
Allopatric Lineage Divergence of the East Asian Endemic Herb Conandron ramondioides Inferred from Low-Copy Nuclear and Plastid Markers

 , and
, and
Abstract
:1. Introduction
2. Results
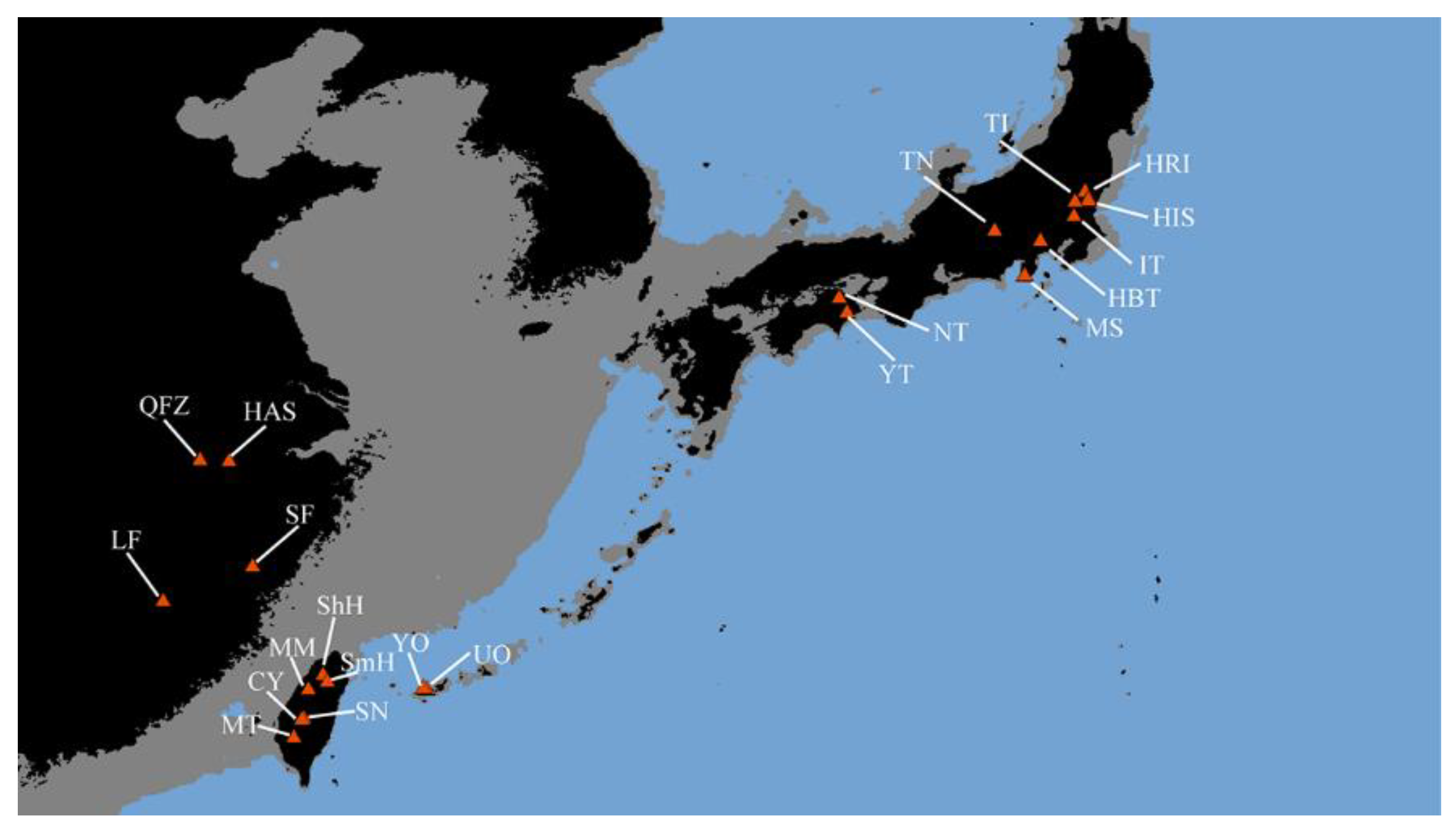
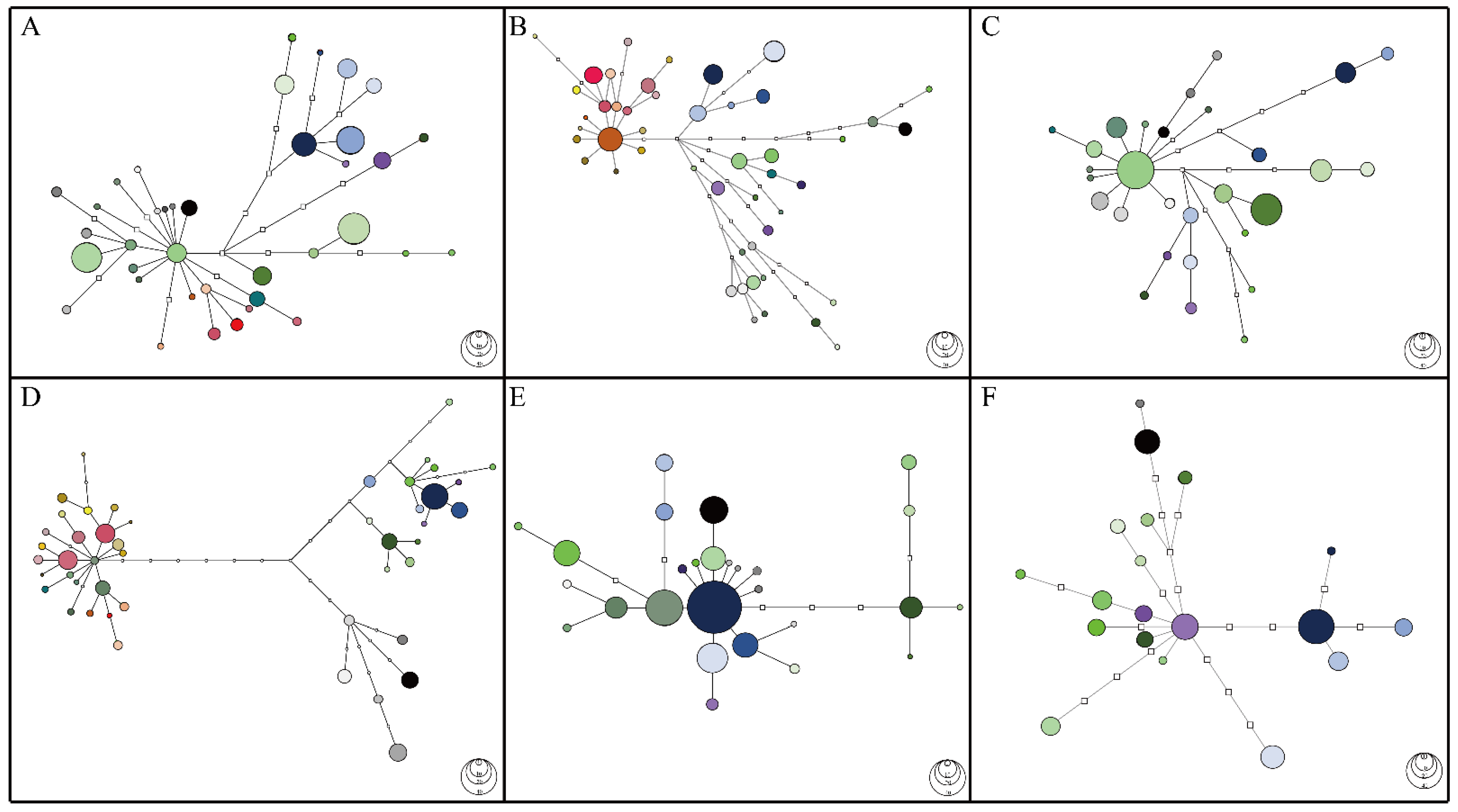
2.1. Haplotype Networks Reconstructed from Four Low-Copy Markers Reveal Three Groups of C. ramondioides Haplotypes Which Correspond to Geographical Locations
