
Impact of Siponimod on Enteric and Central Nervous System Pathology in Late-Stage Experimental Autoimmune Encephalomyelitis
 , ,
, ,  ,
,  and
and 
Abstract
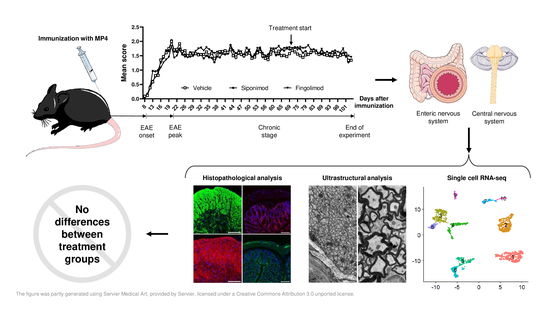
:
1. Introduction
2. Results
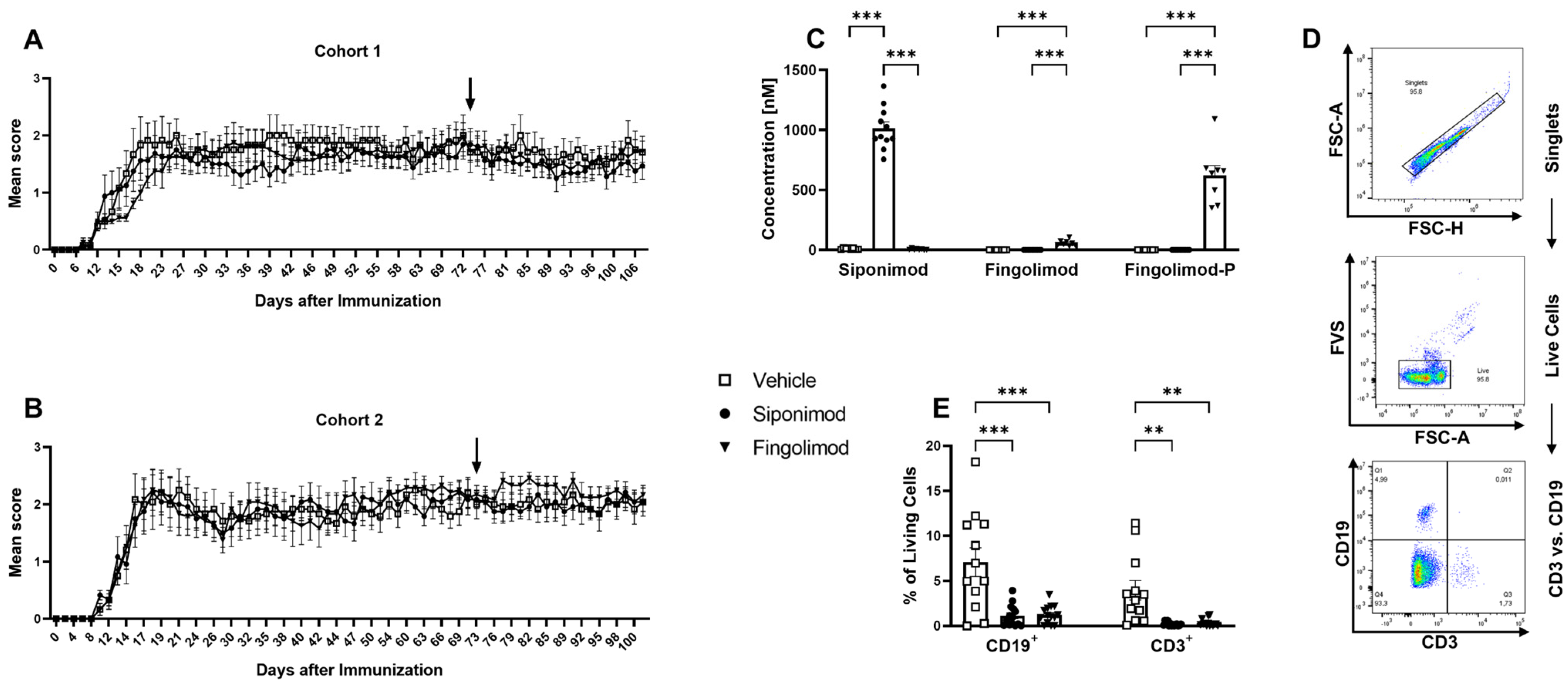
2.1. Siponimod and Fingolimod Induce Peripheral Lymphopenia in MP4-Immunized Experimental Autoimmune Encephalomyelitis (EAE) Mice
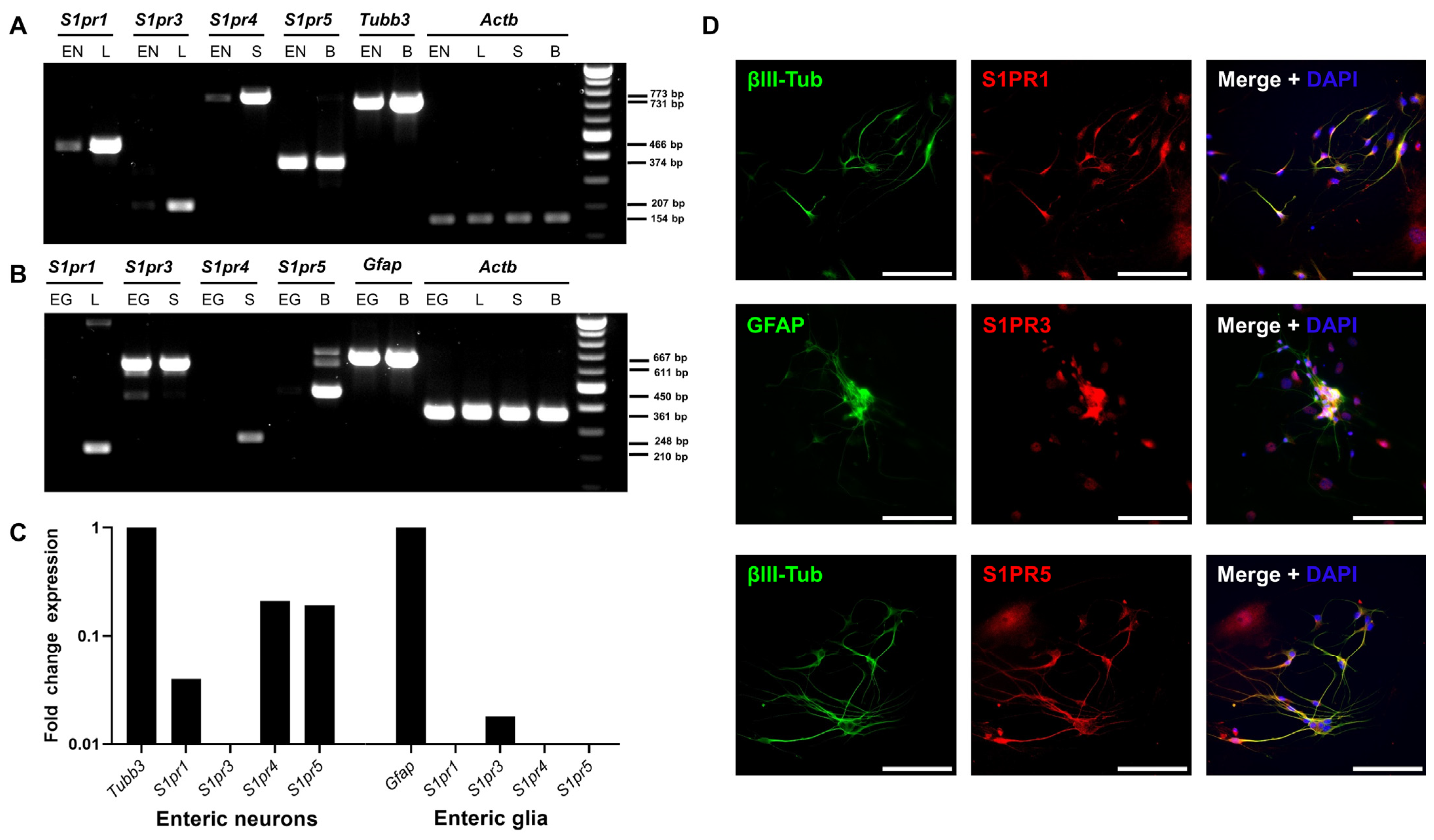
2.2. The Enteric Nervous System Expresses Sphingosine-1-Phosphate (S1P) Receptors
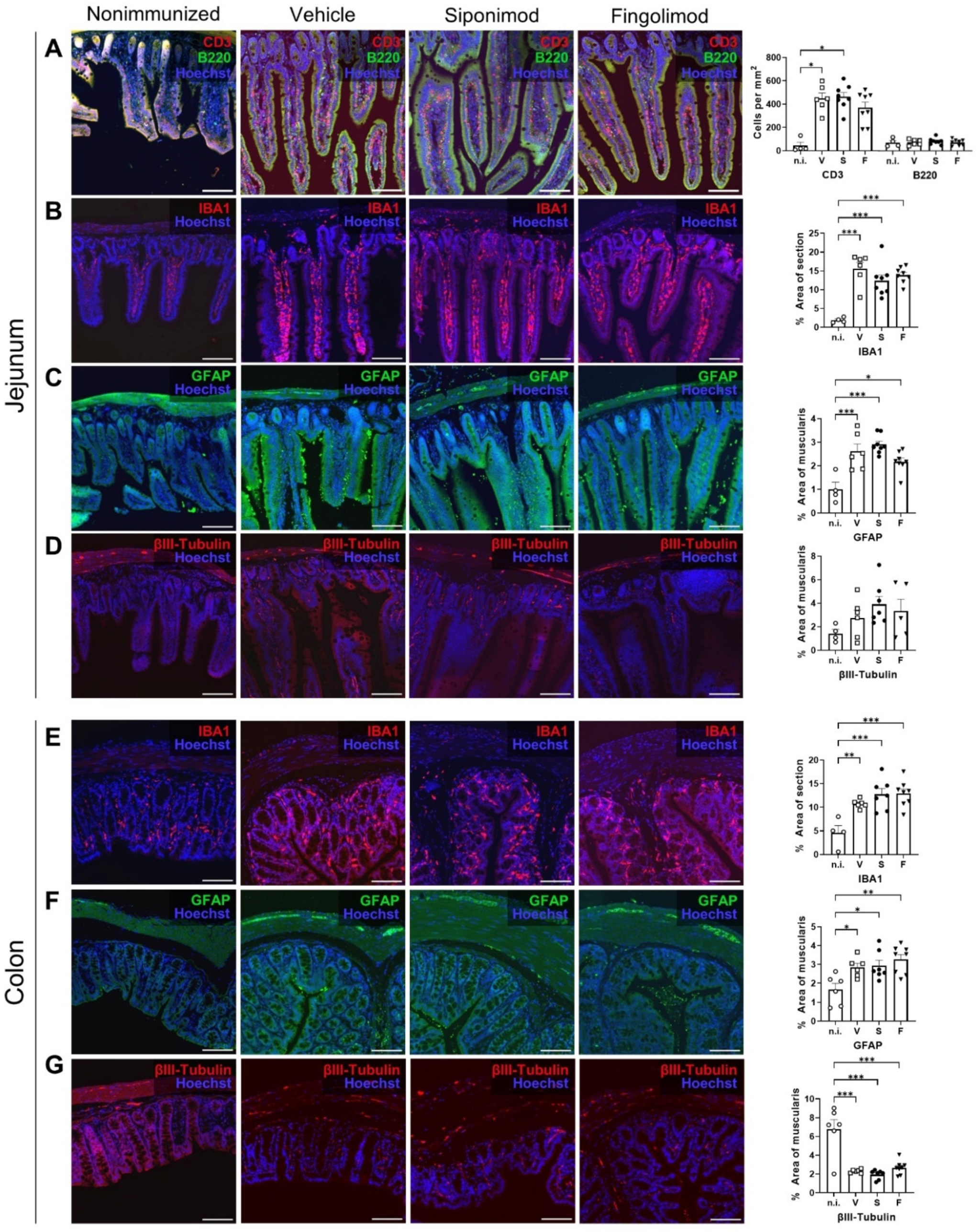
2.3. Siponimod and Fingolimod Do Not Have an Impact on the Pathology of the Enteric Nervous System (ENS) in Chronic Experimental Autoimmune Encephalomyelitis (EAE)
2.4. Siponimod Has Limited Effects on Gene Expression in Enteric Neurons and Glial Cells
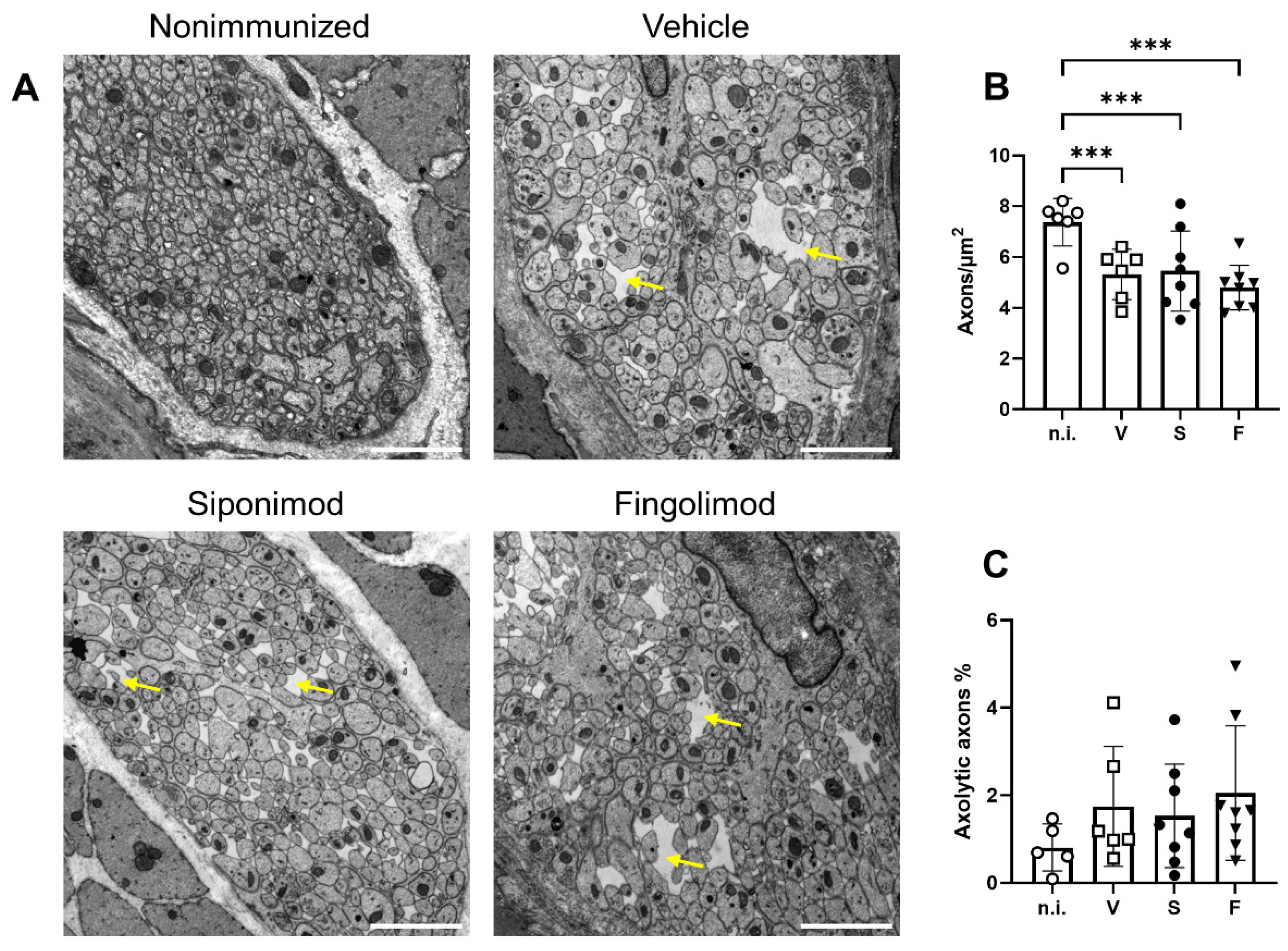
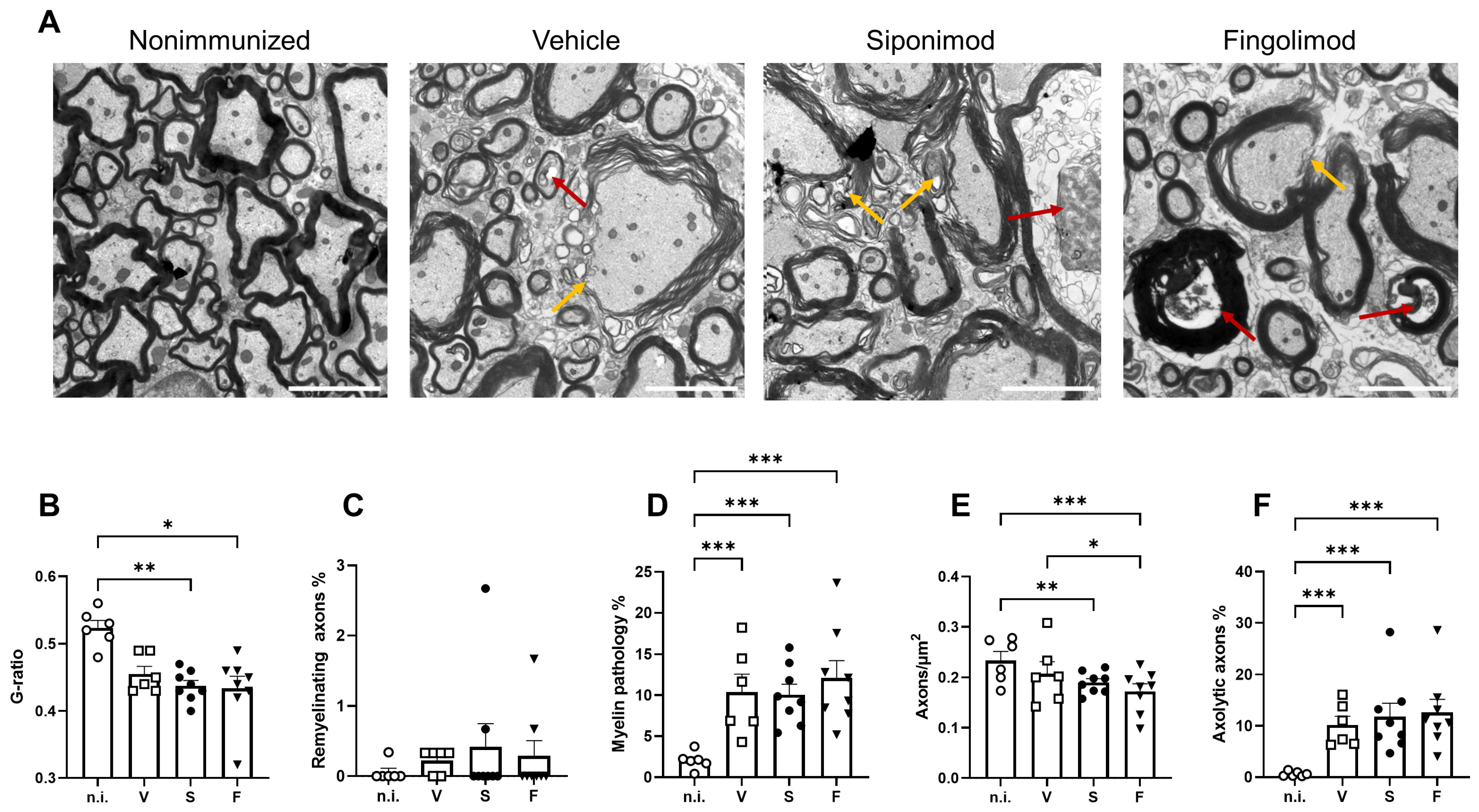
2.5. Siponimod and Fingolimod Do Not Affect Central Nervous System Pathology in Chronic Experimental Autoimmune Encephalomyelitis (EAE)
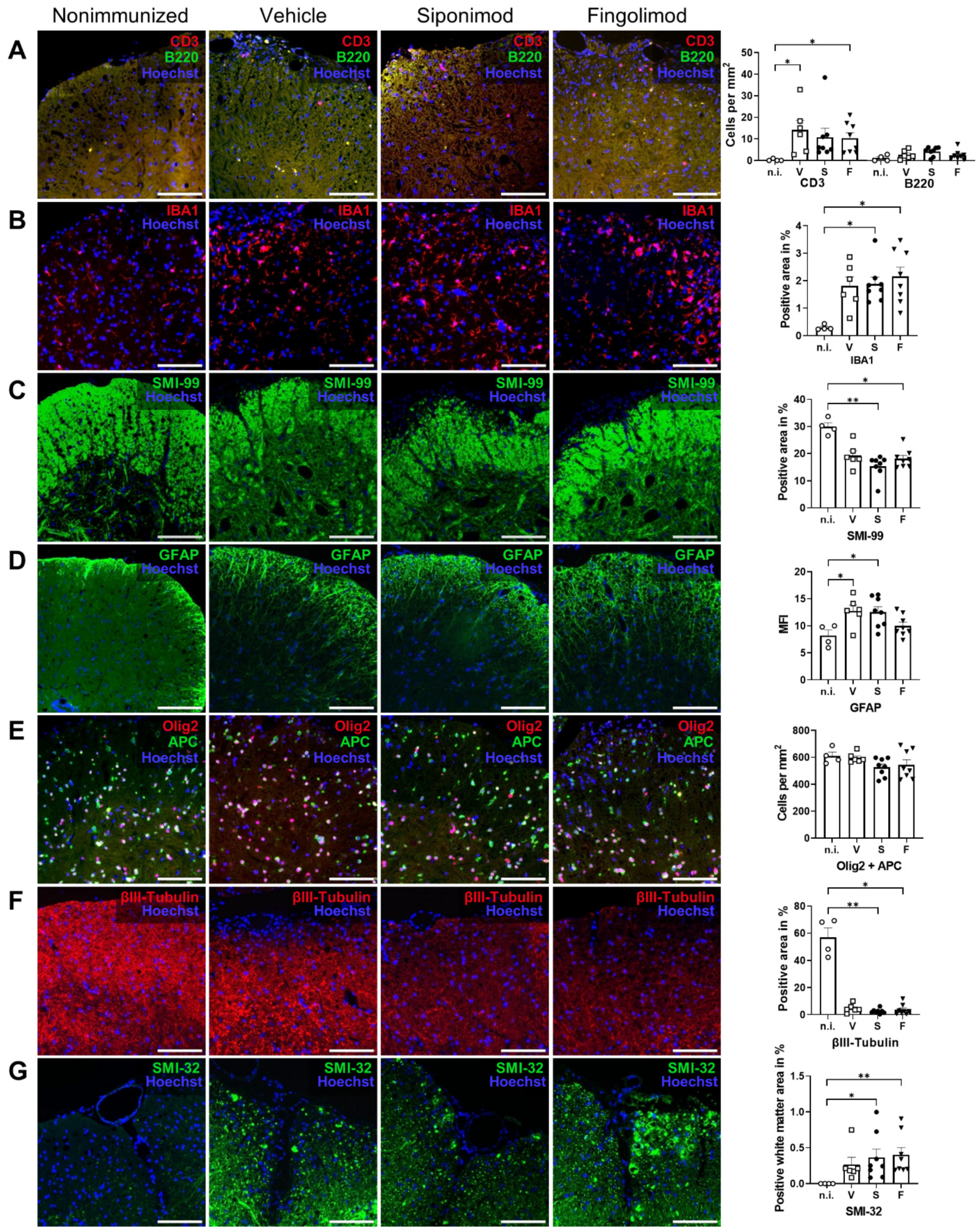
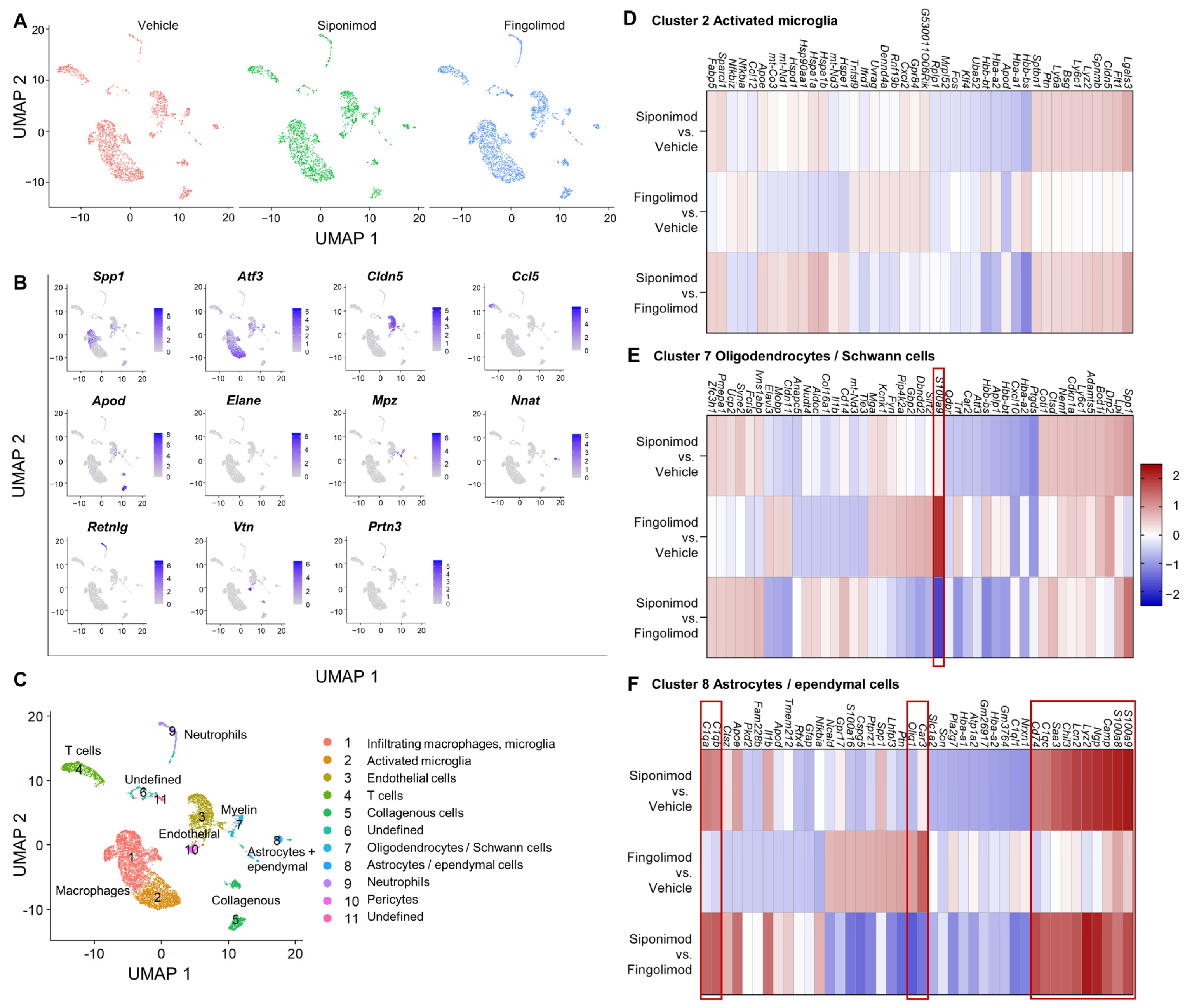
2.6. Siponimod and Fingolimod Do Not Induce Pro-Myelinating Gene Expression but Affect Immune Regulation
3. Discussion
4. Materials and Methods
4.1. Experimental Autoimmune Encephalomyelitis (EAE) Induction, Treatment and Verification of Treatment Success
4.2. Perfusion Fixation and Tissue Embedding
4.3. Mass Spectrometry
4.4. Primary ENS Culture
4.5. PCR
4.6. EM Image Analysis
4.7. IHC
4.8. ScRNA-Seq
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pugliatti, M.; Rosati, G.; Carton, H.; Riise, T.; Drulovic, J.; Vecsei, L.; Milanov, I. The Epidemiology of Multiple Sclerosis in Europe. Eur. J. Neurol. 2006, 13, 700–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walton, C.; King, R.; Rechtman, L.; Kaye, W.; Leray, E.; Marrie, R.A.; Robertson, N.; La Rocca, N.; Uitdehaag, B.; van der Mei, I.; et al. Rising Prevalence of Multiple Sclerosis Worldwide: Insights from the Atlas of MS, Third Edition. Mult. Scler. 2020, 26, 1816–1821. [Google Scholar] [CrossRef] [PubMed]
- Kalincik, T. Multiple Sclerosis Relapses: Epidemiology, Outcomes and Management. A Systematic Review. Neuroepidemiology 2015, 44, 199–214. [Google Scholar] [CrossRef] [PubMed]
- Bjornevik, K.; Cortese, M.; Healy, B.C.; Kuhle, J.; Mina, M.J.; Leng, Y.; Elledge, S.J.; Niebuhr, D.W.; Scher, A.I.; Munger, K.L.; et al. Longitudinal Analysis Reveals High Prevalence of Epstein-Barr Virus Associated with Multiple Sclerosis. Science 2022, 375, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Doshi, A.; Chataway, J. Multiple Sclerosis, a Treatable Disease. Clin. Med. 2016, 16, s53–s59. [Google Scholar] [CrossRef] [PubMed]
- Travers, B.S.; Tsang, B.K.-T.; Barton, J.L. Multiple Sclerosis: Diagnosis, Disease-Modifying Therapy and Prognosis. Aust. J. Gen. Pract. 2022, 51, 199–206. [Google Scholar] [CrossRef]
- Vidal-Jordana, A.; Montalban, X. Multiple Sclerosis. Neuroimaging Clin. N. Am. 2017, 27, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Cree, B.A.C.; Arnold, D.L.; Chataway, J.; Chitnis, T.; Fox, R.J.; Pozo Ramajo, A.; Murphy, N.; Lassmann, H. Secondary Progressive Multiple Sclerosis: New Insights. Neurology 2021, 97, 378–388. [Google Scholar] [CrossRef]
