
Clinical and Molecular Insights into Gastrointestinal Dysfunction in Myotonic Dystrophy Types 1 & 2



Abstract
:1. Introduction
2. Clinical Findings
2.1. Prevalence
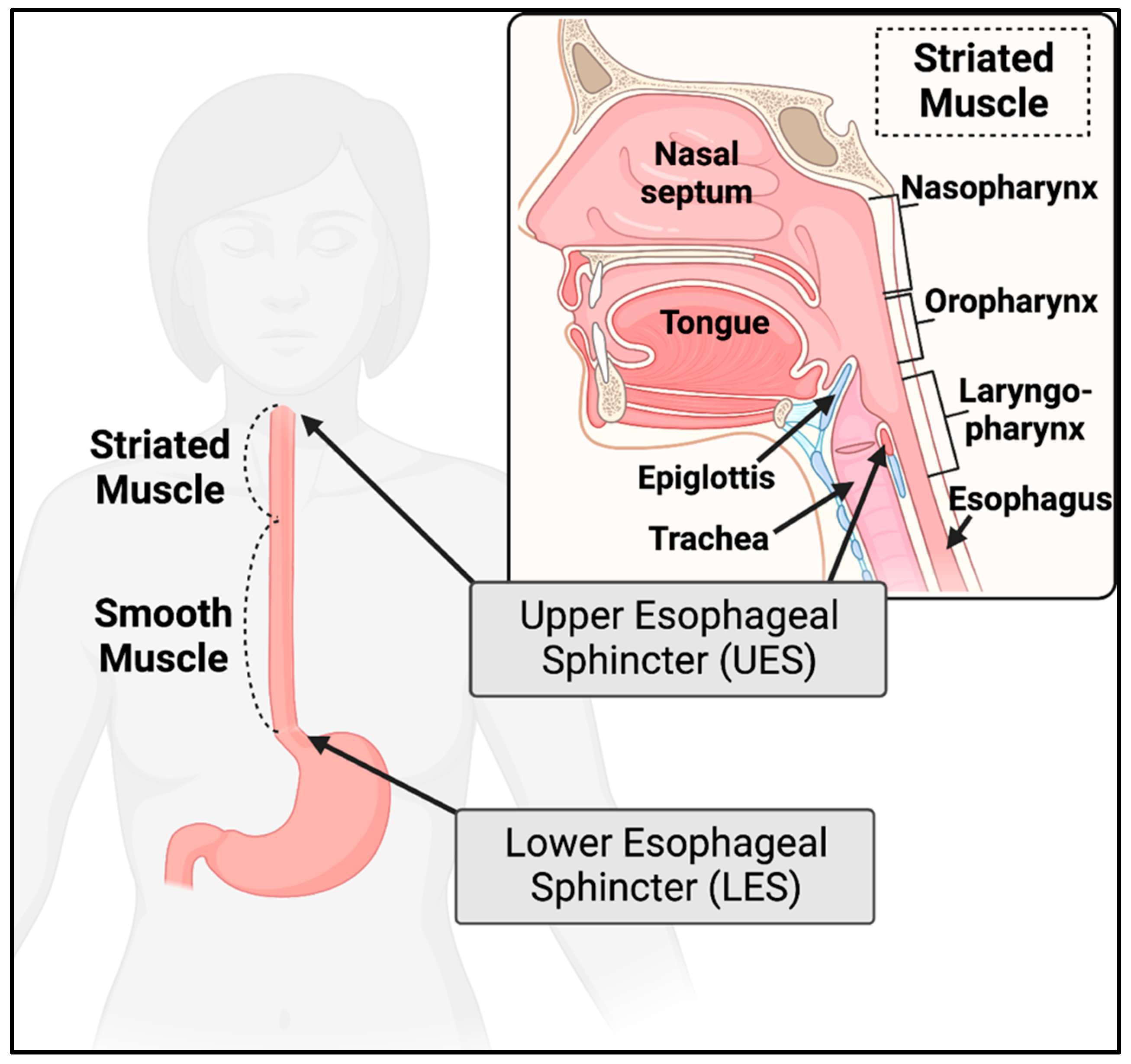
2.2. Esophagus
2.3. Stomach
2.4. Liver & Gallbladder
2.5. Small & Large Intestine
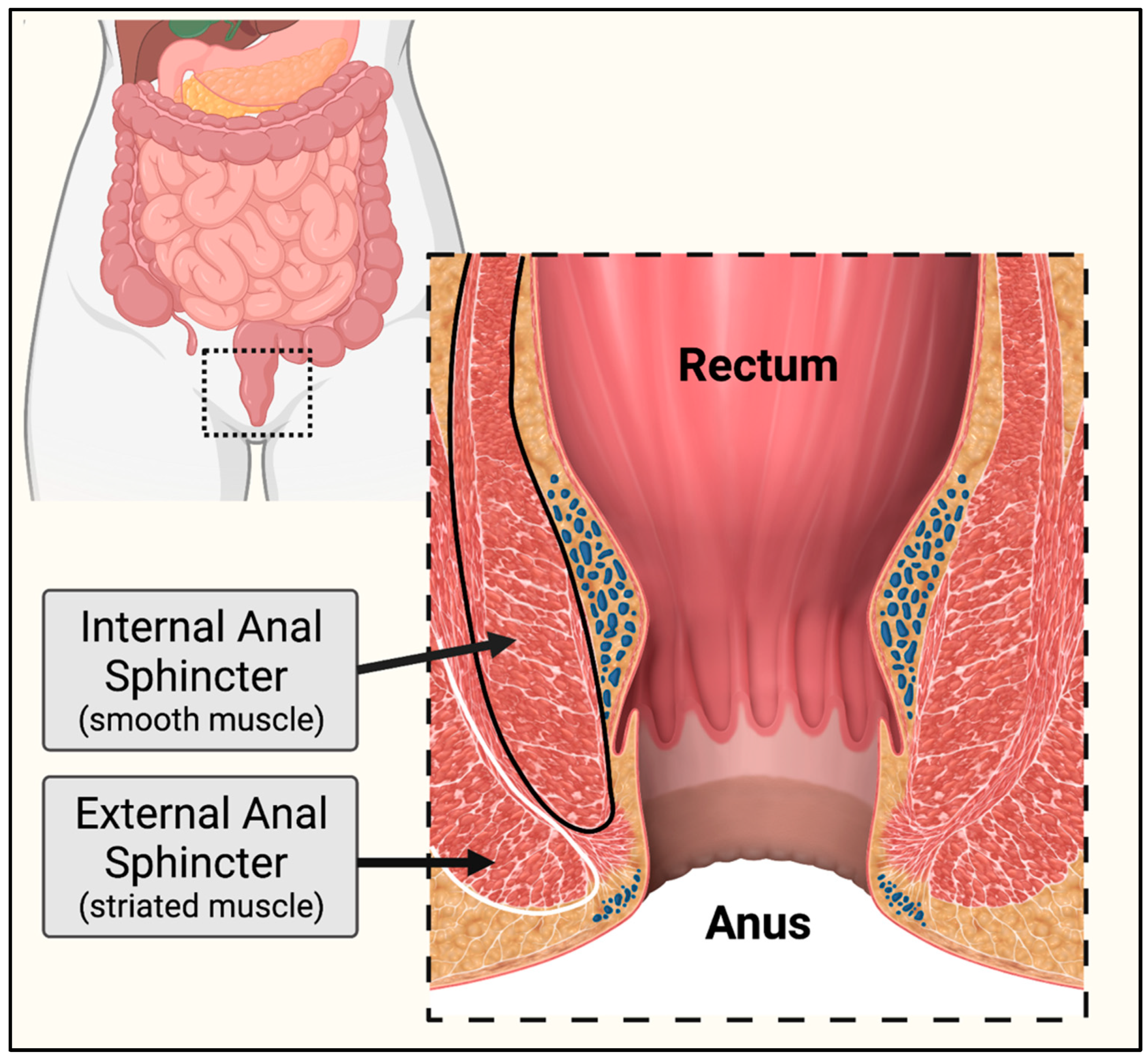
2.6. Rectum & Anal Sphincters
2.7. Enteric Nervous System
2.8. Clinical Conclusions
3. Molecular Findings
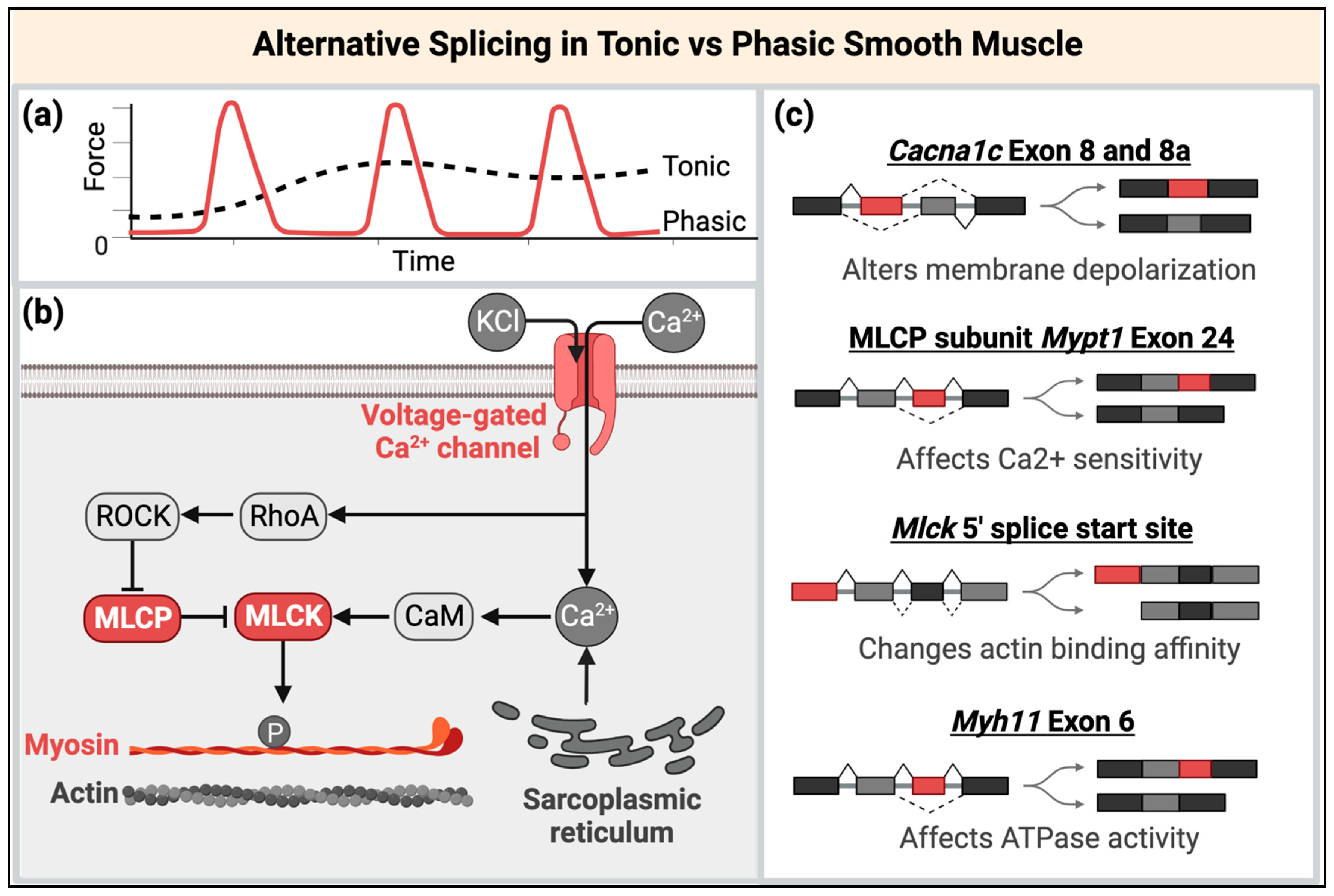
3.1. DMPK and CNBP Expression in the Gastrointestinal Tract Smooth Muscle
3.2. The Potential Roles of Disrupted MBNL and CELF Functions in Smooth Muscle RNA Processing and Regulation
3.3. Molecular Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yum, K.; Wang, E.T.; Kalsotra, A. Myotonic Dystrophy: Disease Repeat Range, Penetrance, Age of Onset, and Relationship between Repeat Size and Phenotypes. Curr. Opin. Genet. Dev. 2017, 44, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Suominen, T.; Bachinski, L.L.; Auvinen, S.; Hackman, P.; Baggerly, K.A.; Angelini, C.; Peltonen, L.; Krahe, R.; Udd, B. Population Frequency of Myotonic Dystrophy: Higher than Expected Frequency of Myotonic Dystrophy Type 2 (DM2) Mutation in Finland. Eur. J. Hum. Genet. 2011, 19, 776–782. [Google Scholar] [CrossRef]
- Johnson, N.E.; Butterfield, R.J.; Mayne, K.; Newcomb, T.; Imburgia, C.; Dunn, D.; Duval, B.; Feldkamp, M.L.; Weiss, R.B. Population Based Prevalence of Myotonic Dystrophy Type 1 Using Genetic Analysis of State-Wide Blood Screening Program. Neurology 2021, 96, e1045–e1053. [Google Scholar] [CrossRef]
- Kumar, A.; Agarwal, S.; Agarwal, D.; Phadke, S.R. Myotonic Dystrophy Type 1 (DM1): A Triplet Repeat Expansion Disorder. Gene 2013, 522, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Brook, J.D.; McCurrach, M.E.; Harley, H.G.; Buckler, A.J.; Church, D.; Aburatani, H.; Hunter, K.; Stanton, V.P.; Thirion, J.-P.; Hudson, T.; et al. Molecular Basis of Myotonic Dystrophy: Expansion of a Trinucleotide (CTG) Repeat at the 3′ End of a Transcript Encoding a Protein Kinase Family Member. Cell 1992, 68, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, M.; Tsilfidis, C.; Sabourin, L.; Shutler, G.; Amemiya, C.; Jansen, G.; Neville, C.; Narang, M.; Barceló, J.; O’Hoy, K.; et al. Myotonic Dystrophy Mutation: An Unstable CTG Repeat in the 3′ Untranslated Region of the Gene. Science 1992, 255, 1253–1255. [Google Scholar] [CrossRef] [PubMed]
- Pizzuti, A.; Friedman, D.L.; Caskey, C.T. The Myotonic Dystrophy Gene. Arch. Neurol. 1993, 50, 1173–1179. [Google Scholar] [CrossRef] [PubMed]
- Liquori, C.L.; Ricker, K.; Moseley, M.L.; Jacobsen, J.F.; Kress, W.; Naylor, S.L.; Day, J.W.; Ranum, L.P.W. Myotonic Dystrophy Type 2 Caused by a CCTG Expansion in Intron 1 of ZNF9. Science 2001, 293, 864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, J.W.; Urbinati, C.R.; Teng-umnuay, P.; Stenberg, M.G.; Byrne, B.J.; Thornton, C.A.; Swanson, M.S. Recruitment of Human Muscleblind Proteins to (CUG)n Expansions Associated with Myotonic Dystrophy. EMBO J. 2000, 19, 4439–4448. [Google Scholar] [CrossRef] [Green Version]
- Fardaei, M.; Rogers, M.T.; Thorpe, H.M.; Larkin, K.; Hamshere, M.G.; Harper, P.S.; Brook, J.D. Three Proteins, MBNL, MBLL and MBXL, Co-Localize in Vivo with Nuclear Foci of Expanded-Repeat Transcripts in DM1 and DM2 Cells. Hum. Mol. Genet. 2002, 11, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Mankodi, A.; Swanson, M.S.; Moxley, R.T.; Thornton, C.A. Myotonic Dystrophy Type 1 Is Associated with Nuclear Foci of Mutant RNA, Sequestration of Muscleblind Proteins and Deregulated Alternative Splicing in Neurons. Hum. Mol. Genet. 2004, 13, 3079–3088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Zhuang, Y.; Batra, R.; Thomas, J.D.; Li, M.; Nutter, C.A.; Scotti, M.M.; Carter, H.A.; Wang, Z.J.; Huang, X.-S.; et al. HNRNPA1-Induced Spliceopathy in a Transgenic Mouse Model of Myotonic Dystrophy. Proc. Natl. Acad. Sci. USA 2020, 117, 5472–5477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.; Li, M.; Manchanda, M.; Batra, R.; Charizanis, K.; Mohan, A.; Warren, S.A.; Chamberlain, C.M.; Finn, D.; Hong, H.; et al. Compound Loss of Muscleblind-like Function in Myotonic Dystrophy. EMBO Mol. Med. 2013, 5, 1887–1900. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Dansithong, W.; Kim, D.; Rossi, J.; Webster, N.J.; Comai, L.; Reddy, S. Interaction of Musleblind, CUG-BP1 and HnRNP H Proteins in DM1-associated Aberrant IR Splicing. EMBO J. 2006, 25, 4271–4283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, E.T.; Cody, N.A.L.; Jog, S.; Biancolella, M.; Wang, T.T.; Treacy, D.J.; Luo, S.; Schroth, G.P.; Housman, D.E.; Reddy, S.; et al. Transcriptome-Wide Regulation of Pre-MRNA Splicing and MRNA Localization by Muscleblind Proteins. Cell 2012, 150, 710–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ladd, A.N.; Charlet-B., N.; Cooper, T.A. The CELF Family of RNA Binding Proteins Is Implicated in Cell-Specific and Developmentally Regulated Alternative Splicing. Mol. Cell. Biol. 2001, 21, 1285–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taneja, K.L.; McCurrach, M.; Schalling, M.; Housman, D.; Singer, R.H. Foci of Trinucleotide Repeat Transcripts in Nuclei of Myotonic Dystrophy Cells and Tissues. J. Cell Biol. 1995, 128, 995–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mankodi, A.; Urbinati, C.R.; Yuan, Q.-P.; Moxley, R.T.; Sansone, V.; Krym, M.; Henderson, D.; Schalling, M.; Swanson, M.S.; Thornton, C.A. Muscleblind Localizes to Nuclear Foci of Aberrant RNA in Myotonic Dystrophy Types 1 and 2. Hum. Mol. Genet. 2001, 10, 2165–2170. [Google Scholar] [CrossRef] [Green Version]
