

Whole Exome Sequencing Identifies a Heterozygous Variant in the Cav1.3 Gene CACNA1D Associated with Familial Sinus Node Dysfunction and Focal Idiopathic Epilepsy
 , , , ,
, , , ,  and
and
Abstract
:1. Introduction
2. Results
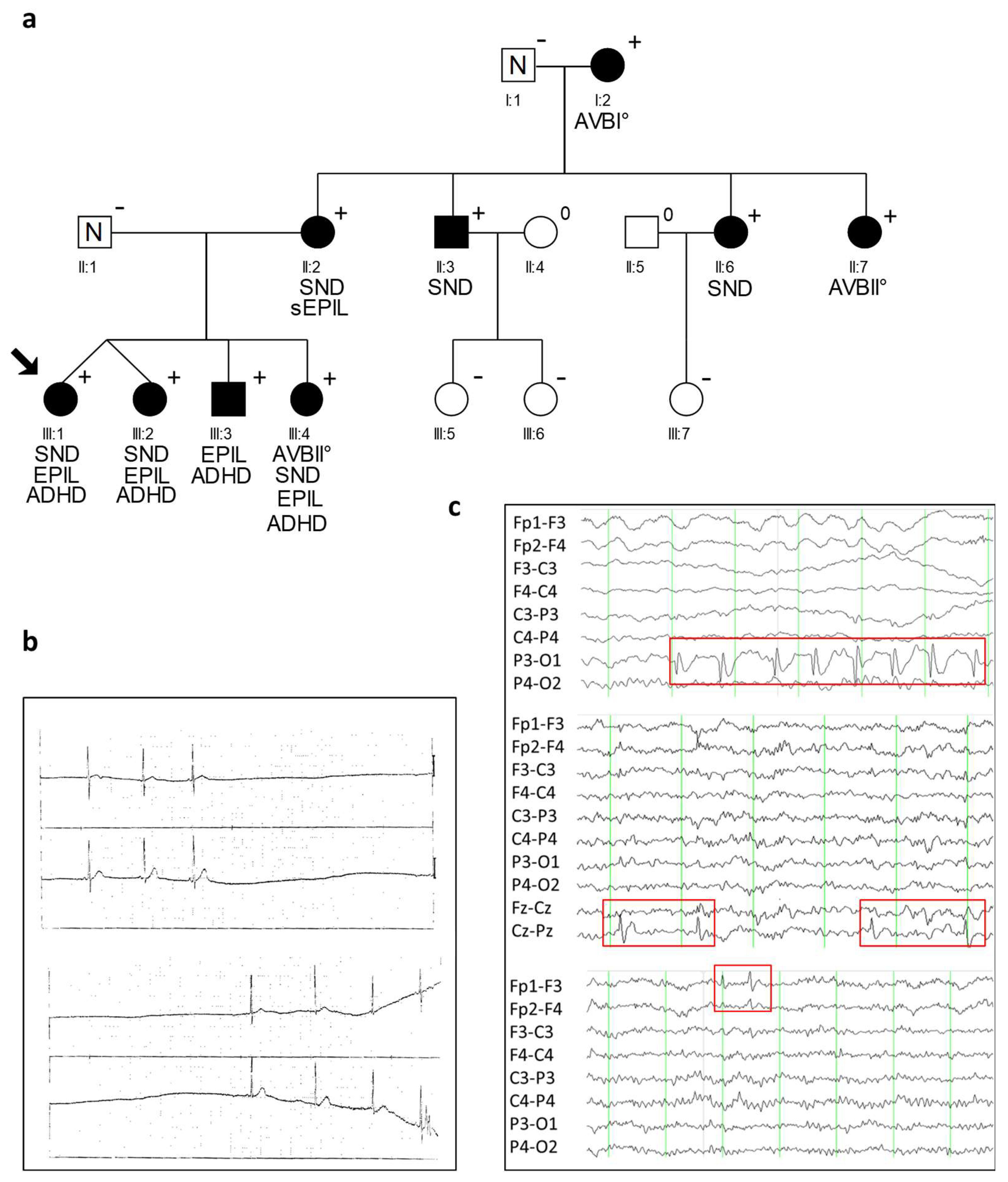
2.1. Clinical Characteristics of CACNA1D R930H Variant Carriers
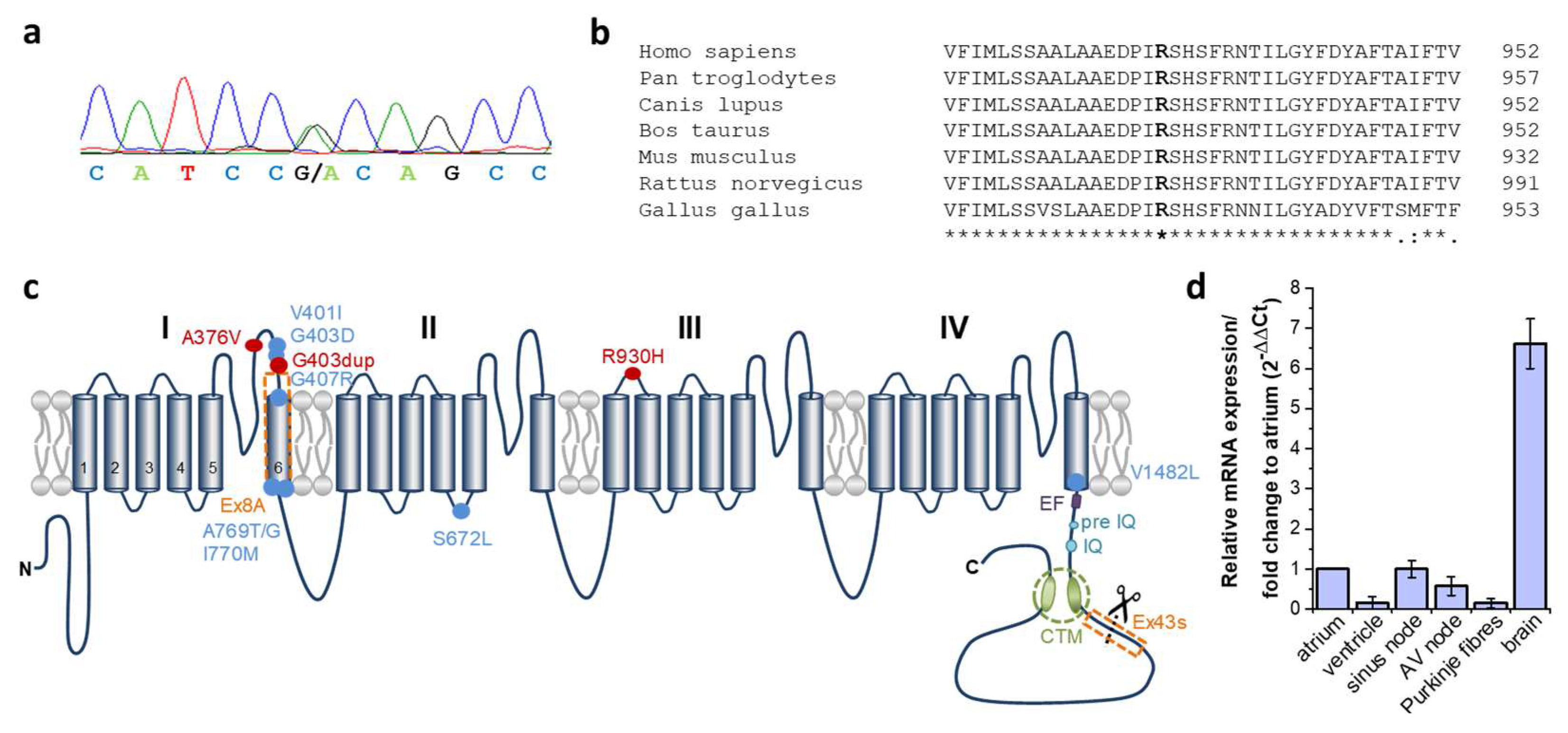
2.2. Identification of a Heterozygous Non-Synonymous Variant (p.R930H) in the CACNA1D Gene Cosegregating in Syndromic Sinus Bradycardia and Epilepsy
2.3. Expression of CACNA1D in Human Cardiac Tissue
2.4. Cellular Localization of Transfected Human Cav1.3-R930H in HEK293 Cells
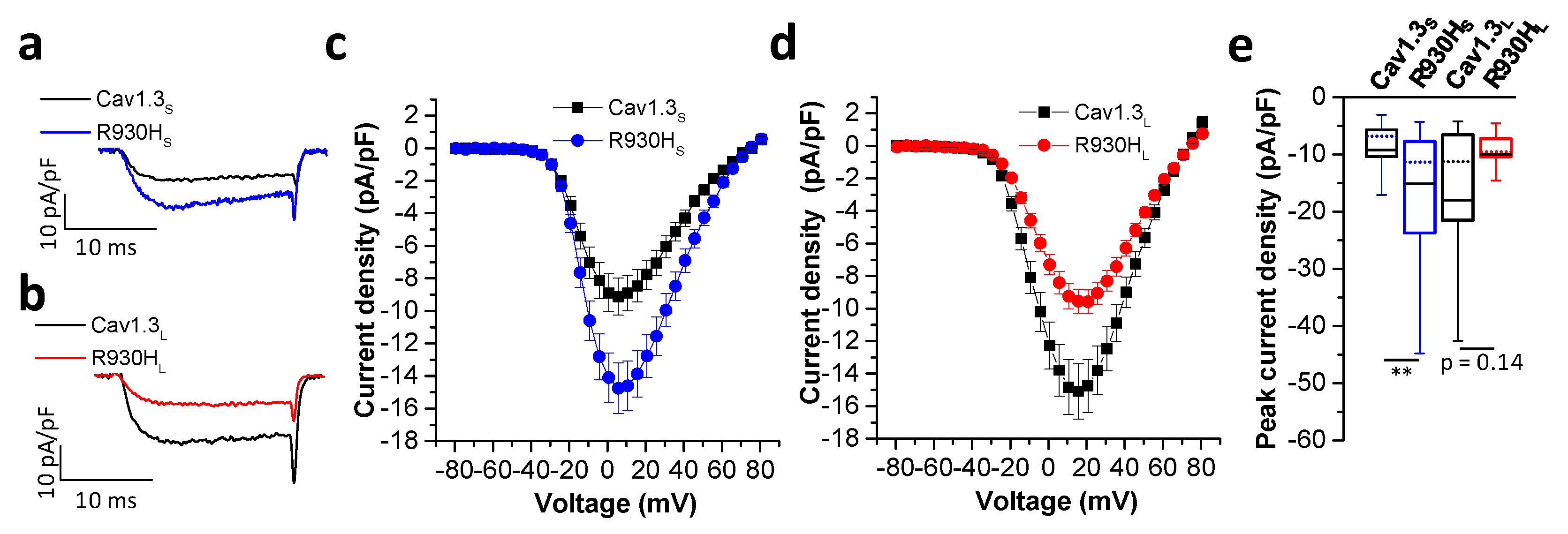
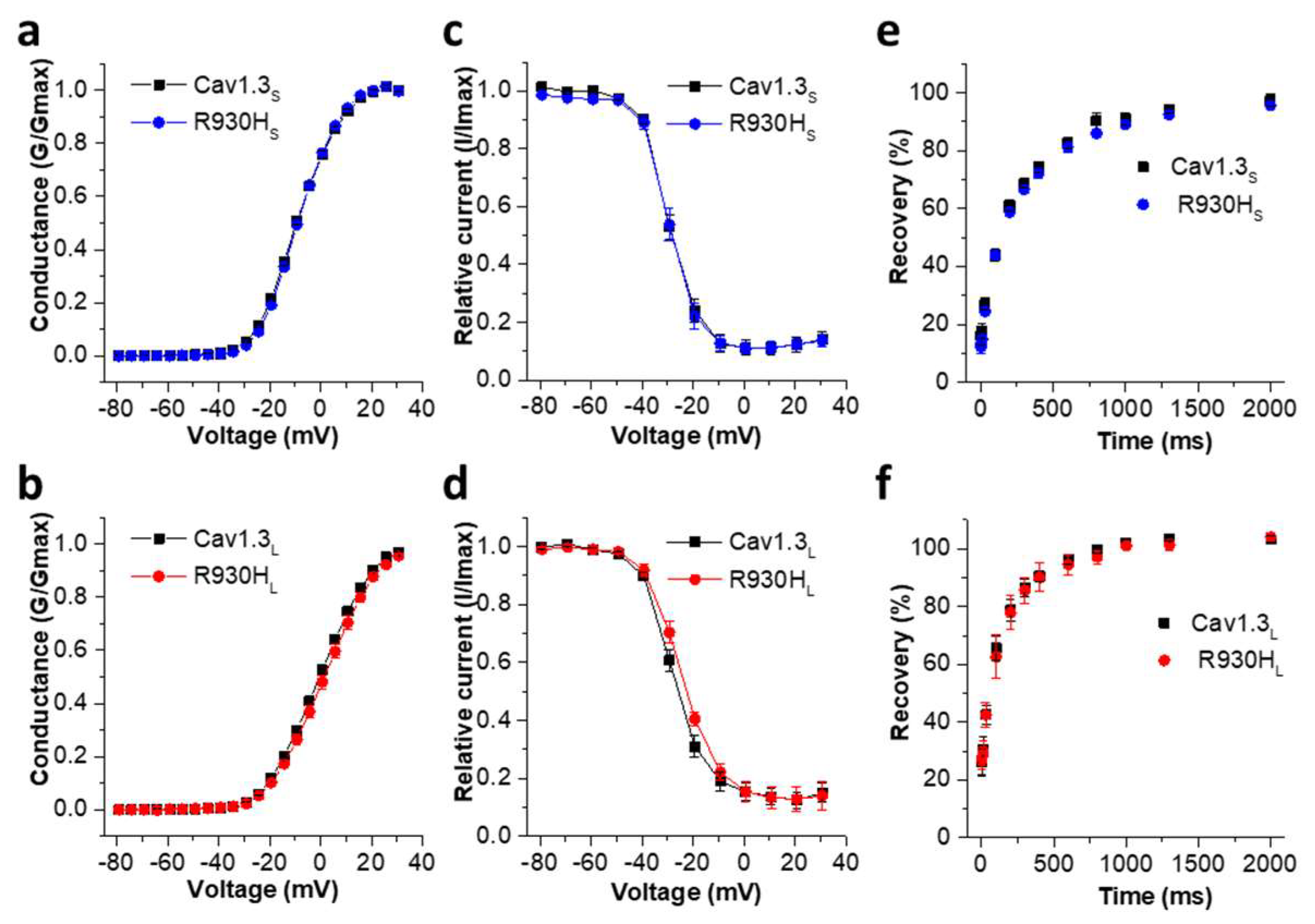
2.5. The CACNA1D R930H Variant Induces Isoform-Dependent Alterations in Cav1.3 Channel Activity and Channel Function
3. Discussion
4. Materials and Methods
4.1. Study Population
4.2. Whole-Exome Sequencing
4.3. Screening for Genetic Variants in the CACNA1D Gene in 39 Additional SND Patients
4.4. Cardiac-Specific Expression of CACNA1D
4.5. Site-Directed Mutagenesis of CACNA1D Constructs
4.6. Immunofluorescence Staining of Cav1.3 R930H Transfected HEK 293T Cells
4.7. Patch-Clamp Recordings of Wild-Type and Mutant Human Cav1.3 Transcripts (Cav1.3S and Cav1.3L) in tsA-201 Cells
4.8. Patch-Clamp Recordings of Rat Cav1.3 Transcripts (Corresponds to the Long Human Cav1.3L Isoform) in CHO Cells
4.9. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schulze-Bahr, E.; Neu, A.; Friederich, P.; Kaupp, U.B.; Breithardt, G.; Pongs, O.; Isbrandt, D. Pacemaker channel dysfunction in a patient with sinus node disease. J. Clin. Investig. 2003, 111, 1537–1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benson, D.W.; Wang, D.W.; Dyment, M.; Knilans, T.K.; Fish, F.A.; Strieper, M.J.; Rhodes, T.H.; George, A.L., Jr. Congenital sick sinus syndrome caused by recessive mutations in the cardiac sodium channel gene (SCN5A). J. Clin. Investig. 