- Kappos, L.; Bar-Or, A.; Cree, B.A.C.; Fox, R.J.; Giovannoni, G.; Gold, R.; Vermersch, P.; Arnold, D.L.; Arnould, S.; Scherz, T.; et al. Siponimod versus Placebo in Secondary Progressive Multiple Sclerosis (EXPAND): A Double-Blind, Randomised, Phase 3 Study. Lancet 2018, 391, 1263–1273. [Google Scholar] [CrossRef]
- Al-Salama, Z.T. Siponimod: First Global Approval. Drugs 2019, 79, 1009–1015. [Google Scholar] [CrossRef]
- Gergely, P.; Nuesslein-Hildesheim, B.; Guerini, D.; Brinkmann, V.; Traebert, M.; Bruns, C.; Pan, S.; Gray, N.; Hinterding, K.; Cooke, N.; et al. The Selective Sphingosine 1-phosphate Receptor Modulator BAF312 Redirects Lymphocyte Distribution and Has Species-specific Effects on Heart Rate. Br. J. Pharmacol. 2012, 167, 1035–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matloubian, M.; Lo, C.G.; Cinamon, G.; Lesneski, M.J.; Xu, Y.; Brinkmann, V.; Allende, M.L.; Proia, R.L.; Cyster, J.G. Lymphocyte Egress from Thymus and Peripheral Lymphoid Organs Is Dependent on S1P Receptor 1. Nature 2004, 427, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Im, D.S.; Heise, C.E.; Ancellin, N.; O’Dowd, B.F.; Shei, G.J.; Heavens, R.P.; Rigby, M.R.; Hla, T.; Mandala, S.; McAllister, G.; et al. Characterization of a Novel Sphingosine 1-Phosphate Receptor, Edg-8. J. Biol. Chem. 2000, 275, 14281–14286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novgorodov, A.S.; El-Alwani, M.; Bielawski, J.; Obeid, L.M.; Gudz, T.I. Activation of Sphingosine-1-Phosphate Receptor S1P5 Inhibits Oligodendrocyte Progenitor Migration. FASEB J. 2007, 21, 1503–1514. [Google Scholar] [CrossRef]
- Mannioui, A.; Vauzanges, Q.; Fini, J.B.; Henriet, E.; Sekizar, S.; Azoyan, L.; Thomas, J.L.; Pasquier, D.D.; Giovannangeli, C.; Demeneix, B.; et al. The Xenopus Tadpole: An in Vivo Model to Screen Drugs Favoring Remyelination. Mult. Scler. 2018, 24, 1421–1432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, E.; Bassani, C.; De Angelis, A.; Ruffini, F.; Ottoboni, L.; Comi, G.; Martino, G.; Farina, C. Siponimod (BAF312) Activates Nrf2 While Hampering NFκB in Human Astrocytes, and Protects From Astrocyte-Induced Neurodegeneration. Front. Immunol. 2020, 11, 635. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, C.; Schubart, A.; Mir, A.K.; Dev, K.K. The Dual S1PR1/S1PR5 Drug BAF312 (Siponimod) Attenuates Demyelination in Organotypic Slice Cultures. J. Neuroinflammation 2016, 13, 31. [Google Scholar] [CrossRef] [Green Version]
- Uosef, A.; Vaughn, N.; Chu, X.; Elshawwaf, M.; Abdelshafy, A.A.A.; Elsaid, K.M.K.; Ghobrial, R.M.; Kloc, M. Siponimod (Mayzent) Downregulates RhoA and Cell Surface Expression of the S1P1 and CX3CR1 Receptors in Mouse RAW 264.7 Macrophages. Arch. Immunol. Ther. Exp. 2020, 68, 19. [Google Scholar] [CrossRef]
- Gentile, A.; Musella, A.; Bullitta, S.; Fresegna, D.; De Vito, F.; Fantozzi, R.; Piras, E.; Gargano, F.; Borsellino, G.; Battistini, L.; et al. Siponimod (BAF312) Prevents Synaptic Neurodegeneration in Experimental Multiple Sclerosis. J. Neuroinflammation 2016, 13, 207. [Google Scholar] [CrossRef] [Green Version]
