
Neuroprotective Effects of Erinacine A on an Experimental Model of Traumatic Optic Neuropathy

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
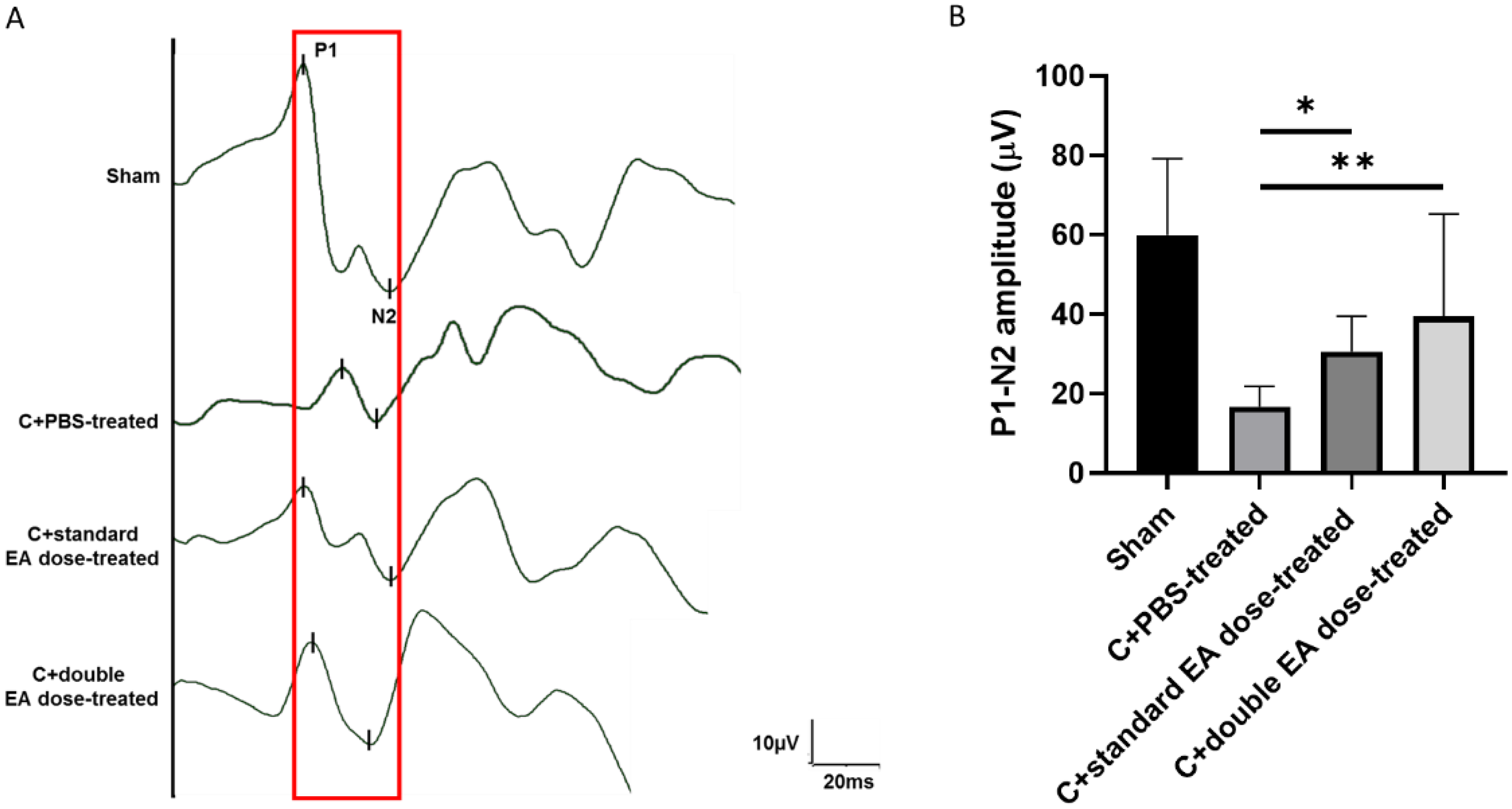
2.1. Treatment with EA Preserves Visual Function
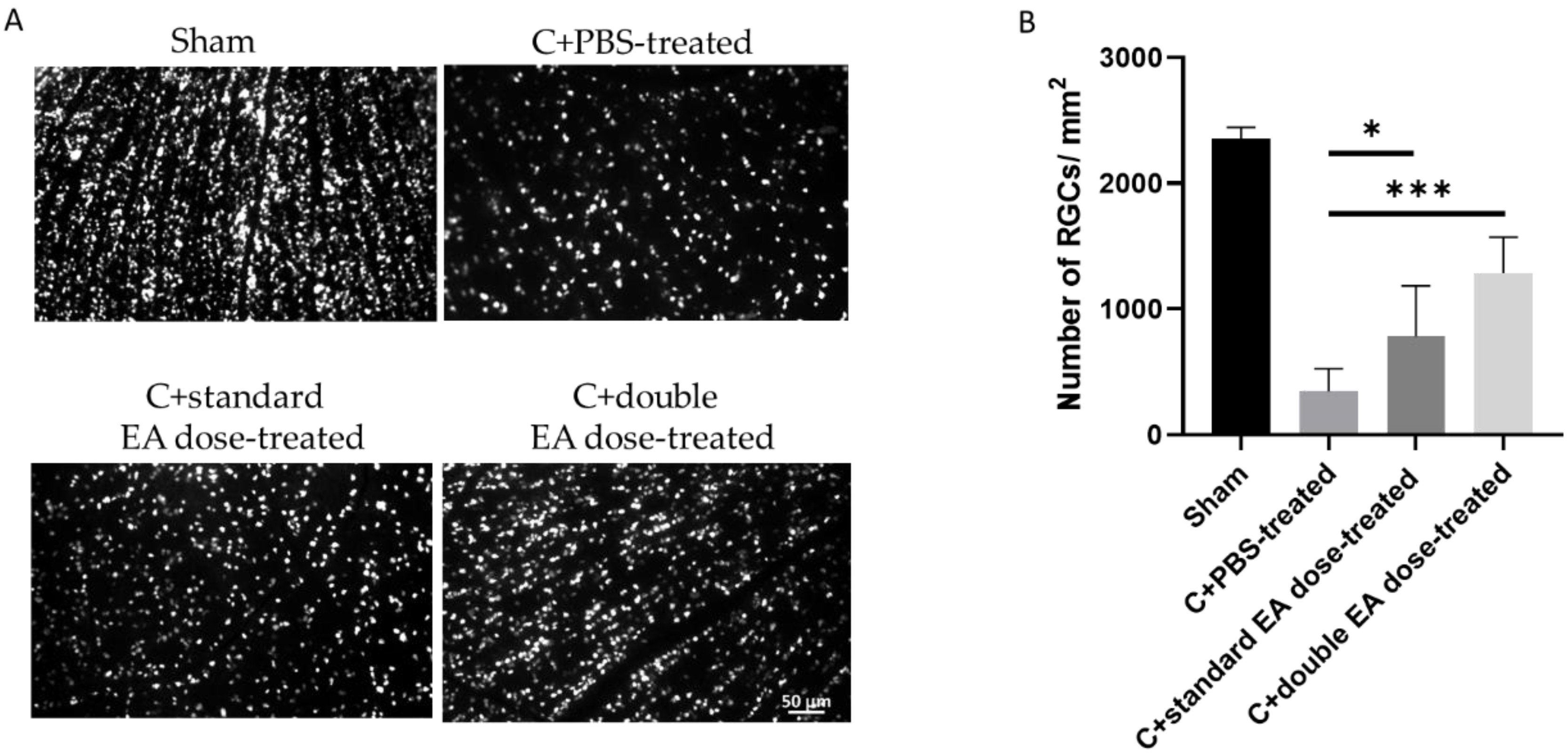
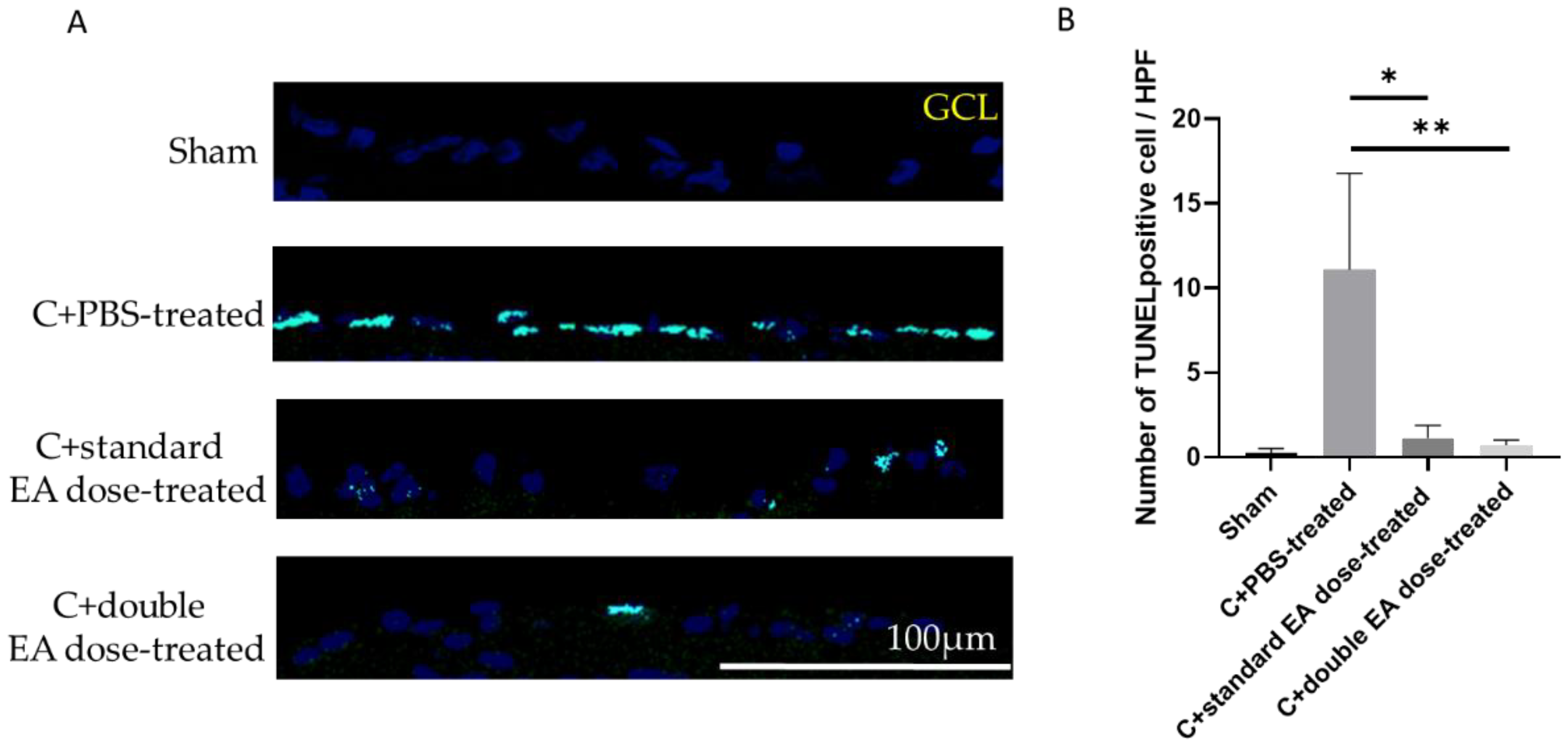
2.2. Treatment with EA Enhances RGC Survival Rate and Reduces RGC Apoptosis
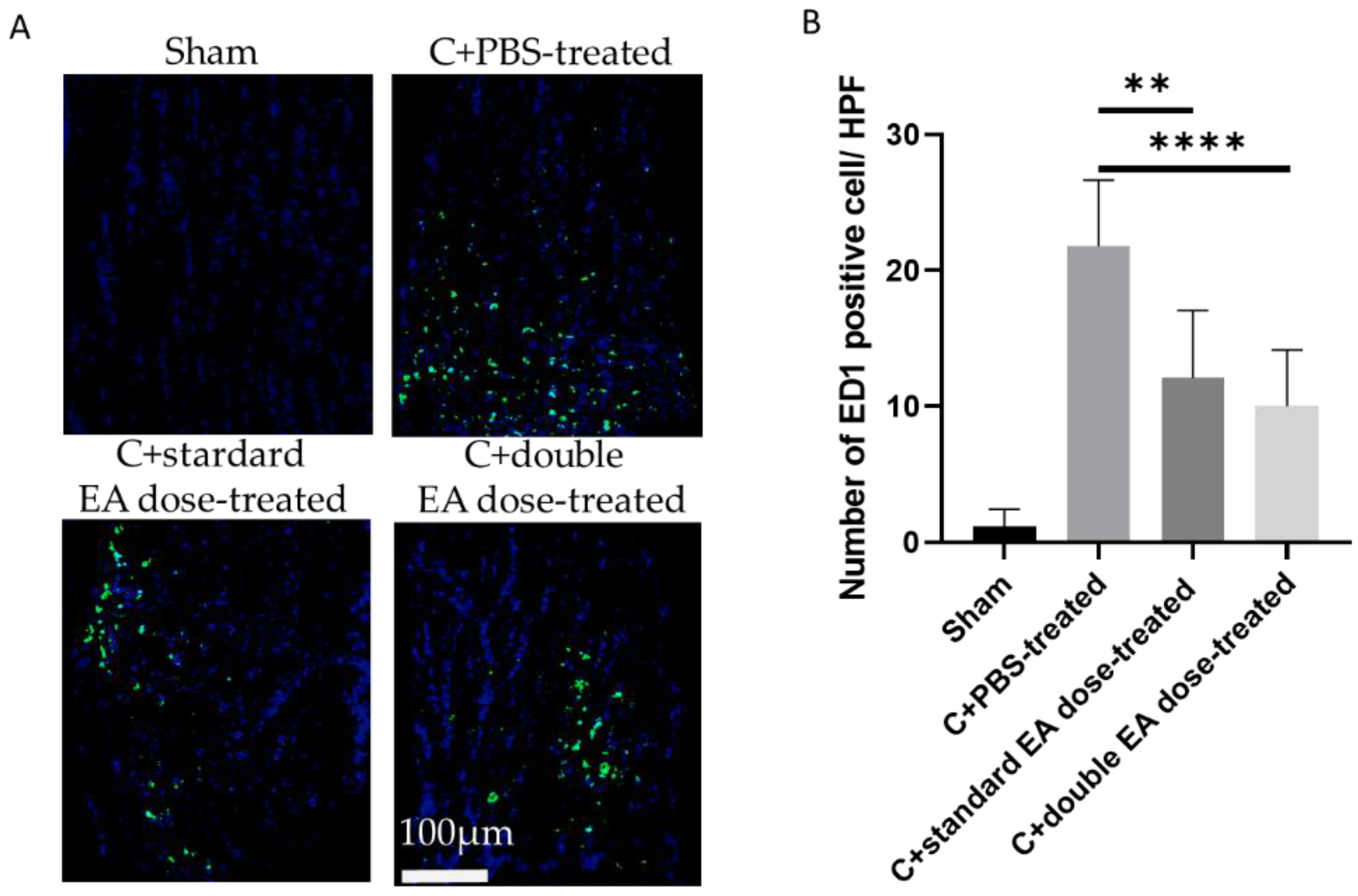
2.3. Treatment with EA Avoids Macrophage Infiltration in the on Tissue
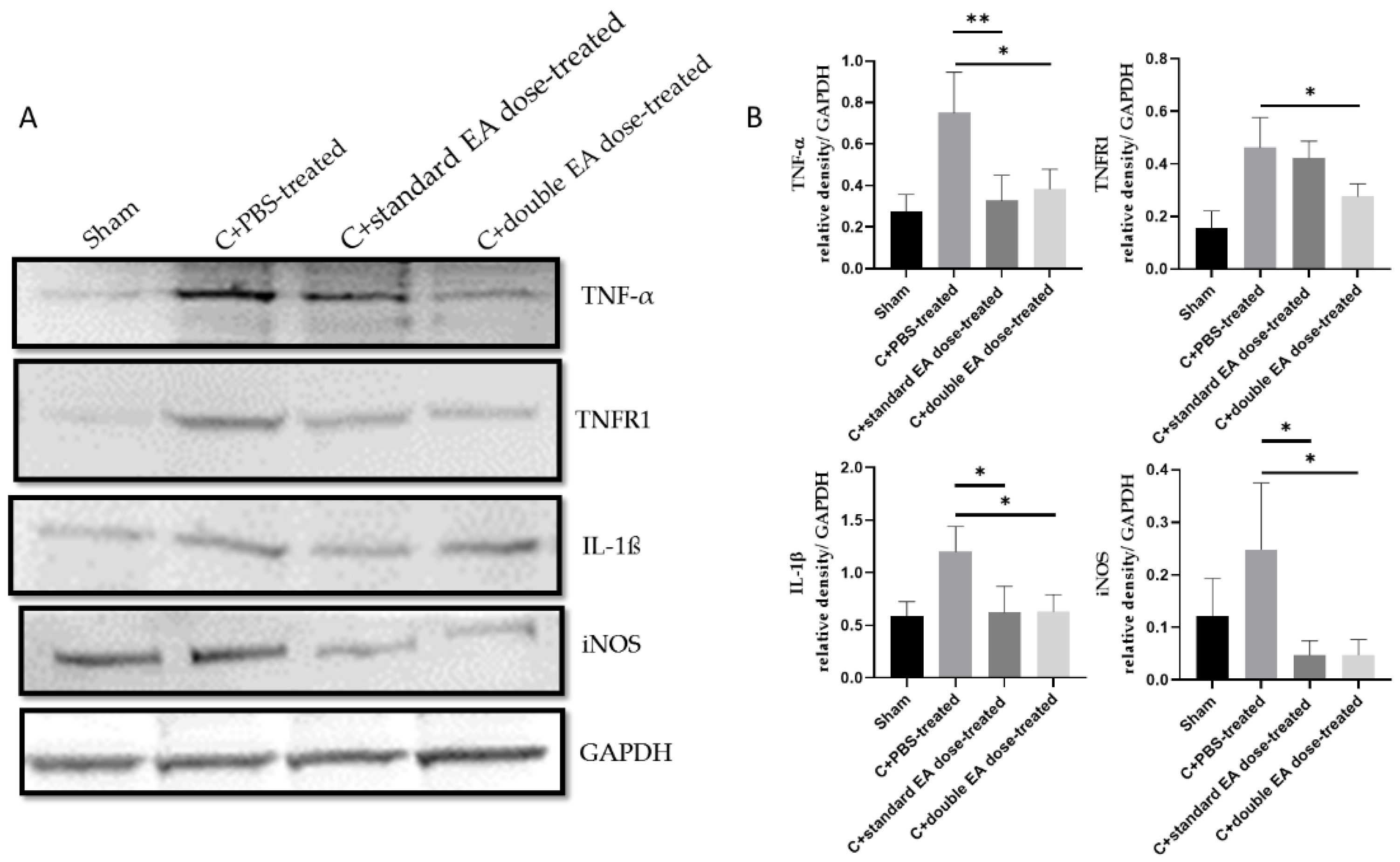
2.4. Treatment with EA Inhibits Apoptosis, Decreases Inflammation, and Enhances Antioxidative Stress Ability
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Study Design
4.3. ON Crush Model in Rats and EA Application
4.4. fVEPs
4.5. Retrograde Labeling of RGCs with Fluoro-Gold
4.6. TUNEL Assay for Apoptotic Cell Measurements
4.7. IHC of ED1
4.8. Western Blotting
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Steinsapir, K.D. Traumatic optic neuropathy. Curr. Opin. Ophthalmol. 1999, 10, 340–342. [Google Scholar] [CrossRef] [PubMed]
- Guy, W.M.; Soparkar, C.N.; Alford, E.L.; Patrinely, J.R.; Sami, M.S.; Parke, R.B. Traumatic optic neuropathy and second optic nerve injuries. JAMA Ophthalmol. 2014, 132, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Kumaran, A.M.; Sundar, G.; Chye, L.T. Traumatic optic neuropathy: A review. Craniomaxillofac. Trauma Reconstr. 2015, 8, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Tao, W.; Dvoriantchikova, G.; Tse, B.C.; Pappas, S.; Chou, T.H.; Tapia, M.; Porciatti, V.; Ivanov, D.; Tse, D.T.; Pelaez, D. A Novel Mouse Model of Traumatic Optic Neuropathy Using External Ultrasound Energy to Achieve Focal, Indirect Optic Nerve Injury. Sci. Rep. 2017, 7, 11779. [Google Scholar] [CrossRef] [PubMed]
- Ohlsson, M.; Mattsson, P.; Svensson, M. A temporal study of axonal degeneration and glial scar formation following a standardized crush injury of the optic nerve in the adult rat. Restor. Neurol. Neurosci. 2004, 22, 1–10. [Google Scholar]
- Ohlsson, M.; Westerlund, U.; Langmoen, I.A.; Svensson, M. Methylprednisolone treatment does not influence axonal regeneration or degeneration following optic nerve injury in the adult rat. J. Neuroophthalmol. 2004, 24, 11–18. [Google Scholar] [CrossRef]
- Kermer, P.; Klöcker, N.; Labes, M.; Thomsen, S.; Srinivasan, A.; Bähr, M. Activation of caspase-3 in axotomized rat retinal ganglion cells in vivo. FEBS Lett. 1999, 453, 361–364. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P. Traumatic optic neuropathy-Clinical features and management issues. Taiwan J. Ophthalmol. 2015, 5, 3–8. [Google Scholar] [CrossRef]