2003, 112, 1019–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuss, J.; Stallmeyer, B.; Goldstein, M.; Rinné, S.; Pees, C.; Zumhagen, S.; Seebohm, G.; Decher, N.; Pott, L.; Kienitz, M.C.; et al. Familial Sinus Node Disease Caused by a Gain of GIRK (G-Protein Activated Inwardly Rectifying K(+) Channel) Channel Function. Circ. Genom. Precis. Med. 2019, 12, e002238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, N.; Asano, Y.; Fujita, M.; Yamazaki, S.; Inanobe, A.; Matsuura, N.; Kobayashi, H.; Ohno, S.; Ebana, Y.; Tsukamoto, O.; et al. Mutant KCNJ3 and KCNJ5 Potassium Channels as Novel Molecular Targets in Bradyarrhythmias and Atrial Fibrillation. Circulation 2019, 139, 2157–2169. [Google Scholar] [CrossRef] [PubMed]
- Kruse, M.; Schulze-Bahr, E.; Corfield, V.; Beckmann, A.; Stallmeyer, B.; Kurtbay, G.; Ohmert, I.; Schulze-Bahr, E.; Brink, P.; Pongs, O. Impaired endocytosis of the ion channel TRPM4 is associated with human progressive familial heart block type I. J. Clin. Investig. 2009, 119, 2737–2744. [Google Scholar] [CrossRef]
- Stallmeyer, B.; Zumhagen, S.; Denjoy, I.; Duthoit, G.; Hebert, J.L.; Ferrer, X.; Maugenre, S.; Schmitz, W.; Kirchhefer, U.; Schulze-Bahr, E.; et al. Mutational spectrum in the Ca2+—Activated cation channel gene TRPM4 in patients with cardiac conductance disturbances. Hum. Mutat. 2012, 33, 109–117. [Google Scholar] [CrossRef]
- Stallmeyer, B.; Kuss, J.; Kotthoff, S.; Zumhagen, S.; Vowinkel, K.; Rinné, S.; Matschke, L.A.; Friedrich, C.; Schulze-Bahr, E.; Rust, S.; et al. A Mutation in the G-Protein Gene GNB2 Causes Familial Sinus Node and Atrioventricular Conduction Dysfunction. Circ. Res. 2017, 120, e33–e44. [Google Scholar] [CrossRef]
- Lodder, E.M.; De Nittis, P.; Koopman, C.D.; Wiszniewski, W.; Moura de Souza, C.F.; Lahrouchi, N.; Guex, N.; Napolioni, V.; Tessadori, F.; Beekman, L.; et al. GNB5 Mutations Cause an Autosomal-Recessive Multisystem Syndrome with Sinus Bradycardia and Cognitive Disability. Am. J. Hum. Genet. 2016, 99, 786. [Google Scholar] [CrossRef] [Green Version]
- Veerman, C.C.; Mengarelli, I.; Koopman, C.D.; Wilders, R.; van Amersfoorth, S.C.; Bakker, D.; Wolswinkel, R.; Hababa, M.; de Boer, T.P.; Guan, K.; et al. Genetic variation in GNB5 causes bradycardia by augmenting the cholinergic response via increased acetylcholine-activated potassium current (IK,ACh). Dis. Model. Mech. 2019, 12, dmm037994. [Google Scholar] [CrossRef] [Green Version]