- Levinthal, D.J.; Rahman, A.; Nusrat, S.; O’Leary, M.; Heyman, R.; Bielefeldt, K. Adding to the Burden: Gastrointestinal Symptoms and Syndromes in Multiple Sclerosis. Mult. Scler. Int. 2013, 2013, 319201. [Google Scholar] [CrossRef]
- Almeida, M.N.; Silvernale, C.; Kuo, B.; Staller, K. Bowel Symptoms Predate the Diagnosis among Many Patients with Multiple Sclerosis: A 14-year Cohort Study. Neurogastroenterol. Motil. 2019, 31, e13592. [Google Scholar] [CrossRef] [PubMed]
- Kurtzke, J.F. Rating Neurologic Impairment in Multiple Sclerosis: An Expanded Disability Status Scale (EDSS). Neurology 1983, 33, 1444–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wunsch, M.; Jabari, S.; Voussen, B.; Enders, M.; Srinivasan, S.; Cossais, F.; Wedel, T.; Boettner, M.; Schwarz, A.; Weyer, L.; et al. The Enteric Nervous System Is a Potential Autoimmune Target in Multiple Sclerosis. Acta Neuropathol. 2017, 134, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.; Gershon, M.D. The Bowel and beyond: The Enteric Nervous System in Neurological Disorders. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 517–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furness, J.B.; Callaghan, B.P.; Rivera, L.R.; Cho, H.-J. The Enteric Nervous System and Gastrointestinal Innervation: Integrated Local and Central Control. Adv. Exp. Med. Biol. 2014, 817, 39–71. [Google Scholar] [CrossRef] [PubMed]
- Kuerten, S.; Lichtenegger, F.S.; Faas, S.; Angelov, D.N.; Tary-Lehmann, M.; Lehmann, P.V. MBP-PLP Fusion Protein-Induced EAE in C57BL/6 Mice. J. Neuroimmunol. 2006, 177, 99–111. [Google Scholar] [CrossRef]
- Kuerten, S.; Gruppe, T.L.; Laurentius, L.-M.; Kirch, C.; Tary-Lehmann, M.; Lehmann, P.V.; Addicks, K. Differential Patterns of Spinal Cord Pathology Induced by MP4, MOG Peptide 35-55, and PLP Peptide 178-191 in C57BL/6 Mice. APMIS 2011, 119, 336–346. [Google Scholar] [CrossRef]
- Prinz, J.; Karacivi, A.; Stormanns, E.R.; Recks, M.S.; Kuerten, S. Time-Dependent Progression of Demyelination and Axonal Pathology in MP4-Induced Experimental Autoimmune Encephalomyelitis. PLoS ONE 2015, 10, e0144847. [Google Scholar] [CrossRef] [Green Version]
- Spear, E.T.; Holt, E.A.; Joyce, E.J.; Haag, M.M.; Mawe, S.M.; Hennig, G.W.; Lavoie, B.; Applebee, A.M.; Teuscher, C.; Mawe, G.M. Altered Gastrointestinal Motility Involving Autoantibodies in the Experimental Autoimmune Encephalomyelitis Model of Multiple Sclerosis. Neurogastroenterol. Motil. 2018, 30, e13349. [Google Scholar] [CrossRef]
- Kicherer, J.; Weier, A.; Enders, M.; Neuhuber, W.; Heider, T.; Kuerten, S. Characterization of Neurochemical Signature Alterations in the Enteric Nervous System in Autoimmune Encephalomyelitis. Appl. Sci. 2022, 12, 5974. [Google Scholar] [CrossRef]
- Lublin, F.; Miller, D.H.; Freedman, M.S.; Cree, B.A.C.; Wolinsky, J.S.; Weiner, H.; Lubetzki, C.; Hartung, H.-P.; Montalban, X.; Uitdehaag, B.M.J.; et al. Oral Fingolimod in Primary Progressive Multiple Sclerosis (INFORMS): A Phase 3, Randomised, Double-Blind, Placebo-Controlled Trial. Lancet 2016, 387, 1075–1084. [Google Scholar] [CrossRef]