- Wang, A.G. How to manage traumatic optic neuropathy? Taiwan J. Ophthalmol. 2015, 5, 1–2. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P.; Griffiths, P.G. Steroids for traumatic optic neuropathy. Cochrane Database Syst. Rev. 2013, 2013, Cd006032. [Google Scholar] [CrossRef]
- Thomas, C.N.; Thompson, A.M.; Ahmed, Z.; Blanch, R.J. Retinal Ganglion Cells Die by Necroptotic Mechanisms in a Site-Specific Manner in a Rat Blunt Ocular Injury Model. Cells 2019, 8, 1517. [Google Scholar] [CrossRef]
- Degterev, A.; Ofengeim, D.; Yuan, J. Targeting RIPK1 for the treatment of human diseases. Proc. Natl. Acad. Sci. USA 2019, 116, 9714–9722. [Google Scholar] [CrossRef]
- Amin, P.; Florez, M.; Najafov, A.; Pan, H.; Geng, J.; Ofengeim, D.; Dziedzic, S.A.; Wang, H.; Barrett, V.J.; Ito, Y.; et al. Regulation of a distinct activated RIPK1 intermediate bridging complex I and complex II in TNFα-mediated apoptosis. Proc. Natl. Acad. Sci. USA 2018, 115, E5944–E5953. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Amin, P.; Ofengeim, D. Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat. Rev. Neurosci. 2019, 20, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Christofferson, D.E.; Li, Y.; Hitomi, J.; Zhou, W.; Upperman, C.; Zhu, H.; Gerber, S.A.; Gygi, S.; Yuan, J. A novel role for RIP1 kinase in mediating TNFα production. Cell Death Dis. 2012, 3, e320. [Google Scholar] [CrossRef] [PubMed]
- Saleh, D.; Najjar, M.; Zelic, M.; Shah, S.; Nogusa, S.; Polykratis, A.; Paczosa, M.K.; Gough, P.J.; Bertin, J.; Whalen, M.; et al. Kinase Activities of RIPK1 and RIPK3 Can Direct IFN-β Synthesis Induced by Lipopolysaccharide. J. Immunol. 2017, 198, 4435–4447. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.N.; Berry, M.; Logan, A.; Blanch, R.J.; Ahmed, Z. Caspases in retinal ganglion cell death and axon regeneration. Cell Death Discov. 2017, 3, 17032. [Google Scholar] [CrossRef]
- Dröge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef]
- Nita, M.; Grzybowski, A. The Role of the Reactive Oxygen Species and Oxidative Stress in the Pathomechanism of the Age-Related Ocular Diseases and Other Pathologies of the Anterior and Posterior Eye Segments in Adults. Oxid. Med. Cell. Longev. 2016, 2016, 3164734. [Google Scholar] [CrossRef]
- Lieven, C.J.; Hoegger, M.J.; Schlieve, C.R.; Levin, L.A. Retinal ganglion cell axotomy induces an increase in intracellular superoxide anion. Invest. Ophthalmol. Vis. Sci. 2006, 47, 1477–1485. [Google Scholar] [CrossRef]