- André, L.M.; van Cruchten, R.T.P.; Willemse, M.; Wansink, D.G. (CTG)n Repeat-Mediated Dysregulation of MBNL1 and MBNL2 Expression during Myogenesis in DM1 Occurs Already at the Myoblast Stage. PLoS ONE 2019, 14, e0217317. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Miller, J.W.; Mankodi, A.; Kanadia, R.N.; Yuan, Y.; Moxley, R.T.; Swanson, M.S.; Thornton, C.A. Failure of MBNL1-Dependent Post-Natal Splicing Transitions in Myotonic Dystrophy. Hum. Mol. Genet. 2006, 15, 2087–2097. [Google Scholar] [CrossRef]
- Heatwole, C.; Bode, R.; Johnson, N.; Quinn, C.; Martens, W.; McDermott, M.P.; Rothrock, N.; Thornton, C.; Vickrey, B.; Victorson, D.; et al. Patient-Reported Impact of Symptoms in Myotonic Dystrophy Type 1 (PRISM-1). Neurology 2012, 79, 348–357. [Google Scholar] [CrossRef] [Green Version]
- Peric, S.; Heatwole, C.; Durovic, E.; Kacar, A.; Nikolic, A.; Basta, I.; Marjanovic, A.; Stevic, Z.; Lavrnic, D.; Stojanovic, V.R. Prospective Measurement of Quality of Life in Myotonic Dystrophy Type 1. Acta Neurol. Scand. 2017, 136, 694–697. [Google Scholar] [CrossRef] [PubMed]
- Perna, A.; Maccora, D.; Rossi, S.; Nicoletti, T.F.; Zocco, M.A.; Riso, V.; Modoni, A.; Petrucci, A.; Valenza, V.; Grieco, A.; et al. High Prevalence and Gender-Related Differences of Gastrointestinal Manifestations in a Cohort of DM1 Patients: A Perspective, Cross-Sectional Study. Front. Neurol. 2020, 11, 394. [Google Scholar] [CrossRef] [PubMed]
- Hilbert, J.E.; Barohn, R.J.; Clemens, P.R.; Luebbe, E.A.; Martens, W.B.; McDermott, M.P.; Parkhill, A.L.; Tawil, R.; Thornton, C.A.; Moxley, R.T.; et al. High Frequency of Gastrointestinal Manifestations in Myotonic Dystrophy Type 1 and Type 2. Neurology 2017, 89, 1348–1354. [Google Scholar] [CrossRef] [PubMed]
- Tieleman, A.A.; van Vliet, J.; Jansen, J.B.M.J.; van der Kooi, A.J.; Borm, G.F.; van Engelen, B.G.M. Gastrointestinal Involvement Is Frequent in Myotonic Dystrophy Type 2. Neuromuscul. Disord. 2008, 18, 646–649. [Google Scholar] [CrossRef] [PubMed]
- Petty, R.K.H.; Eugenicos, M.P.; Hamilton, M.J.; Farrugia, M.E.; Robb, Y.; Ballantyne, R.; Gregory, H.; McWilliam, C.; Longman, C. The Prevalence of Faecal Incontinence in Myotonic Dystrophy Type 1. Neuromuscul. Disord. 2019, 29, 562–566. [Google Scholar] [CrossRef]
- Cheng, L.K.; O’Grady, G.; Du, P.; Egbuji, J.U.; Windsor, J.A.; Pullan, A.J. Gastrointestinal System. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010, 2, 65–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degraeuwe, J.; Laecke, E.V.; Muynck, M.D.; Biervliet, S.V.; Velde, S.V.; Winckel, M.V. Faecal Incontinence Due to Atrophy of the Anal Sphincter in Myotonic Dystrophy: A Case Report. Acta Gastro-Enterol. Belg. 2011, 74, 88–90. [Google Scholar]
- Fisette-Paulhus, I.; Gagnon, C.; Girard-Côté, L.; Morin, M. Genitourinary and Lower Gastrointestinal Conditions in Patients with Myotonic Dystrophy Type 1: A Systematic Review of Evidence and Implications for Clinical Practice. Neuromuscul. Disord. 2022, 32, 361–376. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, J.; Allard, P.; Potvin, L.; Prévost, C.; Bégin, P. A 10-Year Study of Mortality in a Cohort of Patients with Myotonic Dystrophy. Neurology 1999, 52, 1658–1662. [Google Scholar] [CrossRef]
- Gagnon, C.; Chouinard, M.C.; Laberge, L.; Veillette, S.; Bégin, P.; Breton, R.; Jean, S.; Brisson, D.; Gaudet, D.; Mathieu, J.; et al. Health Supervision and Anticipatory Guidance in Adult Myotonic Dystrophy Type 1. Neuromuscul. Disord. 2010, 20, 847–851. [Google Scholar] [CrossRef] [PubMed]
- Pilz, W.; Passos, V.L.; Verdonschot, R.J.; Meijers, J.; Roodenburg, N.; Halmans, Y.; Faber, C.G.; Kremer, B.; Baijens, L.W.J. Swallow-Related Quality of Life and Oropharyngeal Dysphagia in Myotonic Dystrophy. Eur. Arch. Oto-Rhino-Laryngol. 2020, 277, 2357–2362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motlagh, B.; MacDonald, J.R.; Tarnopolsky, M.A. Nutritional Inadequacy in Adults with Muscular Dystrophy. Muscle Nerve 2005, 31, 713–718. [Google Scholar] [CrossRef] [PubMed]
- Malik, H.Z.; Sharma, G.; Moreno, C.; Parcha, S.P. A Medley of Malnutrition and Myotonic Dystrophy: Twice Unlucky. Cureus 2022, 14, e21180. [Google Scholar] [CrossRef]
- Tarnopolsky, M.A.; Pearce, E.; Matteliano, A.; James, C.; Armstrong, D. Bacterial Overgrowth Syndrome in Myotonic Muscular Dystrophy Is Potentially Treatable. Muscle Nerve 2010, 42, 853–855. [Google Scholar] [CrossRef]
- Ishizawa, Y.; Yamaguchi, H.; Dohi, S.; Koyama, K. A Serious Complication Due to Gastrointestinal Malfunction in a Patient with Myotonic Dystrophy. Anesth. Analg. 1986, 65, 1066. [Google Scholar] [CrossRef]