- Bigaud, M.; Rudolph, B.; Briard, E.; Beerli, C.; Hofmann, A.; Hermes, E.; Muellershausen, F.; Schubart, A.; Gardin, A. Siponimod (BAF312) Penetrates, Distributes, and Acts in the Central Nervous System: Preclinical Insights. Mult. Scler. J. Exp. Transl. Clin. 2021, 7, 20552173211049170. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.H.; Ngwainmbi, J.; Grider, J.R.; Dewey, W.L.; Akbarali, H.I. An In-Vitro Preparation of Isolated Enteric Neurons and Glia from the Myenteric Plexus of the Adult Mouse. JoVE 2013, 78, 50688. [Google Scholar] [CrossRef]
- Veríssimo, C.P.; Carvalho, J.D.S.; da Silva, F.J.M.; Campanati, L.; Moura-Neto, V.; Coelho-Aguiar, J. de M. Laminin and Environmental Cues Act in the Inhibition of the Neuronal Differentiation of Enteric Glia in Vitro. Front. Neurosci. 2019, 13, 914. [Google Scholar] [CrossRef] [PubMed]
- Neill, T.; Schaefer, L.; Iozzo, R.V. Decorin: A Guardian from the Matrix. Am. J. Pathol. 2012, 181, 380–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuerten, S.; Kostova-Bales, D.A.; Frenzel, L.P.; Tigno, J.T.; Tary-Lehmann, M.; Angelov, D.N.; Lehmann, P.V. MP4- and MOG:35-55-Induced EAE in C57BL/6 Mice Differentially Targets Brain, Spinal Cord and Cerebellum. J. Neuroimmunol. 2007, 189, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Lang, J.; Maeda, Y.; Bannerman, P.; Xu, J.; Horiuchi, M.; Pleasure, D.; Guo, F. Adenomatous Polyposis Coli Regulates Oligodendroglial Development. J. Neurosci. 2013, 33, 3113–3130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsang, Y.M.; Chiong, F.; Kuznetsov, D.; Kasarskis, E.; Geula, C. Motor Neurons Are Rich in Non-Phosphorylated Neurofilaments: Cross-Species Comparison and Alterations in ALS. Brain Res. 2000, 861, 45–58. [Google Scholar] [CrossRef]
- Mancardi, G.; Hart, B.; Roccatagliata, L.; Brok, H.; Giunti, D.; Bontrop, R.; Massacesi, L.; Capello, E.; Uccelli, A. Demyelination and Axonal Damage in a Non-Human Primate Model of Multiple Sclerosis. J. Neurol. Sci. 2001, 184, 41–49. [Google Scholar] [CrossRef]
- Wu, M.; Xu, L.; Wang, Y.; Zhou, N.; Zhen, F.; Zhang, Y.; Qu, X.; Fan, H.; Liu, S.; Chen, Y.; et al. S100A8/A9 Induces Microglia Activation and Promotes the Apoptosis of Oligodendrocyte Precursor Cells by Activating the NF-ΚB Signaling Pathway. Brain Res. Bull. 2018, 143, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Bhusal, A.; Nam, Y.; Seo, D.; Rahman, M.H.; Hwang, E.M.; Kim, S.; Lee, W.; Suk, K. Cathelicidin-related Antimicrobial Peptide Promotes Neuroinflammation through Astrocyte–Microglia Communication in Experimental Autoimmune Encephalomyelitis. Glia 2022, 70, 1902–1926. [Google Scholar] [CrossRef] [PubMed]