- Cansler, S.M.; Evanson, N.K. Connecting endoplasmic reticulum and oxidative stress to retinal degeneration, TBI, and traumatic optic neuropathy. J. Neurosci. Res. 2020, 98, 571–574. [Google Scholar] [CrossRef]
- Gupta, R.; Saha, P.; Sen, T.; Sen, N. An augmentation in histone dimethylation at lysine nine residues elicits vision impairment following traumatic brain injury. Free Radic. Biol. Med. 2019, 134, 630–643. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.D. Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug Metab. Rev. 2006, 38, 769–789. [Google Scholar] [CrossRef]
- Yang, J.H.; Shin, B.Y.; Han, J.Y.; Kim, M.G.; Wi, J.E.; Kim, Y.W.; Cho, I.J.; Kim, S.C.; Shin, S.M.; Ki, S.H. Isorhamnetin protects against oxidative stress by activating Nrf2 and inducing the expression of its target genes. Toxicol. Appl. Pharmacol. 2014, 274, 293–301. [Google Scholar] [CrossRef]
- Himori, N.; Yamamoto, K.; Maruyama, K.; Ryu, M.; Taguchi, K.; Yamamoto, M.; Nakazawa, T. Critical role of Nrf2 in oxidative stress-induced retinal ganglion cell death. J. Neurochem. 2013, 127, 669–680. [Google Scholar] [CrossRef]
- Koriyama, Y.; Chiba, K.; Yamazaki, M.; Suzuki, H.; Muramoto, K.; Kato, S. Long-acting genipin derivative protects retinal ganglion cells from oxidative stress models in vitro and in vivo through the Nrf2/antioxidant response element signaling pathway. J. Neurochem. 2010, 115, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Cho, H.; Hartsock, M.J.; Mitchell, K.L.; Gong, J.; Wu, L.; Wei, Y.; Wang, S.; Thimmulappa, R.K.; Sporn, M.B.; et al. Neuroprotective role of Nrf2 for retinal ganglion cells in ischemia-reperfusion. J. Neurochem. 2015, 133, 233–241. [Google Scholar] [CrossRef]
- Fujita, K.; Nishiguchi, K.M.; Shiga, Y.; Nakazawa, T. Spatially and Temporally Regulated NRF2 Gene Therapy Using Mcp-1 Promoter in Retinal Ganglion Cell Injury. Mol. Ther. Methods Clin. Dev. 2017, 5, 130–141. [Google Scholar] [CrossRef]
- Cao, Y.; Wang, L.; Zhao, J.; Zhang, H.; Tian, Y.; Liang, H.; Ma, Q. Serum Response Factor Protects Retinal Ganglion Cells Against High-Glucose Damage. J. Mol. Neurosci. 2016, 59, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Hartsock, M.J.; Xu, Z.; He, M.; Duh, E.J. Monomethyl fumarate promotes Nrf2-dependent neuroprotection in retinal ischemia-reperfusion. J. Neuroinflammation 2015, 12, 239. [Google Scholar] [CrossRef]
- Tezel, G. Oxidative stress in glaucomatous neurodegeneration: Mechanisms and consequences. Prog. Retin. Eye Res. 2006, 25, 490–513. [Google Scholar] [CrossRef]
- Buendia, I.; Michalska, P.; Navarro, E.; Gameiro, I.; Egea, J.; León, R. Nrf2-ARE pathway: An emerging target against oxidative stress and neuroinflammation in neurodegenerative diseases. Pharmacol. Ther. 2016, 157, 84–104. [Google Scholar] [CrossRef]