- Rönnblom, A.; Forsberg, H.; Danielsson, A. Gastrointestinal Symptoms in Myotonic Dystrophy. Scand. J. Gastroenterol. 1996, 31, 654–657. [Google Scholar] [CrossRef]
- Tieleman, A.A.; Knuijt, S.; van Vliet, J.; de Swart, B.J.M.; Ensink, R.; Engelen, B.G.M. van Dysphagia Is Present but Mild in Myotonic Dystrophy Type 2. Neuromuscul. Disord. 2009, 19, 196–198. [Google Scholar] [CrossRef]
- Wenninger, S.; Stahl, K.; Montagnese, F.; Schoser, B. Utility and Results from a Patient-Reported Online Survey in Myotonic Dystrophies Types 1 and 2. Eur. Neurol. 2020, 83, 523–533. [Google Scholar] [CrossRef]
- Hunter, M.; Ekstrom, A.; Campbell, C.; Hung, M.; Bounsanga, J.; Bates, K.; Adams, H.R.; Luebbe, E.; Moxley, R.T.; Heatwole, C.; et al. Patient-reported Study of the Impact of Pediatric-onset Myotonic Dystrophy. Muscle Nerve 2019, 60, 392–399. [Google Scholar] [CrossRef]
- Kerr, T.P.; Robb, S.A.; Clayden, G.S. Lower Gastrointestinal Tract Disturbance in Congenital Myotonic Dystrophy. Eur. J. Pediatr. 2002, 161, 468–469. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, W.E. Diagnosing and Managing Fecal Incontinence: If You Don’t Ask, They Won’t Tell. Gastroenterology 2005, 129, 6. [Google Scholar] [CrossRef] [PubMed]
- Garrett, J.M.; DuBose, T.D.; Jackson, J.E.; Norman, J.R. Esophageal and Pulmonary Disturbances in Myotonia Dystrophica. Arch. Intern. Med. 1969, 123, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Goyal, R.K.; Chaudhury, A. Physiology of Normal Esophageal Motility. J. Clin. Gastroenterol. 2008, 42, 610–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pruzanski, W.; Profis, A. Dysfunction of the Alimentary Tract in Myotonic Dystrophy. Isr. J. Med Sci. 1966, 2, 59–64. [Google Scholar]
- Schuman, B.M.; Rinaldo, J.A.; Darnley, J.D. Visceral Changes in Myotonic Dystrophy. Ann. Intern. Med. 1965, 63, 793. [Google Scholar] [CrossRef]
- Horowitz, M.; Maddox, A.; Maddern, G.J.; Wishart, J.; Collins, P.J.; Shearman, D.J.C. Gastric and Esophageal Emptying in Dystrophia Myotonica Effect of Metoclopramide. Gastroenterology 1987, 92, 570–577. [Google Scholar] [CrossRef]
- Costantini, M.; Zaninotto, G.; Anselmino, M.; Marcon, M.; Iurilli, V.; Boccù, C.; Feltrin, G.P.; Angelini, C.; Ancona, E. Esophageal Motor Function in Patients with Myotonic Dystrophy. Am. J. Dig. Dis. 1996, 41, 2032–2038. [Google Scholar] [CrossRef]
- Harvey, J.C.; Sherbourne, D.H.; Siegel, C.I. Smooth Muscle Involvement in Myotonic Dystrophy. Am. J. Med. 1965, 39, 81–90. [Google Scholar] [CrossRef]
- Hughes, D.T.D.; Swann, J.C.; Gleeson, J.A.; Lee, F.I. Abnormalities In Swallowing Associated With Dystrophia Myotonica. Brain 1965, 88, 1037–1042. [Google Scholar] [CrossRef] [PubMed]
- Kramer, P.; Atkinson, M.; Wyman, S.M.; Ingelfinger, F.J. The Dynamics of Swallowing. II. Neuromuscular Dysphagia of Pharynx. J. Clin. Investig. 1957, 36, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, H.I.; Sheft, D.J. Esophageal and Colon Changes in Myotonia Dystrophica. Gastroenterology 1972, 63, 134–139. [Google Scholar] [CrossRef]
- Nowak, T.V.; Ionasescu, V.; Anuras, S. Gastrointestinal Manifestations of the Muscular Dystrophies. Gastroenterology 1982, 82, 800–810. [Google Scholar] [CrossRef] [PubMed]
- André, L.M.; Ausems, C.R.M.; Wansink, D.G.; Wieringa, B. Abnormalities in Skeletal Muscle Myogenesis, Growth, and Regeneration in Myotonic Dystrophy. Front. Neurol. 2018, 9, 368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mankodi, A.; Takahashi, M.P.; Jiang, H.; Beck, C.L.; Bowers, W.J.; Moxley, R.T.; Cannon, S.C.; Thornton, C.A. Expanded CUG Repeats Trigger Aberrant Splicing of ClC-1 Chloride Channel Pre-MRNA and Hyperexcitability of Skeletal Muscle in Myotonic Dystrophy. Mol. Cell 2002, 10, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.Z.; Yarotskyy, V.; Wei, L.; Sobczak, K.; Nakamori, M.; Eichinger, K.; Moxley, R.T.; Dirksen, R.T.; Thornton, C.A. Muscle Weakness in Myotonic Dystrophy Associated with Misregulated Splicing and Altered Gating of CaV1.1 Calcium Channel. Hum. Mol. Genet. 2011, 21, 1312–1324. [Google Scholar] [CrossRef] [Green Version]
- Bachinski, L.L.; Baggerly, K.A.; Neubauer, V.L.; Nixon, T.J.; Raheem, O.; Sirito, M.; Unruh, A.K.; Zhang, J.; Nagarajan, L.; Timchenko, L.T.; et al. Most Expression and Splicing Changes in Myotonic Dystrophy Type 1 and Type 2 Skeletal Muscle Are Shared with Other Muscular Dystrophies. Neuromuscul. Disord. 2014, 24, 227–240. [Google Scholar] [CrossRef] [Green Version]
- Swick, H.M.; Werlin, S.L.; Dodds, W.J.; Hogan, W.J. Pharyngoesophageal Motor Function in Patients with Myotonic Dystrophy. Ann. Neurol. 1981, 10, 454–457. [Google Scholar] [CrossRef] [PubMed]
- Lecointe-Besancon, I.; Leroy, F.; Devroede, G.; Chevrollier, M.; Lebeurier, F.; Congard, P.; Arhan, P. A Comparative Study of Esophageal and Anorectal Motility in Myotonic Dystrophy. Dig. Dis. Sci. 1999, 44, 1090–1099. [Google Scholar] [CrossRef] [PubMed]