- Fryer, R.M.; Muthukumarana, A.; Harrison, P.C.; Nodop Mazurek, S.; Chen, R.R.; Harrington, K.E.; Dinallo, R.M.; Horan, J.C.; Patnaude, L.; Modis, L.K.; et al. The Clinically-Tested S1P Receptor Agonists, FTY720 and BAF312, Demonstrate Subtype-Specific Bradycardia (S1P1) and Hypertension (S1P3) in Rat. PLoS ONE 2012, 7, e52985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spampinato, S.F.; Merlo, S.; Costantino, G.; Sano, Y.; Kanda, T.; Sortino, M.A. Decreased Astrocytic CCL2 Accounts for BAF-312 Effect on PBMCs Transendothelial Migration Through a Blood Brain Barrier in Vitro Model. J. Neuroimmune Pharmacol. 2021, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Gudi, V.; Gingele, S.; Skripuletz, T.; Stangel, M. Glial Response during Cuprizone-Induced de- and Remyelination in the CNS: Lessons Learned. Front. Cell. Neurosci. 2014, 8, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Sullivan, C.; Dev, K.K. Galactosylsphingosine (Psychosine)-Induced Demyelination Is Attenuated by Sphingosine 1-Phosphate Signalling. J. Cell Sci. 2015, 128, 3878–3887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietrich, M.; Hecker, C.; Martin, E.; Langui, D.; Gliem, M.; Stankoff, B.; Lubetzki, C.; Gruchot, J.; Göttle, P.; Issberner, A.; et al. Increased Remyelination and Proregenerative Microglia Under Siponimod Therapy in Mechanistic Models. Neurol. Neuroimmunol. Neuroinflamm. 2022, 9, 131–145. [Google Scholar] [CrossRef]
- Hundehege, P.; Cerina, M.; Eichler, S.; Thomas, C.; Herrmann, A.; Göbel, K.; Müntefering, T.; Fernandez-Orth, J.; Bock, S.; Narayanan, V.; et al. The Next-Generation Sphingosine-1 Receptor Modulator BAF312 (Siponimod) Improves Cortical Network Functionality in Focal Autoimmune Encephalomyelitis. Neural Regen. Res. 2019, 14, 1950. [Google Scholar] [CrossRef]
- Brand, R.M.; Diddens, J.; Friedrich, V.; Pfaller, M.; Radbruch, H.; Hemmer, B.; Steiger, K.; Lehmann-Horn, K. Siponimod Inhibits the Formation of Meningeal Ectopic Lymphoid Tissue in Experimental Autoimmune Encephalomyelitis. Neurol. Neuroimmunol. Neuroinflamm. 2022, 9, e1117. [Google Scholar] [CrossRef]
- Ward, L.A.; Lee, D.S.W.; Sharma, A.; Wang, A.; Naouar, I.; Ma, X.I.; Pikor, N.; Nuesslein-Hildesheim, B.; Ramaglia, V.; Gommerman, J.L. Siponimod Therapy Implicates Th17 Cells in a Preclinical Model of Subpial Cortical Injury. JCI Insight 2020, 5, e132522. [Google Scholar] [CrossRef] [Green Version]
- Kuerten, S.; Javeri, S.; Tary-Lehmann, M.; Lehmann, P.V.; Angelov, D.N. Fundamental Differences in the Dynamics of CNS Lesion Development and Composition in MP4- and MOG Peptide 35-55-Induced Experimental Autoimmune Encephalomyelitis. Clin. Immunol. 2008, 129, 256–267. [Google Scholar] [CrossRef]
- Bail, K.; Notz, Q.; Rovituso, D.M.; Schampel, A.; Wunsch, M.; Koeniger, T.; Schropp, V.; Bharti, R.; Scholz, C.-J.; Foerstner, K.U.; et al. Differential Effects of FTY720 on the B Cell Compartment in a Mouse Model of Multiple Sclerosis. J. Neuroinflammation 2017, 14, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haist, V.; Ulrich, R.; Kalkuhl, A.; Deschl, U.; Baumgärtner, W. Distinct Spatio-Temporal Extracellular Matrix Accumulation within Demyelinated Spinal Cord Lesions in Theiler’s Murine Encephalomyelitis. Brain Pathol. 2012, 22, 188–204. [Google Scholar] [CrossRef]