- Schlieve, C.R.; Lieven, C.J.; Levin, L.A. Biochemical activity of reactive oxygen species scavengers do not predict retinal ganglion cell survival. Invest. Ophthalmol. Vis. Sci. 2006, 47, 3878–3886. [Google Scholar] [CrossRef]
- Kanamori, A.; Catrinescu, M.M.; Kanamori, N.; Mears, K.A.; Beaubien, R.; Levin, L.A. Superoxide is an associated signal for apoptosis in axonal injury. Brain 2010, 133, 2612–2625. [Google Scholar] [CrossRef] [PubMed]
- Thongbai, B.; Rapior, S.; Hyde, K.D.; Wittstein, K.; Stadler, M. Hericium erinaceus, an amazing medicinal mushroom. Mycol. Prog. 2015, 14, 91. [Google Scholar] [CrossRef]
- He, X.; Wang, X.; Fang, J.; Chang, Y.; Ning, N.; Guo, H.; Huang, L.; Huang, X.; Zhao, Z. Structures, biological activities, and industrial applications of the polysaccharides from Hericium erinaceus (Lion’s Mane) mushroom: A review. Int. J. Biol. Macromol. 2017, 97, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Tzeng, T.T.; Chen, C.C.; Ni, C.L.; Lee, L.Y.; Chen, W.P.; Shiao, Y.J.; Shen, C.C. Erinacine S, a Rare Sesterterpene from the Mycelia of Hericium erinaceus. J. Nat. Prod. 2016, 79, 438–441. [Google Scholar] [CrossRef]
- Kawagishi, H.; Masui, A.; Tokuyama, S.; Nakamura, T. Erinacines J and K from the mycelia of Hericium erinaceum. Tetrahedron 2006, 62, 8463–8466. [Google Scholar] [CrossRef]
- Mori, K.; Obara, Y.; Hirota, M.; Azumi, Y.; Kinugasa, S.; Inatomi, S.; Nakahata, N. Nerve growth factor-inducing activity of Hericium erinaceus in 1321N1 human astrocytoma cells. Biol. Pharm. Bull. 2008, 31, 1727–1732. [Google Scholar] [CrossRef]
- Phan, C.W.; David, P.; Naidu, M.; Wong, K.H.; Sabaratnam, V. Therapeutic potential of culinary-medicinal mushrooms for the management of neurodegenerative diseases: Diversity, metabolite, and mechanism. Crit. Rev. Biotechnol. 2015, 35, 355–368. [Google Scholar] [CrossRef]
- Tsai-Teng, T.; Chin-Chu, C.; Li-Ya, L.; Wan-Ping, C.; Chung-Kuang, L.; Chien-Chang, S.; Chi-Ying, H.F.; Chien-Chih, C.; Shiao, Y.J. Erinacine A-enriched Hericium erinaceus mycelium ameliorates Alzheimer’s disease-related pathologies in APPswe/PS1dE9 transgenic mice. J. Biomed. Sci. 2016, 23, 49. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Lee, S.H.; Jang, H.D.; Ma, J.Y.; Kim, Y.H. Antioxidant and Anti-Osteoporotic Activities of Aromatic Compounds and Sterols from Hericium erinaceum. Molecules 2017, 22, 108. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.H.; Ye, J.M.; Wang, G.F. Evaluation of in vivo antioxidant activity of Hericium erinaceus polysaccharides. Int. J. Biol. Macromol. 2013, 52, 66–71. [Google Scholar] [CrossRef]