2.2. Two Groups of C. ramondioides Are Proposed from the Reconstructed ITS Ribotype Network
2.3. Two Groups of C. ramondioides Are Shown by the Reconstructed cpDNA Haplotype Network
2.4. Significant MK Test Results Suggest That CrCYC1 Deviates from the Neutral Evolution Hypothesis
2.5. Multiple ITS Haplotypes Were Identified from Selected Conandron Individuals
2.6. Significant Tajima’s D and Fu’s Fs Suggest cpDNA Amplified from Southeast China Deviate from Neutral Evolution
2.7. Genetic Diversity Measured in Conandron Populations
2.8. Significant Fst Suggests Population Differentiation among Assigned Geographical Regions
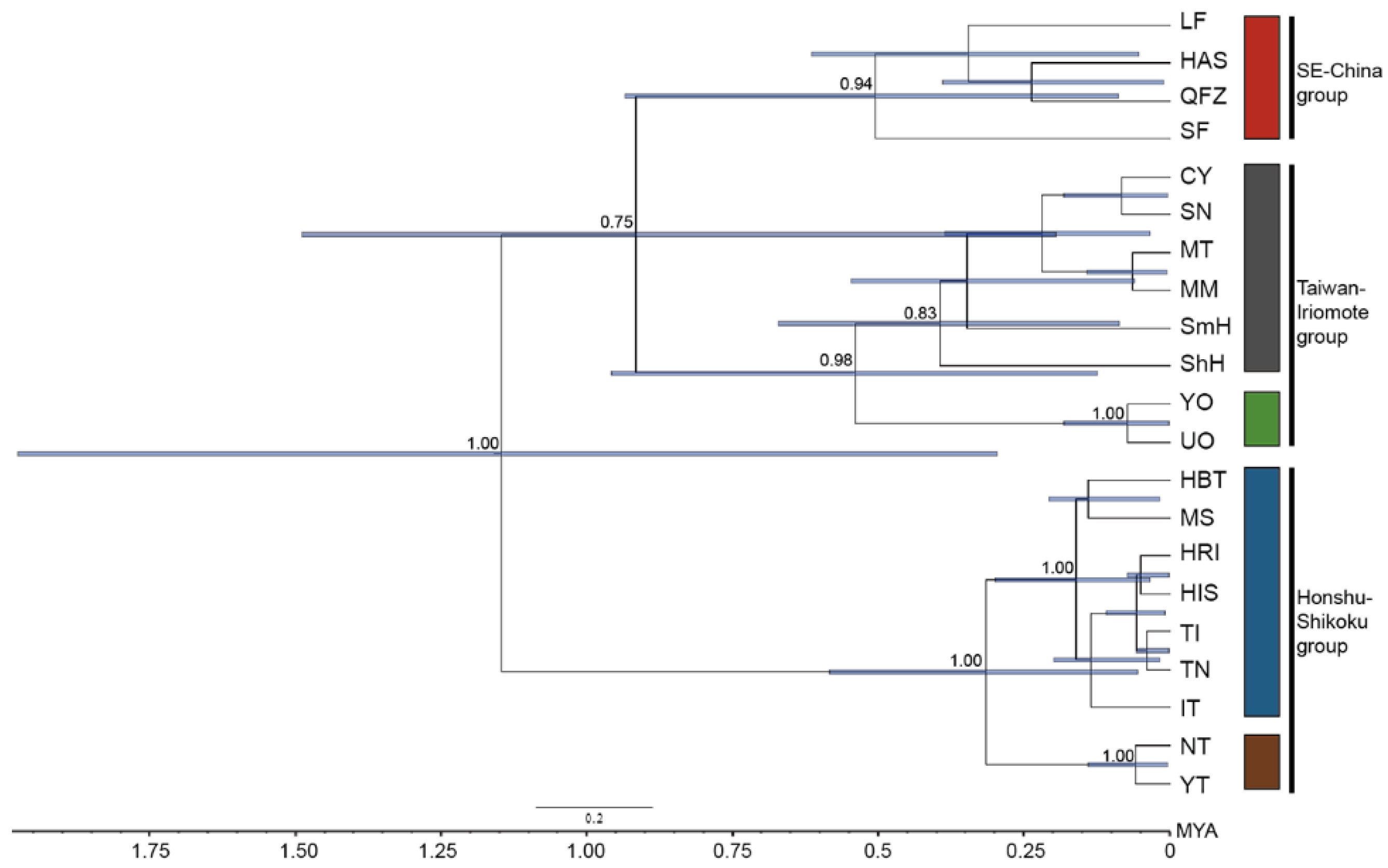
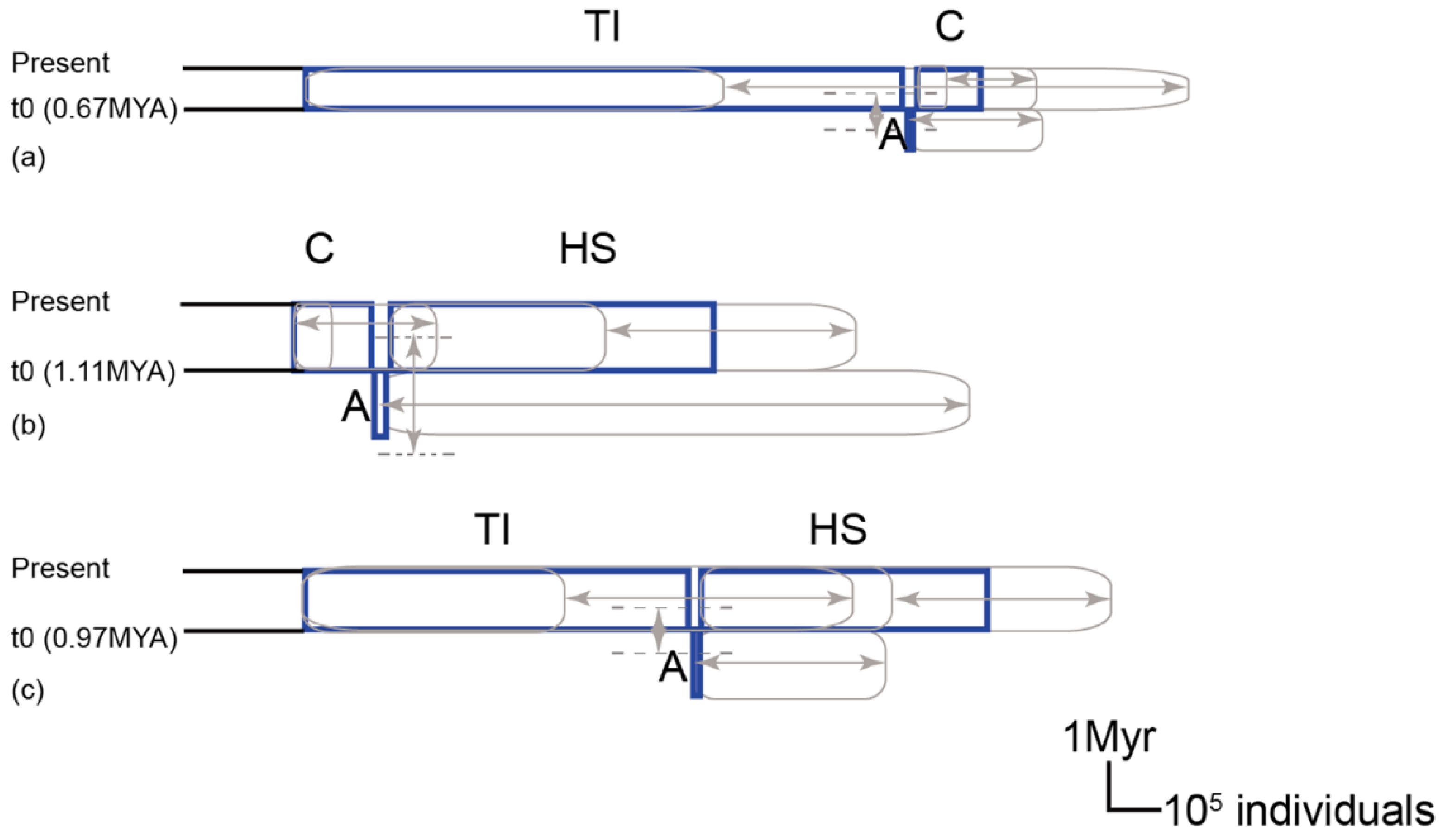
2.9. Lineage Divergence Order and Divergence Time among C. ramondioides Lineages Revealed by nrDNA
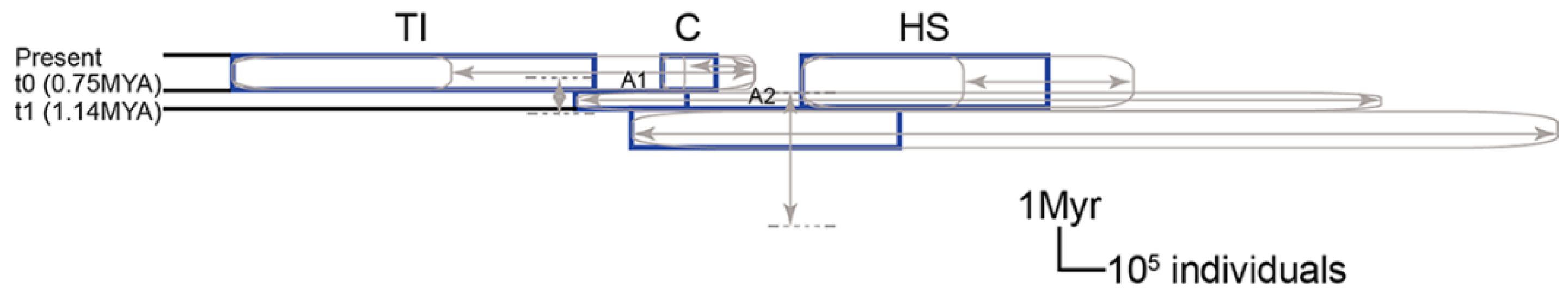