- Goncalves, M.B.; Wu, Y.; Clarke, E.; Grist, J.; Hobbs, C.; Trigo, D.; Jack, J.; Corcoran, J.P.T. Regulation of Myelination by Exosome Associated Retinoic Acid Release from NG2-Positive Cells. J. Neurosci. 2019, 39, 3013–3027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starossom, S.C.; Campo Garcia, J.; Woelfle, T.; Romero-Suarez, S.; Olah, M.; Watanabe, F.; Cao, L.; Yeste, A.; Tukker, J.J.; Quintana, F.J.; et al. Chi3l3 Induces Oligodendrogenesis in an Experimental Model of Autoimmune Neuroinflammation. Nat. Commun. 2019, 10, 217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassatella, M.A.; Östberg, N.K.; Tamassia, N.; Soehnlein, O. Biological Roles of Neutrophil-Derived Granule Proteins and Cytokines. Trends Immunol. 2019, 40, 648–664. [Google Scholar] [CrossRef] [PubMed]
- Mollinedo, F. Neutrophil Degranulation, Plasticity, and Cancer Metastasis. Trends Immunol. 2019, 40, 228–242. [Google Scholar] [CrossRef] [PubMed]
- Cougnoux, A.; Yerger, J.C.; Fellmeth, M.; Serra-Vinardell, J.; Wassif, C.A.; Cawley, N.X.; Porter, F.D. Toll-like Receptor Mediated Lysozyme Expression in Niemann-Pick Disease, Type C1. Mol. Genet. Metab. 2020, 131, 364–366. [Google Scholar] [CrossRef]
- Berard, J.L.; Zarruk, J.G.; Arbour, N.; Prat, A.; Yong, V.W.; Jacques, F.H.; Akira, S.; David, S. Lipocalin 2 Is a Novel Immune Mediator of Experimental Autoimmune Encephalomyelitis Pathogenesis and Is Modulated in Multiple Sclerosis. Glia 2012, 60, 1145–1159. [Google Scholar] [CrossRef]
- Al Nimer, F.; Elliott, C.; Bergman, J.; Khademi, M.; Dring, A.M.; Aeinehband, S.; Bergenheim, T.; Romme Christensen, J.; Sellebjerg, F.; Svenningsson, A.; et al. Lipocalin-2 Is Increased in Progressive Multiple Sclerosis and Inhibits Remyelination. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e191. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-Y.; Hall, J.A.; Kroehling, L.; Wu, L.; Najar, T.; Nguyen, H.H.; Lin, W.-Y.; Yeung, S.T.; Silva, H.M.; Li, D.; et al. Serum Amyloid A Proteins Induce Pathogenic Th17 Cells and Promote Inflammatory Disease. Cell 2020, 180, 79–91.e16. [Google Scholar] [CrossRef]
- Prineas, J.W.; Kwon, E.E.; Goldenberg, P.Z.; Ilyas, A.A.; Quarles, R.H.; Benjamins, J.A.; Sprinkle, T.J. Multiple Sclerosis. Oligodendrocyte Proliferation and Differentiation in Fresh Lesions. Lab. Investig. 1989, 61, 489–503. [Google Scholar] [PubMed]
- Nógrádi, A.; Domoki, F.; Dégi, R.; Borda, S.; Pákáski, M.; Szabó, A.; Bari, F. Up-Regulation of Cerebral Carbonic Anhydrase by Anoxic Stress in Piglets. J. Neurochem. 2003, 85, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Selmaj, K.; Li, D.K.B.; Hartung, H.-P.; Hemmer, B.; Kappos, L.; Freedman, M.S.; Stüve, O.; Rieckmann, P.; Montalban, X.; Ziemssen, T.; et al. Siponimod for Patients with Relapsing-Remitting Multiple Sclerosis (BOLD): An Adaptive, Dose-Ranging, Randomised, Phase 2 Study. Lancet Neurol. 2013, 12, 756–767. [Google Scholar] [CrossRef]
- Kappos, L.; Radue, E.-W.; O’Connor, P.; Polman, C.; Hohlfeld, R.; Calabresi, P.; Selmaj, K.; Agoropoulou, C.; Leyk, M.; Zhang-Auberson, L.; et al. A Placebo-Controlled Trial of Oral Fingolimod in Relapsing Multiple Sclerosis. N. Engl. J. Med. 2010, 362, 387–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving Bioscience Research Reporting: The ARRIVE Guidelines for Reporting Animal Research. PLoS Biol. 2010, 8, e1000412. [Google Scholar] [CrossRef] [PubMed]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M.; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e21. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cohort 1 | Cohort 2 | ||||||
