J.; Slater, S.L.; Wagner, N.; Glegola-Madejska, I.; et al. Type III secretion system effectors form robust and flexible intracellular virulence networks. Science 2021, 371, eabc9531. [Google Scholar] [CrossRef]
- Mullineaux-Sanders, C.; Sanchez-Garrido, J.; Hopkins, E.G.D.; Shenoy, A.R.; Barry, R.; Frankel, G. Citrobacter rodentium-host-microbiota interactions: Immunity, bioenergetics and metabolism. Nat. Rev. Microbiol. 2019, 17, 701–715. [Google Scholar] [CrossRef] [PubMed]
- Lupp, C.; Robertson, M.L.; Wickham, M.E.; Sekirov, I.; Champion, O.L.; Gaynor, E.C.; Finlay, B.B. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe 2007, 2, 119–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borenshtein, D.; McBee, M.E.; Schauer, D.B. Utility of the Citrobacter rodentium infection model in laboratory mice. Curr. Opin. Gastroenterol. 2008, 24, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Borenshtein, D.; Nambiar, P.R.; Groff, E.B.; Fox, J.G.; Schauer, D.B. Development of fatal colitis in FVB mice infected with Citrobacter rodentium. Infect. Immun. 2007, 75, 3271–3281. [Google Scholar] [CrossRef] [Green Version]
- Caruso, R.; Lo, B.C.; Nunez, G. Host-microbiota interactions in inflammatory bowel disease. Nat. Rev. Immunol. 2020, 20, 411–426. [Google Scholar] [CrossRef]
- Basu, R.; O’Quinn, D.B.; Silberger, D.J.; Schoeb, T.R.; Fouser, L.; Ouyang, W.; Hatton, R.D.; Weaver, C.T. Th22 cells are an important source of IL-22 for host protection against enteropathogenic bacteria. Immunity 2012, 37, 1061–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinnebrew, M.A.; Buffie, C.G.; Diehl, G.E.; Zenewicz, L.A.; Leiner, I.; Hohl, T.M.; Flavell, R.A.; Littman, D.R.; Pamer, E.G. Interleukin 23 production by intestinal CD103(+)CD11b(+) dendritic cells in response to bacterial flagellin enhances mucosal innate immune defense. Immunity 2012, 36, 276–287. [Google Scholar] [CrossRef] [Green Version]
- Seo, S.U.; Kuffa, P.; Kitamoto, S.; Nagao-Kitamoto, H.; Rousseau, J.; Kim, Y.G.; Nunez, G.; Kamada, N. Intestinal macrophages arising from CCR2(+) monocytes control pathogen infection by activating innate lymphoid cells. Nat. Commun. 2015, 6, 8010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonnenberg, G.F.; Fouser, L.A.; Artis, D. Border patrol: Regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat. Immunol. 2011, 12, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Schmalzl, A.; Leupold, T.; Kreiss, L.; Waldner, M.; Schurmann, S.; Neurath, M.F.; Becker, C.; Wirtz, S. Interferon regulatory factor 1 (IRF-1) promotes intestinal group 3 innate lymphoid responses during Citrobacter rodentium infection. Nat. Commun. 2022, 13, 5730. [Google Scholar] [CrossRef] [PubMed]
- Aychek, T.; Mildner, A.; Yona, S.; Kim, K.W.; Lampl, N.; Reich-Zeliger, S.; Boon, L.; Yogev, N.; Waisman, A.; Cua, D.J.; et al. IL-23-mediated mononuclear phagocyte crosstalk protects mice from Citrobacter rodentium-induced colon immunopathology. Nat. Commun. 2015, 6, 6525. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.D.; Arora, J.; Diehl, K.; Bora, S.A.; Cantorna, M.T. Vitamin D Is Required for ILC3 Derived IL-22 and Protection From Citrobacter rodentium Infection. Front. Immunol. 2019, 10, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.B.; Yang, H.; Allaire, J.M.; Ma, C.; Graef, F.A.; Mortha, A.; Liang, Q.; Bosman, E.S.; Reid, G.S.; Waschek, J.A.; et al. Vasoactive intestinal peptide promotes host defense against enteric pathogens by modulating the recruitment of group 3 innate lymphoid cells. Proc. Natl. Acad. Sci. USA 2021, 118, e2106634118. [Google Scholar] [CrossRef]