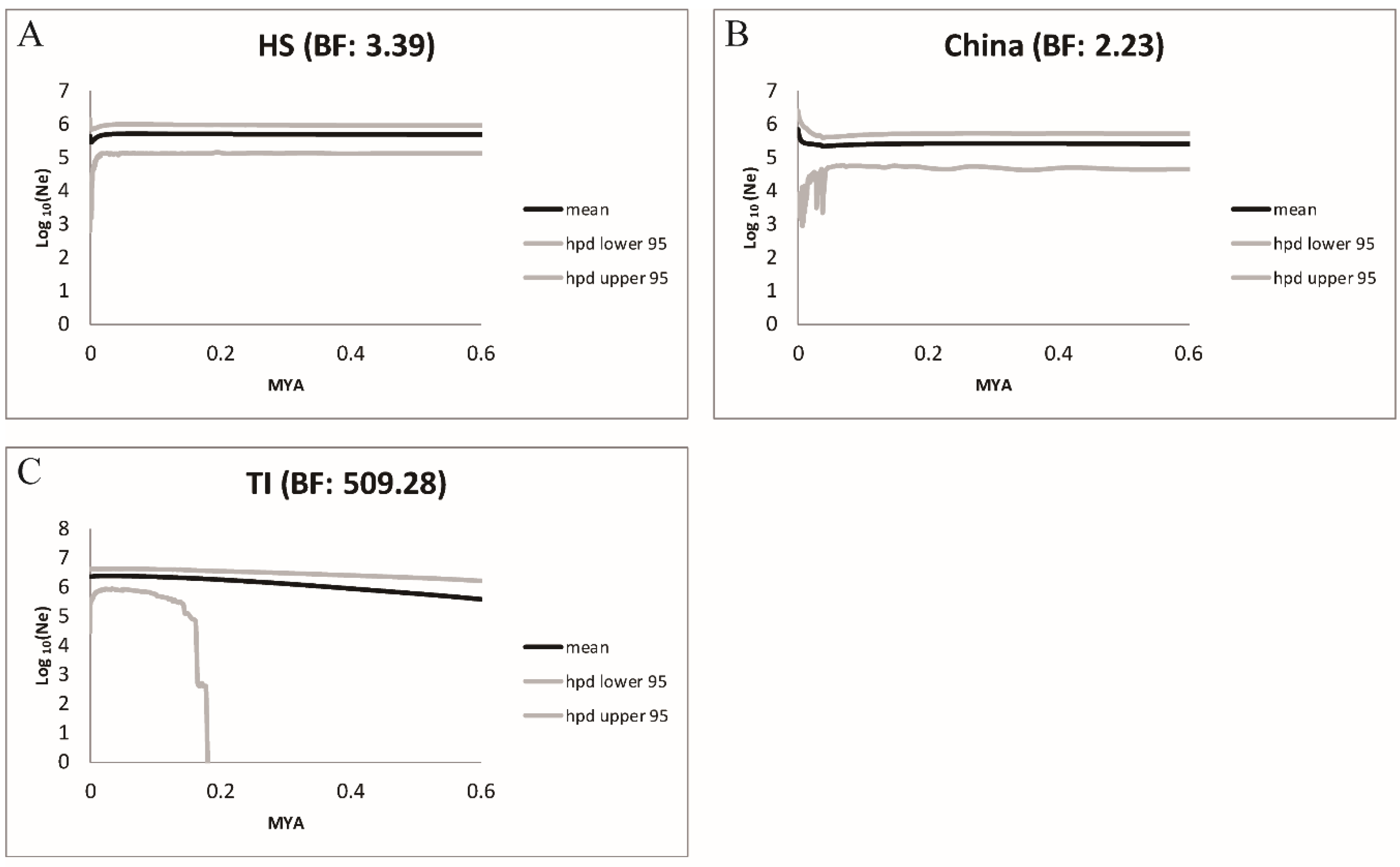
2.10. Population Dynamic through Time of Conandron
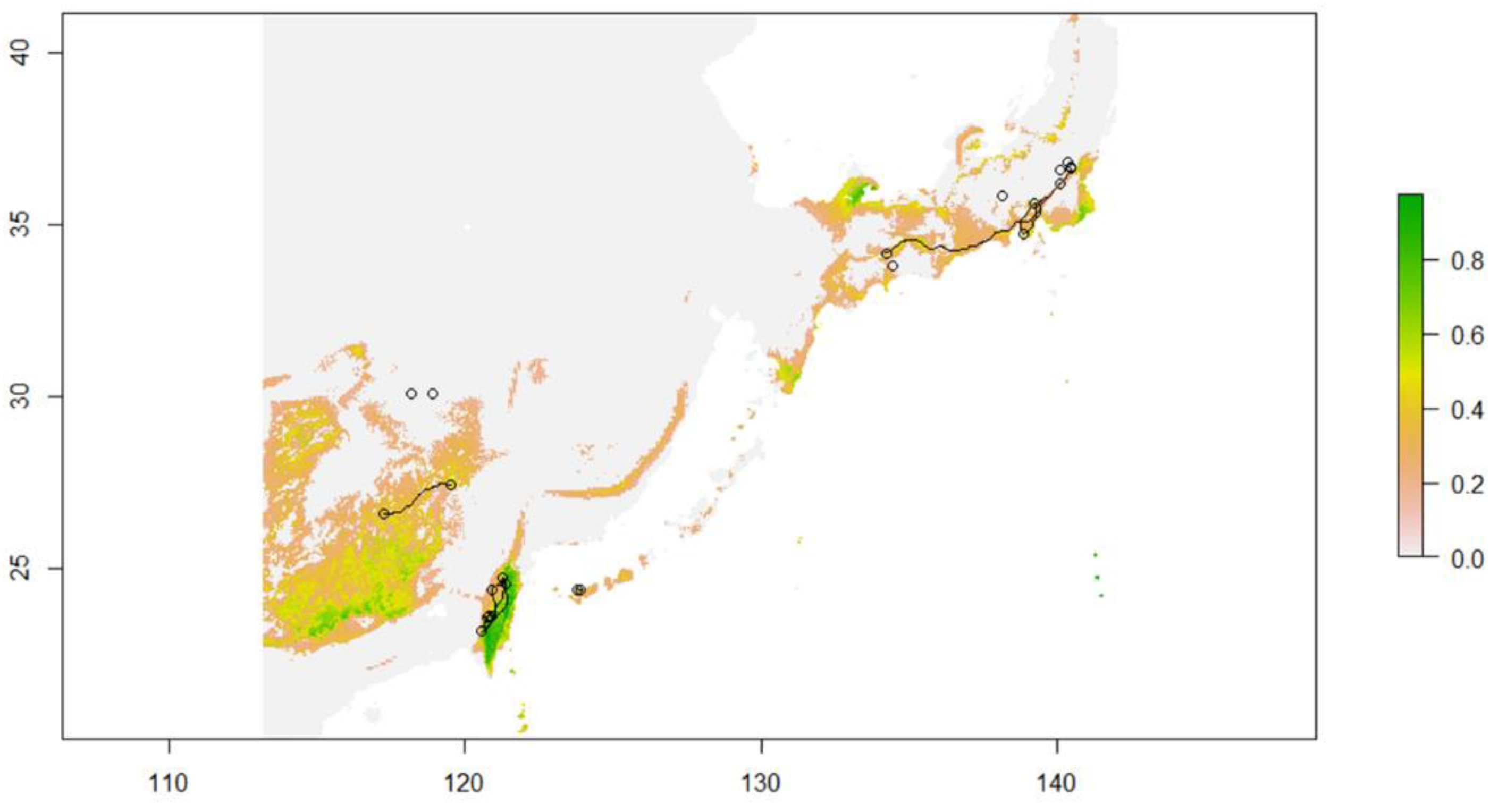
2.11. Modeled Migration Route of C. ramondioides during the LGM (Least-Cost Path)
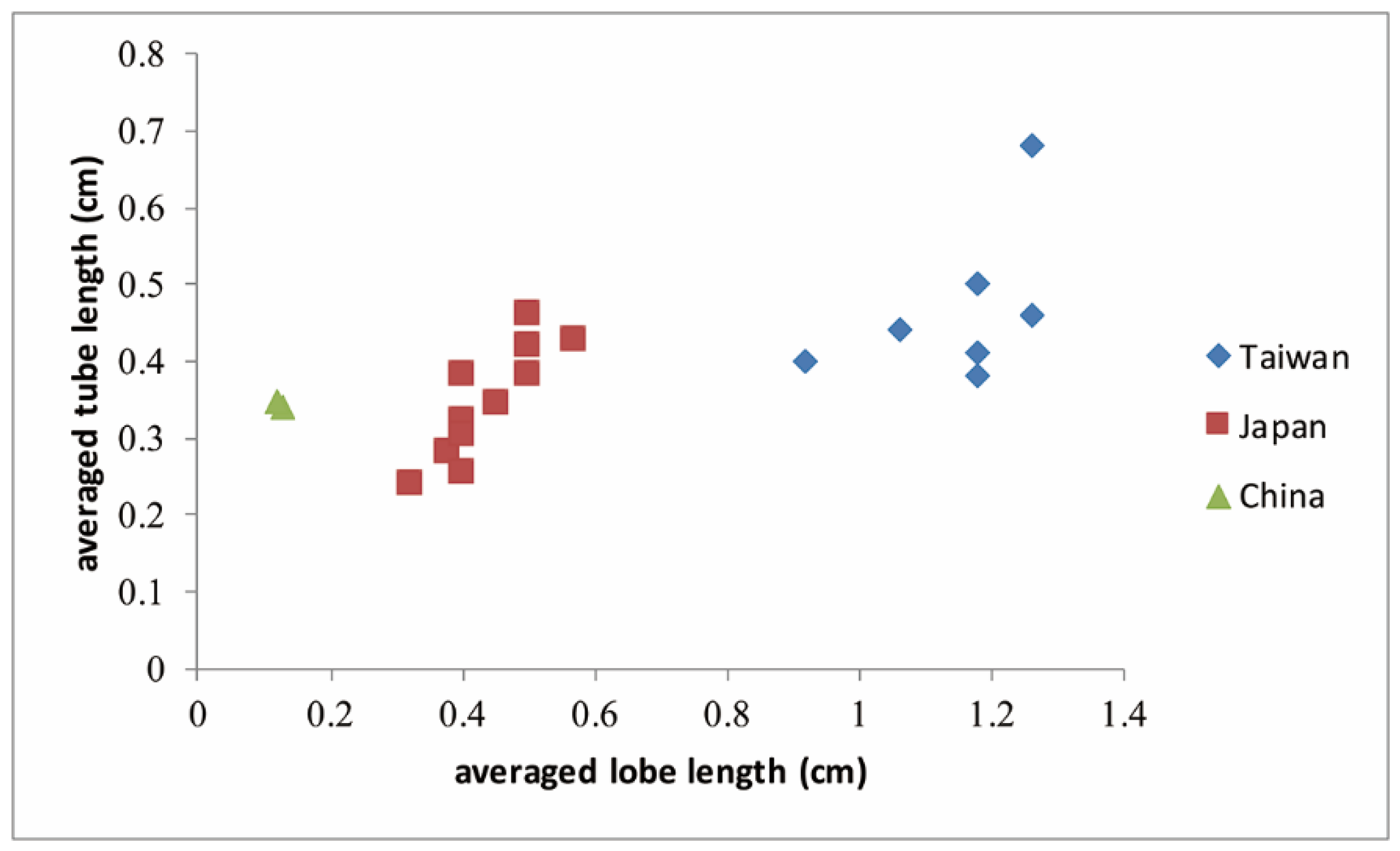
2.12. Morphological Variations Measured in C. ramondioides Individuals Distributed on the Continent and Islands
3. Discussion