- Lewis, T.D.; Daniel, E.E. Gastroduodenal Motility in a Case of Dystrophia Myotonica. Gastroenterology 1981, 81, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Ghazaleh, S.; Nehme, C.; Khader, Y.; Hasan, S.; Nawras, A. Combined Achalasia and Cricopharyngeal Achalasia in a Patient with Type 1 Myotonic Dystrophy: A Case Report. Gastroenterol. Hepatol. Bed Bench 2020, 13, 181–183. [Google Scholar] [PubMed]
- Sato, H.; Mizuno, K.; Hashimoto, S.; Takatsuna, M.; Terai, S. Achalasia in a Patient with Myotonic Dystrophy. Intern. Med. 2020, 59, 67–68. [Google Scholar] [CrossRef] [PubMed]
- Ogasawara, N.; Sato, K.; Tsutsumiuchi, M.; Kanzaki, M.; Uesaka, Y. Steakhouse Syndrome in Myotonic Dystrophy. Intern. Med. 2017, 56, 3179–3181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuiper, D.H. Gastric Bezoar in a Patient with Myotonic Dystrophy. Am. J. Dig. Dis. 1971, 16, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Bodensteiner, J.B.; Grunow, J.E. Gastroparesis in Neonatal Myotonic Dystrophy. Muscle Nerve 1984, 7, 486–487. [Google Scholar] [CrossRef]
- Rönnblom, A.; Andersson, S.; Hellström, P.M.; Danielsson, Å. Gastric Emptying in Myotonic Dystrophy. Eur. J. Clin. Investig. 2002, 32, 570–574. [Google Scholar] [CrossRef] [Green Version]
- Bellini, M.; Alduini, P.; Costa, F.; Tosetti, C.; Pasquali, L.; Pucciani, F.; Tornar, A.; Mammini, C.; Siciliano, G.; Maltinti, G.; et al. Gastric Emptying in Myotonic Dystrophic Patients. Dig. Liver Dis. 2002, 34, 484–488. [Google Scholar] [CrossRef]
- Tanaka, Y.; Kato, T.; Nishida, H.; Yamada, M.; Koumura, A.; Sakurai, T.; Hayashi, Y.; Kimura, A.; Hozumi, I.; Araki, H.; et al. Is There a Difference in Gastric Emptying between Myotonic Dystrophy Type 1 Patients with and without Gastrointestinal Symptoms? J. Neurol. 2013, 260, 1611–1616. [Google Scholar] [CrossRef]
- Braden, B.; Adams, S.; Duan, L.-P.; Orth, K.-H.; Maul, F.-D.; Lembcke, B.; Hör, G.; Caspary, W.F. The [13C]Acetate Breath Test Accurately Reflects Gastric Emptying of Liquids in Both Liquid and Semisolid Test Meals. Gastroenterology 1995, 108, 1048–1055. [Google Scholar] [CrossRef] [PubMed]
- Housset, C.; Chrétien, Y.; Debray, D.; Chignard, N. Functions of the Gallbladder. Compr. Physiol. 2016, 6, 1549–1577. [Google Scholar] [CrossRef] [PubMed]
- Schwindt, W.D.; Bernhardt, L.C.; Peters, H.A. Cholelithiasis and Associated Complications of Myotonia Dystrophica. Postgrad. Med. 1969, 46, 80–83. [Google Scholar] [CrossRef]
- Chiu, V.S.W. Gastrointestinal Disturbances in Myotonic Dystrophica. Aga Annu. Meet. Abstr. 1962, 42, 745–746. [Google Scholar]
- Achiron, A.; Barak, Y.; Magal, N.; Shohat, M.; Cohen, M.; Barar, R.; Gadoth, N. Abnormal Liver Test Results in Myotonic Dystrophy. J. Clin. Gastroenterol. 1998, 26, 292–295. [Google Scholar] [CrossRef] [PubMed]
- Rönnemaa, T.; Alaranta, H.; Viikari, J.; Tilvis, R.; Falck, B. Increased Activity Of Serum Γ-Glutamyltransferase In Myotonic Dystrophy. Acta Med. Scand. 1987, 222, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Heatwole, C.R.; Miller, J.; Martens, B.; Moxley, R.T. Laboratory Abnormalities in Ambulatory Patients With Myotonic Dystrophy Type 1. Arch. Neurol. 2006, 63, 1149–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruinenberg, J.; Rieu, P.; Gabreëls, F.; Tolboom, J. Intestinal Pseudo-obstruction Syndrome in a Child with Myotonic Dystrophy. Acta Paediatr. 1996, 85, 121–123. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.M.; Krishnamurthy, S.; Wattchow, D.A.; Furness, J.B.; Schuffler, M.D. Megacolon in Myotonic Dystrophy Caused by a Degenerative Neuropathy of the Myenteric Plexus. Gastroenterology 1988, 95, 820–827. [Google Scholar] [CrossRef]
- Brunner, H.G.; Hamel, B.C.; Rieu, P.; Höweler, C.J.; Peters, F.T. Intestinal Pseudo-Obstruction in Myotonic Dystrophy. J. Med. Genet. 1992, 29, 791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sartoretti, C.; Sartoretti, S.; DeLorenzi, D.; Buchmann, P. Intestinal Non-Rotation and Pseudoobstruction in Myotonic Dystrophy: Case Report and Review of the Literature. Int. J. Color. Dis. 1996, 11, 10–14. [Google Scholar] [CrossRef]
- Glaser, A.M.; Johnston, J.H.; Gleason, W.A.; Rhoads, J.M. Myotonic Dystrophy as a Cause of Colonic Pseudoobstruction: Not Just Another Constipated Child. Clin. Case Rep. 2015, 3, 424–426. [Google Scholar] [CrossRef]
- Pelizzo, G.; Calcaterra, V.; Villanacci, V.; Mura, G.B.; Bassotti, G. Myotonic Dystrophy Type 1 and Pseudo-Obstruction in a Child with Smooth Muscle α-Actin Deficiency and Eosinophilic Myenteric Plexitis. Turk. J. Gastroenterol. 2018, 29, 226–229. [Google Scholar] [CrossRef] [PubMed]
- Nowak, T.V.; Anuras, S.; Brown, B.P.; Ionasescu, V.; Green, J.B. Small Intestinal Motility in Myotonic Dystrophy Patients. Gastroenterology 1984, 86, 808–813. [Google Scholar] [PubMed]
- Cheng, H.M.; Mah, K.K.; Seluakumaran, K. Defining Physiology: Principles, Themes, Concepts. Volume 2, Neurophysiology and Gastrointestinal Systems; Springer Nature: Berlin/Heidelberg, Germany, 2020. [Google Scholar] [CrossRef]
- Weiner, M. Myotonic Megacolon in Myotonic Dystrophy. Am. J. Roentgenol. 1978, 130, 177–179. [Google Scholar] [CrossRef]
- Vanuytsel, T.; Tack, J.; Farre, R. The Role of Intestinal Permeability in Gastrointestinal Disorders and Current Methods of Evaluation. Front. Nutr. 2021, 8, 717925. [Google Scholar] [CrossRef]