- Kushairi, N.; Phan, C.W.; Sabaratnam, V.; David, P.; Naidu, M. Lion’s Mane Mushroom, Hericium erinaceus (Bull.: Fr.) Pers. Suppresses H(2)O(2)-Induced Oxidative Damage and LPS-Induced Inflammation in HT22 Hippocampal Neurons and BV2 Microglia. Antioxidants 2019, 8, 261. [Google Scholar] [CrossRef]
- Kawagishi, H.; Shimada, A.; Shirai, R.; Okamoto, K.; Ojima, F.; Sakamoto, H.; Ishiguro, Y.; Furukawa, S. Erinacines A, B and C, strong stimulators of nerve growth factor (NGF)-synthesis, from the mycelia of Hericium erinaceum. Tetrahedron Lett. 1994, 35, 1569–1572. [Google Scholar] [CrossRef]
- Shimbo, M.; Kawagishi, H.; Yokogoshi, H. Erinacine A increases catecholamine and nerve growth factor content in the central nervous system of rats. Nutr. Res. 2005, 25, 617–623. [Google Scholar] [CrossRef]
- Ma, B.-J.; Shen, J.-W.; Yu, H.-Y.; Ruan, Y.; Wu, T.-T.; Zhao, X. Hericenones and erinacines: Stimulators of nerve growth factor (NGF) biosynthesis in Hericium erinaceus. Mycology 2010, 1, 92–98. [Google Scholar] [CrossRef]
- Zhang, C.C.; Cao, C.Y.; Kubo, M.; Harada, K.; Yan, X.T.; Fukuyama, Y.; Gao, J.M. Chemical Constituents from Hericium erinaceus Promote Neuronal Survival and Potentiate Neurite Outgrowth via the TrkA/Erk1/2 Pathway. Int. J. Mol. Sci. 2017, 18, 1659. [Google Scholar] [CrossRef]
- Kuo, H.C.; Lu, C.C.; Shen, C.H.; Tung, S.Y.; Hsieh, M.C.; Lee, K.C.; Lee, L.Y.; Chen, C.C.; Teng, C.C.; Huang, W.S.; et al. Hericium erinaceus mycelium and its isolated erinacine A protection from MPTP-induced neurotoxicity through the ER stress, triggering an apoptosis cascade. J. Transl. Med. 2016, 14, 78. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.F.; Tung, S.Y.; Teng, C.C.; Shen, C.H.; Hsieh, M.C.; Huang, C.Y.; Lee, K.C.; Lee, L.Y.; Chen, W.P.; Chen, C.C.; et al. Post-Treatment with Erinacine A, a Derived Diterpenoid of H. erinaceus, Attenuates Neurotoxicity in MPTP Model of Parkinson’s Disease. Antioxidants 2020, 9, 137. [Google Scholar] [CrossRef]
- Chang, C.-H.; Chen, Y.; Yew, X.-X.; Chen, H.-X.; Kim, J.-X.; Chang, C.-C.; Peng, C.-C.; Peng, R.Y. Improvement of erinacine A productivity in Hericium erinaceus mycelia and its neuroprotective bioactivity against the glutamate-insulted apoptosis. LWT-Food Sci. Technol. 2016, 65, 1100–1108. [Google Scholar] [CrossRef]
- Lee, K.F.; Chen, J.H.; Teng, C.C.; Shen, C.H.; Hsieh, M.C.; Lu, C.C.; Lee, K.C.; Lee, L.Y.; Chen, W.P.; Chen, C.C.; et al. Protective effects of Hericium erinaceus mycelium and its isolated erinacine A against ischemia-injury-induced neuronal cell death via the inhibition of iNOS/p38 MAPK and nitrotyrosine. Int. J. Mol. Sci. 2014, 15, 15073–15089. [Google Scholar] [CrossRef] [PubMed]