3.1. High Genetic Diversity and Strong Genetic Differentiation of C. ramondioides
3.2. Middle Pleistocene Lineage Diversification of Conandron
3.3. Cryptic Diversification within C. ramondioides var. Taiwanensis
3.4. Diversifying Selection May Contribute to Shape CrCYC1 Lineage Diversification
3.5. Divergent ITS Sequences Suggest That Conandron ITS Resulted from Incomplete Concerted Evolution
3.6. Lineage Discordance between cpDNA and nrDNA in C. ramondioides
4. Materials and Methods
4.1. Sample Collection and Genomic DNA Extraction
4.2. Amplification of cpDNA Markers and Nuclear Markers
4.3. Genetic Structure of Populations and Geographical Structure Analysis
4.4. Inferring Lineage Divergence of C. ramondioides Populations from Nuclear Markers
4.5. Population Demographical History
4.6. Simulation of the Least-Cost Path to Visualize Potential Dispersal Routes of Conandron Populations during the Last Glacial Maximum in East Asia
4.7. Identification of Cryptic Species of C. ramondioides via Floral Morphology Measurements
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Volckaert, E.; Gobin, B. Ornamental Plants and Floriculture. Encycl. Life Support Syst. (EOLLSS) 2014. Sample Chapters C 10. Available online: https://www.eolss.net/sample-chapters/c10/E1-05A-51.pdf (accessed on 13 March 2022).