- Sjaastad, O. Intestinal Absorption in Myotonic Dystrophy. Acta Neurol. Scand. 1975, 51, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Bjarnason, I.; Macpherson, A.; Hollander, D. Intestinal Permeability: An Overview. Gastroenterology 1995, 108, 1566–1581. [Google Scholar] [CrossRef]
- Shieh, K.; Gilchrist, J.M.; Promrat, K. Frequency and Predictors of Nonalcoholic Fatty Liver Disease in Myotonic Dystrophy. Muscle Nerve 2010, 41, 197–201. [Google Scholar] [CrossRef]
- Miele, L.; Valenza, V.; Torre, G.L.; Montalto, M.; Cammarota, G.; Ricci, R.; Mascianà, R.; Forgione, A.; Gabrieli, M.L.; Perotti, G.; et al. Increased Intestinal Permeability and Tight Junction Alterations in Nonalcoholic Fatty Liver Disease. Hepatology 2009, 49, 1877–1887. [Google Scholar] [CrossRef]
- Eckardt, V.F.; Nix, W. The Anal Sphincter in Patients with Myotonic Muscular Dystrophy. Gastroenterology 1991, 100, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Hamel-Roy, J.; Devroede, G.; Arhan, P.; Tetreault, J.; Lemieux, B.; Scott, H. Functional Abnormalities of the Anal Sphincters in Patients with Myotonic Dystrophy. Gastroenterology 1984, 86, 1469–1474. [Google Scholar] [CrossRef] [PubMed]
- Abercrombie, J.F.; Rogers, J.; Swash, M. Faecal Incontinence in Myotonic Dystrophy. J. Neurol. Neurosurg. Psychiatry 1998, 64, 128–130. [Google Scholar] [CrossRef] [PubMed]
- Sanders, K.M.; Koh, S.D.; Ro, S.; Ward, S.M. Regulation of Gastrointestinal Motility—Insights from Smooth Muscle Biology. Nat. Rev. Gastroenterol. 2012, 9, 633–645. [Google Scholar] [CrossRef] [PubMed]
- Camilleri, M. Gastrointestinal Motility Disorders in Neurologic Disease. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Sanders, K.M.; Ward, S.M. Nitric Oxide and Its Role as a Non-adrenergic, Non-cholinergic Inhibitory Neurotransmitter in the Gastrointestinal Tract. Brit. J. Pharmacol. 2019, 176, 212–227. [Google Scholar] [CrossRef] [Green Version]
- Bellini, M.; Biagi, S.; Stasi, C.; Costa, F.; Mumolo, M.G.; Ricchiuti, A.; Marchi, S. Gastrointestinal Manifestations in Myotonic Muscular Dystrophy. World J. Gastroenterol. 2006, 12, 1821. [Google Scholar] [CrossRef]
- Browning, K.N.; Verheijden, S.; Boeckxstaens, G.E. The Vagus Nerve in Appetite Regulation, Mood, and Intestinal Inflammation. Gastroenterology 2017, 152, 730–744. [Google Scholar] [CrossRef] [Green Version]
- Aminoff, M.J.; Beckley, D.J.; McIlroy, M.B. Autonomic Function in Myotonic Dystrophy. Arch. Neurol. 1985, 42, 16. [Google Scholar] [CrossRef]
- Olofsson, B.-O.; Niklasson, U.; Forsberg, H.; Bjerle, P.; Andersson, S.; Henriksson, A. Assessment of Autonomic Nerve Function in Myotonic Dystrophy. J. Auton. Nerv. Syst. 1990, 29, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Fairhurst, A.S.; Lorenzen, L.; Reavie, D. Altered Tracheal Smooth Muscle Activities in an Animal Model of Human Myotonic Dystrophy. Life Sci. 1976, 18, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhou, H.; Mahavadi, S.; Sriwai, W.; Lyall, V.; Murthy, K.S. Signaling Pathways Mediating Gastrointestinal Smooth Muscle Contraction and MLC20 Phosphorylation by Motilin Receptors. Am. J. Physiol. Liver Physiol. 2005, 288, G23–G31. [Google Scholar] [CrossRef] [Green Version]
- Asnicar, F.; Leeming, E.R.; Dimidi, E.; Mazidi, M.; Franks, P.W.; Khatib, H.A.; Valdes, A.M.; Davies, R.; Bakker, E.; Francis, L.; et al. Blue Poo: Impact of Gut Transit Time on the Gut Microbiome Using a Novel Marker. Gut 2021, 70, 1665–1674. [Google Scholar] [CrossRef] [PubMed]
- Graham, H.K.; Maina, I.; Goldstein, A.M.; Nagy, N. Intestinal Smooth Muscle Is Required for Patterning the Enteric Nervous System. J. Anat. 2017, 230, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Hao, M.M.; Foong, J.P.P.; Bornstein, J.C.; Li, Z.L.; Berghe, P.V.; Boesmans, W. Enteric Nervous System Assembly: Functional Integration within the Developing Gut. Dev. Biol. 2016, 417, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Jansen, G.; Groenen, P.J.T.A.; Bächner, D.; Jap, P.H.K.; Coerwinkel, M.; Oerlemans, F.; van den Broek, W.; Gohlsch, B.; Pette, D.; Plomp, J.J.; et al. Abnormal Myotonic Dystrophy Protein Kinase Levels Produce Only Mild Myopathy in Mice. Nat. Genet. 1996, 13, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Kaliman, P.; Catalucci, D.; Lam, J.T.; Kondo, R.; Gutiérrez, J.C.P.; Reddy, S.; Palacín, M.; Zorzano, A.; Chien, K.R.; Ruiz-Lozano, P. Myotonic Dystrophy Protein Kinase Phosphorylates Phospholamban and Regulates Calcium Uptake in Cardiomyocyte Sarcoplasmic Reticulum*. J. Biol. Chem. 2005, 280, 8016–8021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardani, R.; Mancinelli, E.; Saino, G.; Bonavina, L.; Meola, G. A Putative Role of Ribonuclear Inclusions and MBNL1 in the Impairment of Gallbladder Smooth Muscle Contractility with Cholelithiasis in Myotonic Dystrophy Type 1. Neuromuscul. Disord. 2008, 18, 641–645. [Google Scholar] [CrossRef] [PubMed]
- Lukáš, Z.; Falk, M.; Feit, J.; Souček, O.; Falková, I.; Štefančíková, L.; Janoušová, E.; Fajkusová, L.; Zaorálková, J.; Hrabálková, R. Sequestration of MBNL1 in Tissues of Patients with Myotonic Dystrophy Type 2. Neuromuscul. Disord. 2012, 22, 604–616. [Google Scholar] [CrossRef]