- Bacos, J.M.; Eagan, J.T.; Orgain, E.S. Congenital familial nodal rhythm. Circulation 1960, 22, 887–895. [Google Scholar] [CrossRef]
- Ishikawa, T.; Ohno, S.; Murakami, T.; Yoshida, K.; Mishima, H.; Fukuoka, T.; Kimoto, H.; Sakamoto, R.; Ohkusa, T.; Aiba, T.; et al. Sick sinus syndrome with HCN4 mutations shows early onset and frequent association with atrial fibrillation and left ventricular noncompaction. Heart Rhythm 2017, 14, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Baig, S.M.; Koschak, A.; Lieb, A.; Gebhart, M.; Dafinger, C.; Nurnberg, G.; Ali, A.; Ahmad, I.; Sinnegger-Brauns, M.J.; Brandt, N.; et al. Loss of Ca(v)1.3 (CACNA1D) function in a human channelopathy with bradycardia and congenital deafness. Nat. Neurosci. 2011, 14, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Mark, M.D.; Herlitze, S. G-protein mediated gating of inward-rectifier K+ channels. Eur. J. Biochem. 2000, 267, 5830–5836. [Google Scholar] [CrossRef]
- Fabbri, A.; Fantini, M.; Wilders, R.; Severi, S. Computational analysis of the human sinus node action potential: Model development and effects of mutations. J. Physiol. 2017, 595, 2365–2396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torrente, A.G.; Mesirca, P.; Bidaud, I.; Mangoni, M.E. Channelopathies of voltage-gated L-type Cav1.3/alpha1D and T-type Cav3.1/alpha1G Ca2+ channels in dysfunction of heart automaticity. Pflugers Arch. 2020, 472, 817–830. [Google Scholar] [CrossRef]
- Mangoni, M.E.; Couette, B.; Bourinet, E.; Platzer, J.; Reimer, D.; Striessnig, J.; Nargeot, J. Functional role of L-type Cav1.3 Ca2+ channels in cardiac pacemaker activity. Proc. Natl. Acad. Sci. USA 2003, 100, 5543–5548. [Google Scholar] [CrossRef] [Green Version]
- Koschak, A.; Reimer, D.; Huber, I.; Grabner, M.; Glossmann, H.; Engel, J.; Striessnig, J. alpha 1D (Cav1.3) subunits can form l-type Ca2+ channels activating at negative voltages. J. Biol. Chem. 2001, 276, 22100–22106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlick, B.; Flucher, B.E.; Obermair, G.J. Voltage-activated calcium channel expression profiles in mouse brain and cultured hippocampal neurons. Neuroscience 2010, 167, 786–798. [Google Scholar] [CrossRef] [Green Version]