|---|---|---|---|---|---|---|---|
| Treatment | Vehicle | Siponimod | Fingolimod | Vehicle | Siponimod | Fingolimod | Total |
| Number of mice (n) | 6 | 8 | 8 | 6 | 6 | 6 | 40 |
| EAE onset (d.p.i.) | 12.00 ± 1.03 | 11.75 ± 0.56 | 11.00 ± 0.65 | 12.00 ± 0.45 | 11.50 ± 1.02 | 12.33 ± 0.92 | 11.73 ± 0.30 |
| Maximum EAE score | 2.75 ± 0.28 | 2.63 ± 0.26 | 2.81 ± 0.27 | 2.79 ± 0.16 | 2.88 ± 0.20 | 2.88 ± 0.15 | 2.78 ± 0.09 |
| Score before treatment | 1.83 ± 0.21 | 1.56 ± 0.24 | 1.81 ± 0.27 | 2.21 ± 0.28 | 1.79 ± 0.16 | 2.29 ± 0.14 | 1.89 ± 0.10 |
| Final score | 1.71 ± 0.28 | 1.47 ± 0.24 | 1.72 ± 0.19 | 2.04 ± 0.25 | 2.04 ± 0.16 | 2.17 ± 0.17 | 1.83 ± 0.09 |
| Score difference | −0.13 ± 0.15 | −0.09 ± 0.25 | −0.09 ± 0.18 | −0.17 ± 0.23 | +0.25 ± 0.13 | −0.13 ± 0.15 | −0.06 ± 0.08 |
| Weight before treatment (g) | 22.17 ± 0.92 | 22.36 ± 0.48 | 22.61 ± 0.43 | 22.85 ± 0.51 | 22.22 ± 0.32 | 22.22 ± 0.64 | 22.41 ± 0.22 |
| Final weight (g) | 22.77 ± 1.02 | 23.96 ± 0.67 | 22.96 ± 0.57 | 23.40 ± 0.48 | 23.72 ± 0.39 | 22.75 ± 0.64 | 23.28 ± 0.26 |
| Weight difference (g) | +0.60 ± 0.40 | +1.60 ± 0.52 | +0.35 ± 0.25 | +0.55 ± 0.31 | +1.50 ± 0.31 | +0.53 ± 0.26 | +0.87 ± 0.163 |
| Gene Name | Protein Name | logFCSiponimod/Vehicle | logFCFingolimod/Vehicle | logFCSiponimod/Fingolimod |
|---|---|---|---|---|
| S100a9 | S100A9, Calprotectin | 2.25 | 0.50 | 1.75 |
| S100a8 | S100A8, Calgranulin-A | 2.18 | 0.85 | 1.33 |
| Camp | Cethelicidin antimicrobial protein | 2.11 | 0.48 | 1.63 * |
| Ngp | Neutrophilic granule peptide | 2.04 | −0.10 | 2.14 * |
| Lyz2 | Lysozyme C2 | 1.95 | −0.31 | 2.26 |
| Lcn2 | Lipocalin-2 | 1.87 | 0.20 | 1.67 |
| Chil3 | Chitinase-like protein 3 | 1.67 | 0.12 | 1.5 |
| Saa3 | Serum amyloid A-3 protein | 1.56 | 0.01 | 1.55 |
| C1qc | Complement C1q subunit C | 1.32 | −0.23 | 1.55 |
| Cd74 | CD74 | 1.29 | −0.49 | 1.78 |
| Car3 | Carbonic anhydrase 3 | 0.19 | 1.54 | −1.35 |
| Olig1 | Oligodendrocyte transcription factor 1 | −0.66 | 0.95 | −1.61 |
| C1qb | Complement C1q subunit B | 1.16 | −0.42 | 1.58 |
| C1qa | Complement C1q subunit A | 1.25 | −0.26 | 1.52 |
| Siponimod | Fingolimod | Fingolimod-P | |
|---|---|---|---|
| Calibration Range | 1–1000 ng/mL | 0.1–1000 ng/mL | 0.5–1000 ng/mL |
| LOQ | 1 ng/mL | 0.1 ng/mL | 0.5 ng/mL |
| Accuracy (Bias) | <1.6% | <10.4% | <10.3% |
| Regression R2 | 0.99999 | 0.99925 | 0.99999 |
| Precision RSD | ±2.7% | ±9.2% | ±2.6% |
| Recovery | 93.2% | 87.8% | 89.3% |
| Initial Denaturation | 95 °C | 3 min | |
| PCR (35 cycles) | Denaturation | 95 °C | 30 s |
| Annealing | See Table 4 | 30 s | |
| Extension | 72 °C | See Table 5 | |
| Final extension | 72 °C | 10 min |
| Species | Gene | Direction | Primer Sequence | Product Size | Annealing Temp. | Extension Time | Positive Control |
|---|---|---|---|---|---|---|---|
| Mus musculus | Actb | Forward | GGCTGTATTCCCCTCCATCG | 154 bp | 55 °C | 12 s | - |