- Maeda, M.; Taft, C.S.; Bush, E.W.; Holder, E.; Bailey, W.M.; Neville, H.; Perryman, M.B.; Bies, R.D. Identification, Tissue-Specific Expression, and Subcellular Localization of the 80- and 71-KDa Forms of Myotonic Dystrophy Kinase Protein (∗). J. Biol. Chem. 1995, 270, 20246–20249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furling, D.; Lam, L.T.; Agbulut, O.; Butler-Browne, G.S.; Morris, G.E. Changes in Myotonic Dystrophy Protein Kinase Levels and Muscle Development in Congenital Myotonic Dystrophy. Am. J. Pathol. 2003, 162, 1001–1009. [Google Scholar] [CrossRef] [Green Version]
- Salvatori, S.; Fanin, M.; Trevisan, C.P.; Furlan, S.; Reddy, S.; Nagy, J.I.; Angelini, C. Decreased Expression of DMPK: Correlation with CTG Repeat Expansion and Fibre Type Composition in Myotonic Dystrophy Type 1. Neurol. Sci. 2005, 26, 235–242. [Google Scholar] [CrossRef]
- Wei, C.; Stock, L.; Schneider-Gold, C.; Sommer, C.; Timchenko, N.A.; Timchenko, L. Reduction of Cellular Nucleic Acid Binding Protein Encoded by a Myotonic Dystrophy Type 2 Gene Causes Muscle Atrophy. Mol. Cell. Biol. 2018, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zu, T.; Gibbens, B.; Doty, N.S.; Gomes-Pereira, M.; Huguet, A.; Stone, M.D.; Margolis, J.; Peterson, M.; Markowski, T.W.; Ingram, M.A.C.; et al. Non-ATG–Initiated Translation Directed by Microsatellite Expansions. Proc. Natl. Acad. Sci. USA 2011, 108, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Ash, P.E.A.; Bieniek, K.F.; Gendron, T.F.; Caulfield, T.; Lin, W.-L.; DeJesus-Hernandez, M.; van Blitterswijk, M.M.; Jansen-West, K.; Paul, J.W.; Rademakers, R.; et al. Unconventional Translation of C9ORF72 GGGGCC Expansion Generates Insoluble Polypeptides Specific to C9FTD/ALS. Neuron 2013, 77, 639–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, K.; Weng, S.-M.; Arzberger, T.; May, S.; Rentzsch, K.; Kremmer, E.; Schmid, B.; Kretzschmar, H.A.; Cruts, M.; Broeckhoven, C.V.; et al. The C9orf72 GGGGCC Repeat Is Translated into Aggregating Dipeptide-Repeat Proteins in FTLD/ALS. Science 2013, 339, 1335–1338. [Google Scholar] [CrossRef] [PubMed]
- Zu, T.; Cleary, J.D.; Liu, Y.; Bañez-Coronel, M.; Bubenik, J.L.; Ayhan, F.; Ashizawa, T.; Xia, G.; Clark, H.B.; Yachnis, A.T.; et al. RAN Translation Regulated by Muscleblind Proteins in Myotonic Dystrophy Type 2. Neuron 2017, 95, 1292–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banez-Coronel, M.; Ranum, L.P.W. Repeat-Associated Non-AUG (RAN) Translation: Insights from Pathology. Lab. Investig. 2019, 99, 929–942. [Google Scholar] [CrossRef] [PubMed]
- Carrell, S.T.; Carrell, E.M.; Auerbach, D.; Pandey, S.K.; Bennett, C.F.; Dirksen, R.T.; Thornton, C.A. Dmpk Gene Deletion or Antisense Knockdown Does Not Compromise Cardiac or Skeletal Muscle Function in Mice. Hum. Mol. Genet. 2016, 25, 4328–4338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Ven, P.F.M.; Jansen, G.; van Kuppevelt, T.H.M.S.M.; Perryman, M.B.; Lupa, M.; Dunne, P.W.; ter Laak, H.J.; Jap, P.H.K.; Veerkamp, J.H.; Epstein, H.F.; et al. Myotonic Dystrophy Kinase Is a Component of Neuromuscular Junctions. Hum. Mol. Genet. 1993, 2, 1889–1894. [Google Scholar] [CrossRef]
- Murányi, A.; Zhang, R.; Liu, F.; Hirano, K.; Ito, M.; Epstein, H.F.; Hartshorne, D.J. Myotonic Dystrophy Protein Kinase Phosphorylates the Myosin Phosphatase Targeting Subunit and Inhibits Myosin Phosphatase Activity. FEBS Lett. 2001, 493, 80–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.Y.; Park, C.; Ha, S.E.; Park, P.J.; Berent, R.M.; Jorgensen, B.G.; Corrigan, R.D.; Grainger, N.; Blair, P.J.; Slivano, O.J.; et al. Serum Response Factor Regulates Smooth Muscle Contractility via Myotonic Dystrophy Protein Kinases and L-Type Calcium Channels. PLoS ONE 2017, 12, e0171262. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Wang, Y.; Abe, Y.; Cheney, L.; Udd, B.; Li, Y.-P. Haploinsuffciency for Znf9 in Znf9+/− Mice Is Associated with Multiorgan Abnormalities Resembling Myotonic Dystrophy. J. Mol. Biol. 2007, 368, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Coni, S.; Falconio, F.A.; Marzullo, M.; Munafò, M.; Zuliani, B.; Mosti, F.; Fatica, A.; Ianniello, Z.; Bordone, R.; Macone, A.; et al. Translational Control of Polyamine Metabolism by CNBP Is Required for Drosophila Locomotor Function. eLife 2021, 10, e69269. [Google Scholar] [CrossRef]
- Margolis, J.M.; Schoser, B.G.; Moseley, M.L.; Day, J.W.; Ranum, L.P.W. DM2 Intronic Expansions: Evidence for CCUG Accumulation without Flanking Sequence or Effects on ZNF9 MRNA Processing or Protein Expression. Hum. Mol. Genet. 2006, 15, 1808–1815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santoro, M.; Fontana, L.; Maiorca, F.; Centofanti, F.; Massa, R.; Silvestri, G.; Novelli, G.; Botta, A. Expanded [CCTG]n Repetitions Are Not Associated with Abnormal Methylation at the CNBP Locus in Myotonic Dystrophy Type 2 (DM2) Patients. Biochim. et Biophys. Acta (BBA)-Mol. Basis Dis. 2018, 1864, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Fisher, S.A. Vascular Smooth Muscle Phenotypic Diversity and Function. Physiol. Genom. 2010, 42A, 169–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, A.T.; Gomez, D.; Bell, R.D.; Campbell, J.H.; Clowes, A.W.; Gabbiani, G.; Giachelli, C.M.; Parmacek, M.S.; Raines, E.W.; Rusch, N.J.; et al. Smooth Muscle Cell Plasticity. Circ. Res. 2013, 112, 17–22. [Google Scholar] [CrossRef] [Green Version]