- Scharinger, A.; Eckrich, S.; Vandael, D.H.; Schonig, K.; Koschak, A.; Hecker, D.; Kaur, G.; Lee, A.; Sah, A.; Bartsch, D.; et al. Cell-type-specific tuning of Cav1.3 Ca2+-channels by a C-terminal automodulatory domain. Front. Cell Neurosci. 2015, 9, 309. [Google Scholar] [CrossRef] [Green Version]
- Bock, G.; Gebhart, M.; Scharinger, A.; Jangsangthong, W.; Busquet, P.; Poggiani, C.; Sartori, S.; Mangoni, M.E.; Sinnegger-Brauns, M.J.; Herzig, S.; et al. Functional properties of a newly identified C-terminal splice variant of Cav1.3 L-type Ca2+ channels. J. Biol. Chem. 2011, 286, 42736–42748. [Google Scholar] [CrossRef]
- Platzer, J.; Engel, J.; Schrott-Fischer, A.; Stephan, K.; Bova, S.; Chen, H.; Zheng, H.; Striessnig, J. Congenital deafness and sinoatrial node dysfunction in mice lacking class D L-type Ca2+ channels. Cell 2000, 102, 89–97. [Google Scholar] [CrossRef] [Green Version]
- Liaqat, K.; Schrauwen, I.; Raza, S.I.; Lee, K.; Hussain, S.; Chakchouk, I.; Nasir, A.; Acharya, A.; Abbe, I.; Umair, M.; et al. Identification of CACNA1D variants associated with sinoatrial node dysfunction and deafness in additional Pakistani families reveals a clinical significance. J. Hum. Genet. 2019, 64, 153–160. [Google Scholar] [PubMed]
- Hofer, N.T.; Tuluc, P.; Ortner, N.J.; Nikonishyna, Y.V.; Fernandes-Quintero, M.L.; Liedl, K.R.; Flucher, B.E.; Cox, H.; Striessnig, J. Biophysical classification of a CACNA1D de novo mutation as a high-risk mutation for a severe neurodevelopmental disorder. Mol. Autism 2020, 11, 4. [Google Scholar] [CrossRef] [Green Version]
- Pinggera, A.; Lieb, A.; Benedetti, B.; Lampert, M.; Monteleone, S.; Liedl, K.R.; Tuluc, P.; Striessnig, J. CACNA1D de novo mutations in autism spectrum disorders activate Cav1.3 L-type calcium channels. Biol. Psychiatry 2015, 77, 816–822. [Google Scholar] [CrossRef] [Green Version]
- Pinggera, A.; Mackenroth, L.; Rump, A.; Schallner, J.; Beleggia, F.; Wollnik, B.; Striessnig, J. New gain-of-function mutation shows CACNA1D as recurrently mutated gene in autism spectrum disorders and epilepsy. Hum. Mol. Genet. 2017, 26, 2923–2932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scholl, U.I.; Goh, G.; Stolting, G.; de Oliveira, R.C.; Choi, M.; Overton, J.D.; Fonseca, A.L.; Korah, R.; Starker, L.F.; Kunstman, J.W.; et al. Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat. Genet. 2013, 45, 1050–1054. [Google Scholar] [CrossRef]