- Li, I.C.; Lee, L.Y.; Chen, Y.J.; Chou, M.Y.; Wang, M.F.; Chen, W.P.; Chen, Y.P.; Chen, C.C. Erinacine A-enriched Hericium erinaceus mycelia promotes longevity in Drosophila melanogaster and aged mice. PLoS ONE 2019, 14, e0217226. [Google Scholar] [CrossRef]
- Wang, L.Y.; Huang, C.S.; Chen, Y.H.; Chen, C.C.; Chen, C.C.; Chuang, C.H. Anti-Inflammatory Effect of Erinacine C on NO Production Through Down-Regulation of NF-κB and Activation of Nrf2-Mediated HO-1 in BV2 Microglial Cells Treated with LPS. Molecules 2019, 24, 3317. [Google Scholar] [CrossRef]
- Chang, H.C.; Yang, H.L.; Pan, J.H.; Korivi, M.; Pan, J.Y.; Hsieh, M.C.; Chao, P.M.; Huang, P.J.; Tsai, C.T.; Hseu, Y.C. Hericium erinaceus Inhibits TNF-α-Induced Angiogenesis and ROS Generation through Suppression of MMP-9/NF-κB Signaling and Activation of Nrf2-Mediated Antioxidant Genes in Human EA.hy926 Endothelial Cells. Oxid. Med. Cell. Longev. 2016, 2016, 8257238. [Google Scholar] [CrossRef]
- Kapupara, K.; Wen, Y.T.; Tsai, R.K.; Huang, S.P. Soluble P-selectin promotes retinal ganglion cell survival through activation of Nrf2 signaling after ischemia injury. Cell Death Dis. 2017, 8, e3172. [Google Scholar] [CrossRef]
- Huang, C.T.; Wen, Y.T.; Desai, T.D.; Tsai, R.K. Intravitreal Injection of Long-Acting Pegylated Granulocyte Colony-Stimulating Factor Provides Neuroprotective Effects via Antioxidant Response in a Rat Model of Traumatic Optic Neuropathy. Antioxidants 2021, 10, 1934. [Google Scholar] [CrossRef]
- Tsai, Y.C.; Lin, Y.C.; Huang, C.C.; Villaflores, O.B.; Wu, T.Y.; Huang, S.M.; Chin, T.Y. Hericium erinaceus Mycelium and Its Isolated Compound, Erinacine A, Ameliorate High-Fat High-Sucrose Diet-Induced Metabolic Dysfunction and Spatial Learning Deficits in Aging Mice. J. Med. Food 2019, 22, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.L.; Hsu, J.Y.; Chen, T.C.; Huang, C.C.; Wu, T.Y.; Chin, T.Y. Erinacine A Prevents Lipopolysaccharide-Mediated Glial Cell Activation to Protect Dopaminergic Neurons against Inflammatory Factor-Induced Cell Death In Vitro and In Vivo. Int. J. Mol. Sci. 2022, 23, 810. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, D.M.; Degterev, A.; David, J.; Rosenbaum, P.S.; Roth, S.; Grotta, J.C.; Cuny, G.D.; Yuan, J.; Savitz, S.I. Necroptosis, a novel form of caspase-independent cell death, contributes to neuronal damage in a retinal ischemia-reperfusion injury model. J. Neurosci. Res. 2010, 88, 1569–1576. [Google Scholar] [CrossRef]