- Llorian, M.; Gooding, C.; Bellora, N.; Hallegger, M.; Buckroyd, A.; Wang, X.; Rajgor, D.; Kayikci, M.; Feltham, J.; Ule, J.; et al. The Alternative Splicing Program of Differentiated Smooth Muscle Cells Involves Concerted Non-Productive Splicing of Post-Transcriptional Regulators. Nucleic Acids Res. 2016, 44, 8933–8950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashiwada, K.; Nishida, W.; Hayashi, K.; Ozawa, K.; Yamanaka, Y.; Saga, H.; Yamashita, T.; Tohyama, M.; Shimada, S.; Sato, K.; et al. Coordinate Expression of α-Tropomyosin and Caldesmon Isoforms in Association with Phenotypic Modulation of Smooth Muscle Cells*. J. Biol. Chem. 1997, 272, 15396–15404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagaki-Silva, E.E.; Gooding, C.; Llorian, M.; Jacob, A.G.; Richards, F.; Buckroyd, A.; Sinha, S.; Smith, C.W. Identification of RBPMS as a Mammalian Smooth Muscle Master Splicing Regulator via Proximity of Its Gene with Super-Enhancers. eLife 2019, 8, e46327. [Google Scholar] [CrossRef] [PubMed]
- Kanoldt, V.; Kluger, C.; Barz, C.; Schweizer, A.-L.; Ramanujam, D.; Windgasse, L.; Engelhardt, S.; Chrostek-Grashoff, A.; Grashoff, C. Metavinculin Modulates Force Transduction in Cell Adhesion Sites. Nat. Commun. 2020, 11, 6403. [Google Scholar] [CrossRef]
- Guo, H.; Wang, C.-L.A. Specific Disruption of Smooth Muscle Caldesmon Expression in Mice. Biochem. Biophys. Res. Commun. 2005, 330, 1132–1137. [Google Scholar] [CrossRef]
- Abrams, J.; Davuluri, G.; Seiler, C.; Pack, M. Smooth Muscle Caldesmon Modulates Peristalsis in the Wild Type and Non-innervated Zebrafish Intestine. Neurogastroenterol. Motil. 2012, 24, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Morinaga, A.; Ito, J.; Niimi, T.; Maturana, A.D. RBM20 Regulates CaV1.2 Surface Expression by Promoting Exon 9* Inclusion of CACNA1C in Neonatal Rat Cardiomyocytes. Int. J. Mol. Sci. 2019, 20, 5591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.; Liang, M.C.; Soong, T.W. Alternative Splicing of L-Type CaV1.2 Calcium Channels: Implications in Cardiovascular Diseases. Genes 2017, 8, 344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khatri, J.J.; Joyce, K.M.; Brozovich, F.V.; Fisher, S.A. Role of Myosin Phosphatase Isoforms in CGMP-Mediated Smooth Muscle Relaxation*. J. Biol. Chem. 2001, 276, 37250–37257. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Reho, J.J.; Wirth, B.; Fisher, S.A. TRA2β Controls Mypt1 Exon 24 Splicing in the Developmental Maturation of Mouse Mesenteric Artery Smooth Muscle. Am. J. Physiol. Physiol. 2015, 308, C289–C296. [Google Scholar] [CrossRef] [Green Version]
- Smith, L.; Parizi-Robinson, M.; Zhu, M.-S.; Zhi, G.; Fukui, R.; Kamm, K.E.; Stull, J.T. Properties of Long Myosin Light Chain Kinase Binding to F-Actin in Vitro and in Vivo *. J. Biol. Chem. 2002, 277, 35597–35604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rovner, A.S.; Freyzon, Y.; Trybus, K.M. An Insert in the Motor Domain Determines the Functional Properties of Expressed Smooth Muscle Myosin Isoforms. J. Muscle Res. Cell Motil. 1997, 18, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Kelley, C.A.; Takahashi, M.; Yu, J.H.; Adelstein, R.S. An Insert of Seven Amino Acids Confers Functional Differences between Smooth Muscle Myosins from the Intestines and Vasculature. J. Biol. Chem. 1993, 268, 12848–12854. [Google Scholar] [CrossRef]
- Ho, T.H.; Bundman, D.; Armstrong, D.L.; Cooper, T.A. Transgenic Mice Expressing CUG-BP1 Reproduce Splicing Mis-Regulation Observed in Myotonic Dystrophy. Hum. Mol. Genet. 2005, 14, 1539–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuyumcu-Martinez, N.M.; Wang, G.-S.; Cooper, T.A. Increased Steady-State Levels of CUGBP1 in Myotonic Dystrophy 1 Are Due to PKC-Mediated Hyperphosphorylation. Mol. Cell 2007, 28, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Charlet-B., N.; Savkur, R.S.; Singh, G.; Philips, A.V.; Grice, E.A.; Cooper, T.A. Loss of the Muscle-Specific Chloride Channel in Type 1 Myotonic Dystrophy Due to Misregulated Alternative Splicing. Mol. Cell 2002, 10, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Savkur, R.S.; Philips, A.V.; Cooper, T.A. Aberrant Regulation of Insulin Receptor Alternative Splicing Is Associated with Insulin Resistance in Myotonic Dystrophy. Nat. Genet. 2001, 29, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Pang, P.D.; Alsina, K.M.; Cao, S.; Koushik, A.B.; Wehrens, X.H.T.; Cooper, T.A. CRISPR-Mediated Expression of the Fetal Scn5a Isoform in Adult Mice Causes Conduction Defects and Arrhythmias. J. Am. Heart Assoc. 2018, 7, e010393. [Google Scholar] [CrossRef] [Green Version]