- Fan, L.L.; Chen, Y.Q.; Huang, H.; Yuan, Z.Z.; Jin, J.Y.; Hu, M.; Xiang, R. Exome sequencing identifies a novel nonsense mutation of Ring Finger Protein 207 in a Chinese family with Long QT syndrome and syncope. J. Hum. Genet. 2019, 64, 233–238. [Google Scholar] [CrossRef]
- Li, J.; Shi, L.; Zhang, K.; Zhang, Y.; Hu, S.; Zhao, T.; Teng, H.; Li, X.; Jiang, Y.; Ji, L.; et al. VarCards: An integrated genetic and clinical database for coding variants in the human genome. Nucleic Acids Res. 2018, 46, D1039–D1048. [Google Scholar] [CrossRef] [Green Version]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Tan, B.Z.; Jiang, F.; Tan, M.Y.; Yu, D.; Huang, H.; Shen, Y.; Soong, T.W. Functional characterization of alternative splicing in the C terminus of L-type CaV1.3 channels. J. Biol. Chem. 2011, 286, 42725–42735. [Google Scholar] [CrossRef]
- Ishii, T.M.; Takano, M.; Ohmori, H. Determinants of activation kinetics in mammalian hyperpolarization-activated cation channels. J. Physiol. 2001, 537, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Marsakova, L.; Barvik, I.; Zima, V.; Zimova, L.; Vlachova, V. The First Extracellular Linker Is Important for Several Aspects of the Gating Mechanism of Human TRPA1 Channel. Front. Mol. Neurosci. 2017, 10, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.W.; Park, J.Y.; Jeong, S.W.; Kim, J.A.; Moon, H.J.; Perez-Reyes, E.; Lee, J.H. A molecular determinant of nickel inhibition in Cav3.2 T-type calcium channels. J. Biol. Chem. 2006, 281, 4823–4830. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.W.; Vitko, I.; Lee, S.S.; Perez-Reyes, E.; Lee, J.H. Structural determinants of the high affinity extracellular zinc binding site on Cav3.2 T-type calcium channels. J. Biol. Chem. 2010, 285, 3271–3281. [Google Scholar] [CrossRef] [Green Version]