- Abad, C.; Gomariz, R.; Waschek, J.; Leceta, J.; Martinez, C.; Juarranz, Y.; Arranz, A. VIP in inflammatory bowel disease: State of the art. Endocr. Metab. Immune Disord. Drug Targets 2012, 12, 316–322. [Google Scholar] [CrossRef]
- Kim, C.H.; Hashimoto-Hill, S.; Kim, M. Migration and Tissue Tropism of Innate Lymphoid Cells. Trends Immunol. 2016, 37, 68–79. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.H.; Taparowsky, E.J.; Kim, C.H. Retinoic Acid Differentially Regulates the Migration of Innate Lymphoid Cell Subsets to the Gut. Immunity 2015, 43, 107–119. [Google Scholar] [CrossRef] [Green Version]
- Ibiza, S.; Garcia-Cassani, B.; Ribeiro, H.; Carvalho, T.; Almeida, L.; Marques, R.; Misic, A.M.; Bartow-McKenney, C.; Larson, D.M.; Pavan, W.J.; et al. Glial-cell-derived neuroregulators control type 3 innate lymphoid cells and gut defence. Nature 2016, 535, 440–443. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.W.; Morbe, U.; Lutge, M.; Engetschwiler, C.; Onder, L.; Novkovic, M.; Gil-Cruz, C.; Perez-Shibayama, C.; Hehlgans, T.; Scandella, E.; et al. Intestinal fibroblastic reticular cell niches control innate lymphoid cell homeostasis and function. Nat. Commun. 2022, 13, 2027. [Google Scholar] [CrossRef] [PubMed]
- Serafini, N.; Jarade, A.; Surace, L.; Goncalves, P.; Sismeiro, O.; Varet, H.; Legendre, R.; Coppee, J.Y.; Disson, O.; Durum, S.K.; et al. Trained ILC3 responses promote intestinal defense. Science 2022, 375, 859–863. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.G. The non-H pylori helicobacters: Their expanding role in gastrointestinal and systemic diseases. Gut 2002, 50, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.G.; Ge, Z.; Whary, M.T.; Erdman, S.E.; Horwitz, B.H. Helicobacter hepaticus infection in mice: Models for understanding lower bowel inflammation and cancer. Mucosal Immunol. 2011, 4, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Bostick, J.W.; Wang, Y.; Shen, Z.; Ge, Y.; Brown, J.; Chen, Z.E.; Mohamadzadeh, M.; Fox, J.G.; Zhou, L. Dichotomous regulation of group 3 innate lymphoid cells by nongastric Helicobacter species. Proc. Natl. Acad. Sci. USA 2019, 116, 24760–24769. [Google Scholar] [CrossRef]
- Geremia, A.; Arancibia-Carcamo, C.V.; Fleming, M.P.; Rust, N.; Singh, B.; Mortensen, N.J.; Travis, S.P.; Powrie, F. IL-23-responsive innate lymphoid cells are increased in inflammatory bowel disease. J. Exp. Med. 2011, 208, 1127–1133. [Google Scholar] [CrossRef] [Green Version]
- Eaden, J.A.; Abrams, K.R.; Mayberry, J.F. The risk of colorectal cancer in ulcerative colitis: A meta-analysis. Gut 2001, 48, 526–535. [Google Scholar] [CrossRef] [Green Version]
- Rutter, M.D.; Saunders, B.P.; Wilkinson, K.H.; Rumbles, S.; Schofield, G.; Kamm, M.A.; Williams, C.B.; Price, A.B.; Talbot, I.C.; Forbes, A. Thirty-year analysis of a colonoscopic surveillance program for neoplasia in ulcerative colitis. Gastroenterology 2006, 130, 1030–1038. [Google Scholar] [CrossRef]