| Reverse | TTGAGCGAGGCTGCTGTTTC | ||||||
| Tubb3 | Forward | ATGAGGCCTCCTCTCACAAG | 731 bp | 56 °C | 46 s | Brain | |
| Reverse | ATCGAACATCTGCTGCGTGA | ||||||
| S1pr1 | Forward | TTGAGCGAGGCTGCTGTTTC | 466 bp | 57 °C | 30 s | Lung | |
| Reverse | CGCCTGCTAATAGGTCCGAG | ||||||
| S1pr3 | Forward | CTTCGGATTCTCTGGGGCAG | 207 bp | 56 °C | 12 s | Lung | |
| Reverse | ATAGGCTCTCGTTCTGCAAGG | ||||||
| S1pr4 | Forward | AGCCAATGGGCAGAAGTCTC | 773 bp | 57 °C | 46 s | Spleen | |
| Reverse | ACAGTAGCCTGGGCATTGAC | ||||||
| S1pr5 | Forward | CCGGTTACAGGAGACTTTTGC | 374 bp | 55 °C | 22 s | Brain | |
| Reverse | ACAGTAGGATGTTGGTGGCG | ||||||
| Rattus norvegicus | Actb | Forward | AGCCTTCCTTCCTGGGTATGG | 361 bp | 57 °C | 22 s | - |
| Reverse | GCAGCTCAGTAACAGTCCGC | ||||||
| Gfap | Forward | AACCGCATCACCATTCCTGT | 667 bp | 57 °C | 40 s | Brain | |
| Reverse | TCTGCCCTACCCACTCCTAC | ||||||
| S1pr1 | Forward | TTGAGCGAGGCTGCTGTTTC | 210 bp | 57 °C | 15 s | Lung | |
| Reverse | AGCCTTAACCACTGGGATGC | ||||||
| S1pr3 | Forward | GAACGAGAGCCTGTTTCCAAC | 611 bp | 56 °C | 40 s | Spleen | |
| Reverse | TGCTTCTTGTTGGCGTCGTA | ||||||
| S1pr4 | Forward | AACGGTTAGGCACAAGGAGG | 248 bp | 57 °C | 15 s | Spleen | |
| Reverse | TTATGCTCAAGGTGCCCCAG | ||||||
| S1pr5 | Forward | AGGCGCAAGGTTCGCATA | 450 bp | 56 °C | 30 s | Brain | |
| Reverse | AGGAACATGGGTGCATGGAA |
| UNG Incubation | Hold | 50 °C | 120 s |
| Polymerase Activation | Hold | 95 °C | 120 s |
| PCR (40 cycles) | Denaturation | 95 °C | 3 s |
| Annealing/Extension | 60 °C | 30 s | |
| Ramp Rate | 4.4 °C/s | ||
| Species | Gene | Catalog Number | Assay ID |
|---|---|---|---|
| Mus musculus | Actb | 4453320 | Mm01205647_g1 |
| Tubb3 | 4453320 | Mm00727586_s1 | |
| S1pr1 | 4453320 | Mm00514644_m1 | |
| S1pr3 | 4448892 | Mm00515669_m1 | |
| S1pr4 | 4448892 | Mm00468695_s1 | |
| S1pr5 | 4448892 | Mm00474763_m1 | |
| Rattus norvegicus | Actb | 4453320 | Rn00667869_m1 |
| Gfap | 4453320 | Rn00566603_m1 | |
| S1pr1 | 4448892 | Rn00568869_m1 | |
| S1pr3 | 4448892 | Rn01757498_m1 | |
| S1pr4 | 4448892 | Rn01408095_s1 | |
| S1pr5 | 4448892 | Rn01486961_m1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weier, A.; Enders, M.; Kirchner, P.; Ekici, A.; Bigaud, M.; Kapitza, C.; Wörl, J.; Kuerten, S. Impact of Siponimod on Enteric and Central Nervous System Pathology in Late-Stage Experimental Autoimmune Encephalomyelitis. Int. J. Mol. Sci. 2022, 23, 14209. https://doi.org/10.3390/ijms232214209
Weier A, Enders M, Kirchner P, Ekici A, Bigaud M, Kapitza C, Wörl J, Kuerten S. Impact of Siponimod on Enteric and Central Nervous System Pathology in Late-Stage Experimental Autoimmune Encephalomyelitis. International Journal of Molecular Sciences. 2022; 23(22):14209. https://doi.org/10.3390/ijms232214209
Chicago/Turabian StyleWeier, Alicia, Michael Enders, Philipp Kirchner, Arif Ekici, Marc Bigaud, Christopher Kapitza, Jürgen Wörl, and Stefanie Kuerten. 2022. "Impact of Siponimod on Enteric and Central Nervous System Pathology in Late-Stage Experimental Autoimmune Encephalomyelitis" International Journal of Molecular Sciences 23, no. 22: 14209. https://doi.org/10.3390/ijms232214209