- Shcheglovitov, A.; Vitko, I.; Lazarenko, R.M.; Orestes, P.; Todorovic, S.M.; Perez-Reyes, E. Molecular and biophysical basis of glutamate and trace metal modulation of voltage-gated Cav2.3 calcium channels. J. Gen. Physiol. 2012, 139, 219–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frederickson, C.J.; Koh, J.Y.; Bush, A.I. The neurobiology of zinc in health and disease. Nat. Rev. Neurosci. 2005, 6, 449–462. [Google Scholar] [CrossRef]
- Garza-Lopez, E.; Lopez, J.A.; Hagen, J.; Sheffer, R.; Meiner, V.; Lee, A. Role of a conserved glutamine in the function of voltage-gated Ca(2+) channels revealed by a mutation in human CACNA1D. J. Biol. Chem. 2018, 293, 14444–14454. [Google Scholar] [CrossRef] [Green Version]
- Long, S.; Zhou, H.; Li, S.; Wang, T.; Ma, Y.; Li, C.; Zhou, Y.; Zhou, S.; Wu, B.; Wang, Y. The Clinical and Genetic Features of Co-occurring Epilepsy and Autism Spectrum Disorder in Chinese Children. Front. Neurol. 2019, 10, 505. [Google Scholar] [CrossRef] [Green Version]
- Semenova, N.A.; Ryzhkova, O.R.; Strokova, T.V.; Taran, N.N. [The third case report a patient with primary aldosteronism, seizures, and neurologic abnormalities (PASNA) syndrome de novo variant mutations in the CACNA1D gene]. Zhurnal Nevrol. I Psikhiatrii Im. SS Korsakova 2018, 118, 49–52. [Google Scholar] [CrossRef]
- Strauss, K.A.; Gonzaga-Jauregui, C.; Brigatti, K.W.; Williams, K.B.; King, A.K.; Van Hout, C.; Robinson, D.L.; Young, M.; Praveen, K.; Heaps, A.D.; et al. Genomic diagnostics within a medically underserved population: Efficacy and implications. Genet. Med. 2018, 20, 31–41. [Google Scholar] [CrossRef]
- Flanagan, S.E.; Vairo, F.; Johnson, M.B.; Caswell, R.; Laver, T.W.; Lango Allen, H.; Hussain, K.; Ellard, S. A CACNA1D mutation in a patient with persistent hyperinsulinaemic hypoglycaemia, heart defects, and severe hypotonia. Pediatr. Diabetes 2017, 18, 320–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van den Berg, M.H.; Wilde, A.A.; Robles de Medina, E.O.; Meyer, H.; Geelen, J.L.; Jongbloed, R.J.; Wellens, H.J.; Geraedts, J.P. The long QT syndrome: A novel missense mutation in the S6 region of the KVLQT1 gene. Hum. Genet. 1997, 100, 356–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neyroud, N.; Tesson, F.; Denjoy, I.; Leibovici, M.; Donger, C.; Barhanin, J.; Faure, S.; Gary, F.; Coumel, P.; Petit, C.; et al. A novel mutation in the potassium channel gene KVLQT1 causes the Jervell and Lange-Nielsen cardioauditory syndrome. Nat. Genet. 1997, 15, 186–189. [Google Scholar] [CrossRef] [PubMed]