- Stehle, C.; Hernandez, D.C.; Romagnani, C. Innate lymphoid cells in lung infection and immunity. Immunol. Rev. 2018, 286, 102–119. [Google Scholar] [CrossRef]
- Minodier, L.; Charrel, R.N.; Ceccaldi, P.E.; van der Werf, S.; Blanchon, T.; Hanslik, T.; Falchi, A. Prevalence of gastrointestinal symptoms in patients with influenza, clinical significance, and pathophysiology of human influenza viruses in faecal samples: What do we know? Virol. J. 2015, 12, 215. [Google Scholar] [CrossRef] [Green Version]
- Roach, S.N.; Fiege, J.K.; Shepherd, F.K.; Wiggen, T.D.; Hunter, R.C.; Langlois, R.A. Respiratory Influenza Virus Infection Causes Dynamic Tuft Cell and Innate Lymphoid Cell Changes in the Small Intestine. J. Virol. 2022, 96, e0035222. [Google Scholar] [CrossRef] [PubMed]
- Duerkop, B.A.; Hooper, L.V. Resident viruses and their interactions with the immune system. Nat. Immunol. 2013, 14, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, P.P.; Mahlakoiv, T.; Yang, I.; Schwierzeck, V.; Nguyen, N.; Guendel, F.; Gronke, K.; Ryffel, B.; Hoelscher, C.; Dumoutier, L.; et al. Interferon-lambda and interleukin 22 act synergistically for the induction of interferon-stimulated genes and control of rotavirus infection. Nat. Immunol. 2015, 16, 698–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, C.; Stappenbeck, T.S. Viral interactions with the host and microbiota in the intestine. Curr. Opin. Immunol. 2012, 24, 405–410. [Google Scholar] [CrossRef]
- Ganal-Vonarburg, S.C.; Duerr, C.U. The interaction of intestinal microbiota and innate lymphoid cells in health and disease throughout life. Immunology 2020, 159, 39–51. [Google Scholar] [CrossRef]
- Durbin, R.K.; Kotenko, S.V.; Durbin, J.E. Interferon induction and function at the mucosal surface. Immunol. Rev. 2013, 255, 25–39. [Google Scholar] [CrossRef]
- Katze, M.G.; He, Y.; Gale, M., Jr. Viruses and interferon: A fight for supremacy. Nat. Rev. Immunol. 2002, 2, 675–687. [Google Scholar] [CrossRef]
- Pott, J.; Mahlakoiv, T.; Mordstein, M.; Duerr, C.U.; Michiels, T.; Stockinger, S.; Staeheli, P.; Hornef, M.W. IFN-lambda determines the intestinal epithelial antiviral host defense. Proc. Natl. Acad. Sci. USA 2011, 108, 7944–7949. [Google Scholar] [CrossRef] [Green Version]
- Gil-Cruz, C.; Perez-Shibayama, C.; Onder, L.; Chai, Q.; Cupovic, J.; Cheng, H.W.; Novkovic, M.; Lang, P.A.; Geuking, M.B.; McCoy, K.D.; et al. Fibroblastic reticular cells regulate intestinal inflammation via IL-15-mediated control of group 1 ILCs. Nat. Immunol. 2016, 17, 1388–1396. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leupold, T.; Wirtz, S. ILCs—Crucial Players in Enteric Infectious Diseases. Int. J. Mol. Sci. 2022, 23, 14200. https://doi.org/10.3390/ijms232214200
Leupold T, Wirtz S. ILCs—Crucial Players in Enteric Infectious Diseases. International Journal of Molecular Sciences. 2022; 23(22):14200. https://doi.org/10.3390/ijms232214200
Chicago/Turabian StyleLeupold, Tamara, and Stefan Wirtz. 2022. "ILCs—Crucial Players in Enteric Infectious Diseases" International Journal of Molecular Sciences 23, no. 22: 14200. https://doi.org/10.3390/ijms232214200