- Bailly, C.; Gao, J.M. Erinacine A and related cyathane diterpenoids: Molecular diversity and mechanisms underlying their neuroprotection and anticancer activities. Pharmacol. Res. 2020, 159, 104953. [Google Scholar] [CrossRef]
- Ratto, D.; Corana, F.; Mannucci, B.; Priori, E.C.; Cobelli, F.; Roda, E.; Ferrari, B.; Occhinegro, A.; Di Iorio, C.; De Luca, F.; et al. Hericium erinaceus Improves Recognition Memory and Induces Hippocampal and Cerebellar Neurogenesis in Frail Mice during Aging. Nutrients 2019, 11, 715. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.C.; Kuo, H.C.; Shen, C.H.; Lu, C.C.; Huang, W.S.; Hsieh, M.C.; Huang, C.Y.; Kuo, Y.H.; Hsieh, Y.Y.; Teng, C.C.; et al. A proteomics approach to identifying novel protein targets involved in erinacine A-mediated inhibition of colorectal cancer cells’ aggressiveness. J. Cell. Mol. Med. 2017, 21, 588–599. [Google Scholar] [CrossRef]
- Zhou, L.J.; Mo, Y.B.; Bu, X.; Wang, J.J.; Bai, J.; Zhang, J.W.; Cheng, A.B.; Ma, J.H.; Wang, Y.W.; Xie, Y.X. Erinacine Facilitates the Opening of the Mitochondrial Permeability Transition Pore Through the Inhibition of the PI3K/ Akt/GSK-3β Signaling Pathway in Human Hepatocellular Carcinoma. Cell. Physiol. Biochem. 2018, 50, 851–867. [Google Scholar] [CrossRef]
- Lee, K.C.; Lee, K.F.; Tung, S.Y.; Huang, W.S.; Lee, L.Y.; Chen, W.P.; Chen, C.C.; Teng, C.C.; Shen, C.H.; Hsieh, M.C.; et al. Induction Apoptosis of Erinacine A in Human Colorectal Cancer Cells Involving the Expression of TNFR, Fas, and Fas Ligand via the JNK/p300/p50 Signaling Pathway With Histone Acetylation. Front. Pharmacol. 2019, 10, 1174. [Google Scholar] [CrossRef]
- Kuo, H.C.; Kuo, Y.R.; Lee, K.F.; Hsieh, M.C.; Huang, C.Y.; Hsieh, Y.Y.; Lee, K.C.; Kuo, H.L.; Lee, L.Y.; Chen, W.P.; et al. A Comparative Proteomic Analysis of Erinacine A’s Inhibition of Gastric Cancer Cell Viability and Invasiveness. Cell. Physiol. Biochem. 2017, 43, 195–208. [Google Scholar] [CrossRef]
- Li, I.C.; Chen, Y.L.; Lee, L.Y.; Chen, W.P.; Tsai, Y.T.; Chen, C.C.; Chen, C.S. Evaluation of the toxicological safety of erinacine A-enriched Hericium erinaceus in a 28-day oral feeding study in Sprague-Dawley rats. Food Chem. Toxicol. 2014, 70, 61–67. [Google Scholar] [CrossRef]