- Ortner, N.J.; Kaserer, T.; Copeland, J.N.; Striessnig, J. De novo CACNA1D Ca2+ channelopathies: Clinical phenotypes and molecular mechanism. Pflügers Arch. 2020, 472, 755–773. [Google Scholar] [CrossRef] [PubMed]
- Louradour, J.; Bortolotti, O.; Torre, E.; Bidaud, I.; Lamb, N.; Fernandez, A.; Le Guennec, J.Y.; Mangoni, M.E.; Mesirca, P. L-Type Cav1.3 calcium channels are required for beta-adrenergic triggered automaticity in dormant mouse sinoatrial pacemaker cells. Cells 2022, 11, 1114. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Fagerberg, L.; Hallstrom, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef] [Green Version]
- Richards, C.S.; Bale, S.; Bellissimo, D.B.; Das, S.; Grody, W.W.; Hegde, M.R.; Lyon, E.; Ward, B.E.; Molecular Subcommittee of the ACMG Laboratory Quality Assurance Committee. ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet. Med. 2008, 10, 294–300. [Google Scholar] [CrossRef]
- Lieb, A.; Ortner, N.; Striessnig, J. C-terminal modulatory domain controls coupling of voltage-sensing to pore opening in Cav1.3 L-type Ca2+ channels. Biophys. J. 2014, 106, 1467–1475. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Sex | CACNA1D Variant | Baseline ECG HR [/min]; PQ; QRS; QTc [ms] | Holter ECG | Cardiac Phenotype | Neurologic Phenotype | EEG |
|---|---|---|---|---|---|---|---|
| I:1 | m | − | 75; 148; 100; 400 | n.a. | LAHB | n.a. | |
| I:2 | f | + | 52; 175; 92; 387 | AVB I°; 42–109/min | LAHB | n.a. | |
| II:1 | m | − | 85; 155; 82; 384 | n.a. | − | n.a. | |
| II:2 | f | + | 85; 126; 87; 379 | SND, 40/min; pauses of 2.3 s 48–126/min | TTE: AVB III°; cardiac MRI unremark. | n.a. | |
| II:3 | m | + | 56; 135; 73; 341 | sinus arrhythmia; 48–103/min | − | n.a. | |
| II:6 | f | + | 75; 117; 58; 398 | sinus arrhythmia, 40–143/min | TTE unremark. | n.a. | |
| II:7 | f | + | 70; 135; 89; 390 | AVB I°, AVB II°, sinus arrhythmia; 47–152/min | − | n.a. | |
| III:1 | f | + | n.a. # | SA | cardiac syncope, PM (3 y) * | IFE; ADHD | focal sharp waves: left occipital, left frontal and frontopolar |