- Freyermuth, F.; Rau, F.; Kokunai, Y.; Linke, T.; Sellier, C.; Nakamori, M.; Kino, Y.; Arandel, L.; Jollet, A.; Thibault, C.; et al. Splicing Misregulation of SCN5A Contributes to Cardiac-Conduction Delay and Heart Arrhythmia in Myotonic Dystrophy. Nat. Commun. 2016, 7, 11067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brimacombe, K.R.; Ladd, A.N. Cloning and Embryonic Expression Patterns of the Chicken CELF Family. Dev. Dyn. 2007, 236, 2216–2224. [Google Scholar] [CrossRef] [PubMed]
- Kanadia, R.N.; Johnstone, K.A.; Mankodi, A.; Lungu, C.; Thornton, C.A.; Esson, D.; Timmers, A.M.; Hauswirth, W.W.; Swanson, M.S. A Muscleblind Knockout Model for Myotonic Dystrophy. Science 2003, 302, 1978–1980. [Google Scholar] [CrossRef] [Green Version]
- Pascual, M.; Vicente, M.; Monferrer, L.; Artero, R. The Muscleblind Family of Proteins: An Emerging Class of Regulators of Developmentally Programmed Alternative Splicing. Differentiation 2006, 74, 65–80. [Google Scholar] [CrossRef]
- Gooding, C.; Edge, C.; Lorenz, M.; Coelho, M.B.; Winters, M.; Kaminski, C.F.; Cherny, D.; Eperon, I.C.; Smith, C.W.J. MBNL1 and PTB Cooperate to Repress Splicing of Tpm1 Exon 3. Nucleic Acids Res. 2013, 41, 4765–4782. [Google Scholar] [CrossRef] [Green Version]
- Du, H.; Cline, M.S.; Osborne, R.J.; Tuttle, D.L.; Clark, T.A.; Donohue, J.P.; Hall, M.P.; Shiue, L.; Swanson, M.S.; Thornton, C.A.; et al. Aberrant Alternative Splicing and Extracellular Matrix Gene Expression in Mouse Models of Myotonic Dystrophy. Nat. Struct. Mol. Biol. 2010, 17, 187–193. [Google Scholar] [CrossRef] [Green Version]
- Hinman, M.N.; Richardson, J.I.; Sockol, R.A.; Aronson, E.D.; Stednitz, S.J.; Murray, K.N.; Berglund, J.A.; Guillemin, K. Zebrafish Mbnl Mutants Model Physical and Molecular Phenotypes of Myotonic Dystrophy. Dis. Model. Mech. 2021, 14, dmm045773. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, R.; Hamel, F.; Beaulieu, D.; Patry, L.; Haineault, C.; Tarnopolsky, M.; Schoser, B.; Puymirat, J. Absence of a Differentiation Defect in Muscle Satellite Cells from DM2 Patients. Neurobiol. Dis. 2009, 36, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Sznajder, Ł.J.; Swanson, M.S. Short Tandem Repeat Expansions and RNA-Mediated Pathogenesis in Myotonic Dystrophy. Int. J. Mol. Sci. 2019, 20, 3365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salisbury, E.; Schoser, B.; Schneider-Gold, C.; Wang, G.-L.; Huichalaf, C.; Jin, B.; Sirito, M.; Sarkar, P.; Krahe, R.; Timchenko, N.A.; et al. Expression of RNA CCUG Repeats Dysregulates Translation and Degradation of Proteins in Myotonic Dystrophy 2 Patients. Am. J. Pathol. 2009, 175, 748–762. [Google Scholar] [CrossRef] [Green Version]
- Gromak, N.; Matlin, A.J.; Cooper, T.A.; Smith, C.W.J. Antagonistic Regulation of α-Actinin Alternative Splicing by CELF Proteins and Polypyrimidine Tract Binding Protein. RNA 2003, 9, 443–456. [Google Scholar] [CrossRef] [Green Version]
- Waites, G.T.; Graham, I.R.; Jackson, P.; Millake, D.B.; Patel, B.; Blanchard, A.D.; Weller, P.A.; Eperon, I.C.; Critchley, D.R. Mutually Exclusive Splicing of Calcium-Binding Domain Exons in Chick Alpha-Actinin. J. Biol. Chem. 1992, 267, 6263–6271. [Google Scholar] [CrossRef]
- Louis, I.V.-S.; Dickson, A.M.; Bohjanen, P.R.; Wilusz, C.J. CELFish Ways to Modulate MRNA Decay. Biochim. et Biophys. Acta 2013, 1829, 695–707. [Google Scholar] [CrossRef] [Green Version]
- Kalsotra, A.; Xiao, X.; Ward, A.J.; Castle, J.C.; Johnson, J.M.; Burge, C.B.; Cooper, T.A. A Postnatal Switch of CELF and MBNL Proteins Reprograms Alternative Splicing in the Developing Heart. Proc. Natl. Acad. Sci. USA 2008, 105, 20333–20338. [Google Scholar] [CrossRef] [Green Version]
- Dhaenens, C.M.; Tran, H.; Frandemiche, M.-L.; Carpentier, C.; Schraen-Maschke, S.; Sistiaga, A.; Goicoechea, M.; Eddarkaoui, S.; Brussels, E.V.; Obriot, H.; et al. Mis-Splicing of Tau Exon 10 in Myotonic Dystrophy Type 1 Is Reproduced by Overexpression of CELF2 but Not by MBNL1 Silencing. Biochim. et Biophys. Acta (BBA)-Mol. Basis Dis. 2011, 1812, 732–742. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Serum Enzyme (Abbreviation) | Significance of Elevated Levels |
|---|---|
| Alkaline phosphatase (ALP) | Damage or disease of liver or bone; cholestasis |
| Alanine aminotransferase (ALT or GPT) | Hepatocellular damage |
| Gamma-glutamyl transferase (GGT) | Disease or damage of liver or bile ducts; cholestasis |
| 5′ nucleotidase | Cholestasis, hepatitis, ischemia, or tumor |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peterson, J.A.M.; Cooper, T.A. Clinical and Molecular Insights into Gastrointestinal Dysfunction in Myotonic Dystrophy Types 1 & 2. Int. J. Mol. Sci. 2022, 23, 14779. https://doi.org/10.3390/ijms232314779
Peterson JAM, Cooper TA. Clinical and Molecular Insights into Gastrointestinal Dysfunction in Myotonic Dystrophy Types 1 & 2. International Journal of Molecular Sciences. 2022; 23(23):14779. https://doi.org/10.3390/ijms232314779
Chicago/Turabian StylePeterson, Janel A. M., and Thomas A. Cooper. 2022. "Clinical and Molecular Insights into Gastrointestinal Dysfunction in Myotonic Dystrophy Types 1 & 2" International Journal of Molecular Sciences 23, no. 23: 14779. https://doi.org/10.3390/ijms232314779