- Tsai, R.K.; Chang, C.H.; Wang, H.Z. Neuroprotective effects of recombinant human granulocyte colony-stimulating factor (G-CSF) in neurodegeneration after optic nerve crush in rats. Exp. Eye Res. 2008, 87, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.L.; Chang, C.H.; Lin, K.H.; Sheu, M.M.; Tsai, R.K. Lack of protective effect of local administration of triamcinolone or systemic treatment with methylprednisolone against damages caused by optic nerve crush in rats. Exp. Eye Res. 2011, 92, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Tsai, R.K.; Chang, C.H.; Sheu, M.M.; Huang, Z.L. Anti-apoptotic effects of human granulocyte colony-stimulating factor (G-CSF) on retinal ganglion cells after optic nerve crush are PI3K/AKT-dependent. Exp. Eye Res. 2010, 90, 537–545. [Google Scholar] [CrossRef]
- Chien, J.Y.; Sheu, J.H.; Wen, Z.H.; Tsai, R.K.; Huang, S.P. Neuroprotective effect of 4-(Phenylsulfanyl)butan-2-one on optic nerve crush model in rats. Exp. Eye Res. 2016, 143, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Desai, T.D.; Wen, Y.T.; Daddam, J.R.; Cheng, F.; Chen, C.C.; Pan, C.L.; Lin, K.L.; Tsai, R.K. Long term therapeutic effects of icariin-loaded PLGA microspheres in an experimental model of optic nerve ischemia via modulation of CEBP-β/G-CSF/noncanonical NF-κB axis. Bioeng. Transl. Med. 2022, 7, e10289. [Google Scholar] [CrossRef]
- Schmued, L.C.; Fallon, J.H. Fluoro-Gold: A new fluorescent retrograde axonal tracer with numerous unique properties. Brain Res. 1986, 377, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Milligan, C.E.; Cunningham, T.J.; Levitt, P. Differential immunochemical markers reveal the normal distribution of brain macrophages and microglia in the developing rat brain. J. Comp. Neurol. 1991, 314, 125–135. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, C.-L.; Wen, Y.-T.; Hsu, T.-C.; Chen, C.-C.; Lee, L.-Y.; Chen, W.-P.; Tsai, R.-K. Neuroprotective Effects of Erinacine A on an Experimental Model of Traumatic Optic Neuropathy. Int. J. Mol. Sci. 2023, 24, 1504. https://doi.org/10.3390/ijms24021504
Hsu C-L, Wen Y-T, Hsu T-C, Chen C-C, Lee L-Y, Chen W-P, Tsai R-K. Neuroprotective Effects of Erinacine A on an Experimental Model of Traumatic Optic Neuropathy. International Journal of Molecular Sciences. 2023; 24(2):1504. https://doi.org/10.3390/ijms24021504
Chicago/Turabian StyleHsu, Chiao-Ling, Yao-Tseng Wen, Tzu-Chao Hsu, Chin-Chu Chen, Li-Ya Lee, Wan-Ping Chen, and Rong-Kung Tsai. 2023. "Neuroprotective Effects of Erinacine A on an Experimental Model of Traumatic Optic Neuropathy" International Journal of Molecular Sciences 24, no. 2: 1504. https://doi.org/10.3390/ijms24021504
APA StyleHsu, C.-L., Wen, Y.-T., Hsu, T.-C., Chen, C.-C., Lee, L.-Y., Chen, W.-P., & Tsai, R.-K. (2023). Neuroprotective Effects of Erinacine A on an Experimental Model of Traumatic Optic Neuropathy. International Journal of Molecular Sciences, 24(2), 1504. https://doi.org/10.3390/ijms24021504