| III:2 | f | + | 76; 127; 76; 354 | sinus arrhythmia, AVB I°/II°; 70–154/min | − | IFE; ADHD | focal sharp waves: frontal and central |
| III:3 | m | + | 142; 94; 71; 400 | unremarkable; 110–189/min | − | IFE; ADHD | focal sharp waves: left occipital |
| III:4 | f | + | 86; 157; 63; 387 | AVB I°, AVB II°, SA; 64–142/min | IFE; ADHD | unremarkable |
| Activation | Inactivation | ||||||
|---|---|---|---|---|---|---|---|
| Vrev [mV] | Slope [mV] | V0.5 [mV] | n | V0.5 [mV] | Slope [mV] | n | |
| Cav1.3S | 67.62 ± 1.05 | 7.66 ± 0.11 | −8.94 ± 0.41 | 50 | −29.69 ± 1.06 | 4.45 ± 0.16 | 15 |
| R930HS | 67.63 ± 1.27 | 7.17 ± 0.10 ** | −8.67 ± 0.43 | 39 | −30.24 ± 1.00 | 4.47 ± 0.23 | 9 |
| Cav1.3L | 69.35 ± 0.65 | 9.24 ± 0.11 | −0.14 ± 0.56 | 79 | −25.72 ± 2.08 | 5.56 ± 0.23 | 18 |
| R930HL | 68.80 ± 0.82 | 9.24 ± 0.16 | 1.73 ± 1.06 | 32 | −24.11 ± 1.12 | 6.22 ± 0.35 | 12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rinné, S.; Stallmeyer, B.; Pinggera, A.; Netter, M.F.; Matschke, L.A.; Dittmann, S.; Kirchhefer, U.; Neudorf, U.; Opp, J.; Striessnig, J.; et al. Whole Exome Sequencing Identifies a Heterozygous Variant in the Cav1.3 Gene CACNA1D Associated with Familial Sinus Node Dysfunction and Focal Idiopathic Epilepsy. Int. J. Mol. Sci. 2022, 23, 14215. https://doi.org/10.3390/ijms232214215
Rinné S, Stallmeyer B, Pinggera A, Netter MF, Matschke LA, Dittmann S, Kirchhefer U, Neudorf U, Opp J, Striessnig J, et al. Whole Exome Sequencing Identifies a Heterozygous Variant in the Cav1.3 Gene CACNA1D Associated with Familial Sinus Node Dysfunction and Focal Idiopathic Epilepsy. International Journal of Molecular Sciences. 2022; 23(22):14215. https://doi.org/10.3390/ijms232214215
Chicago/Turabian StyleRinné, Susanne, Birgit Stallmeyer, Alexandra Pinggera, Michael F. Netter, Lina A. Matschke, Sven Dittmann, Uwe Kirchhefer, Ulrich Neudorf, Joachim Opp, Jörg Striessnig, and et al. 2022. "Whole Exome Sequencing Identifies a Heterozygous Variant in the Cav1.3 Gene CACNA1D Associated with Familial Sinus Node Dysfunction and Focal Idiopathic Epilepsy" International Journal of Molecular Sciences 23, no. 22: 14215. https://doi.org/10.3390/ijms232214215