- Odeh, R.; Guy, C.L. Gardening for Therapeutic People-Plant Interactions during Long-Duration Space Missions. Open Agric. 2017, 2, 1–13. [Google Scholar] [CrossRef]
- Manchester, S.R.; Chen, Z.D.; Lu, A.M.; Uemura, K. Eastern Asian endemic seed plant genera and their paleogeographic history throughout the Northern Hemisphere. J. Syst. Evol. 2009, 47, 1–42. [Google Scholar] [CrossRef]
- Cheng, Y.P.; Hwang, S.Y.; Lin, T.P. Potential refugia in Taiwan revealed by the phylogeographical study of Castanopsis carlesii Hayata (Fagaceae). Mol. Ecol. 2005, 14, 2075–2085. [Google Scholar] [CrossRef] [PubMed]
- Chiang, T.Y.; Schaal, B.A. Phylogeography of Plants in Taiwan and the Ryukyu Archipelago. Taxon 2006, 55, 31–41. [Google Scholar] [CrossRef]
- Picó, F.X.; Abdelaziz, M.; Castilla, A.R. Introduction to the Special Issue: The ecology and genetics of population differentiation in plants. AoB Plants 2021, 13, plab057. [Google Scholar] [CrossRef] [PubMed]
- Rieseberg, L.H.; Willis, J.H. Plant speciation. Science 2007, 317, 910–914. [Google Scholar] [CrossRef] [PubMed]
- Chang, F.; Li, T.; Xiong, Z.; Xu, Z. Evidence for sea level and monsoonally driven variations in terrigenous input to the northern East China Sea during the last 24.3 ka. Paleoceanography 2015, 30, 642–658. [Google Scholar] [CrossRef]
- Siddall, M.; Rohling, E.J.; Almogi-Labin, A.; Hemleben, C.; Meischner, D.; Schmelzer, I.; Smeed, D.A. Sea-level fluctuations during the last glacial cycle. Nature 2003, 423, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Zhang, X.; Kang, H.; Sun, X.; Yin, S.; Du, H.; Yamanaka, N.; Gapare, W.; Wu, H.X.; Liu, C. Phylogeography of Quercus variabilis based on chloroplast DNA sequence in East Asia: Multiple glacial refugia and Mainland-migrated island populations. PLoS ONE 2012, 7, e47268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, X.-S.; Chen, C.; Comes, H.P.; Sakaguchi, S.; Liu, Y.-H.; Tanaka, N.; Sakio, H.; Qiu, Y.-X. Molecular data and ecological niche modelling reveal a highly dynamic evolutionary history of the East Asian Tertiary relict Cercidiphyllum (Cercidiphyllaceae). N. Phytol. 2012, 196, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.S.; Yuan, N.; Comes, H.P.; Sakaguchi, S.; Qiu, Y.-X. A strong ‘filter’ effect of the East China Sea land bridge for East Asia’s temperate plant species: Inferences from molecular phylogeography and ecological niche modelling of Platycrater arguta (Hydrangeaceae). BMC Evol. Biol. 2014, 14, 41. [Google Scholar] [CrossRef] [Green Version]
- Sakaguchi, S.; Qiu, Y.-X.; Liu, Y.-H.; Qi, X.S.; Kim, S.H.; Han, J.; Takeuchi, Y.; Worth, J.R.P.; Yamasaki, M.; Sakurai, S.; et al. Climate oscillation during the Quaternary associated with landscape heterogeneity promoted allopatric lineage divergence of a temperate tree Kalopanax septemlobus (Araliaceae) in East Asia. Mol. Ecol. 2012, 21, 3823–3838. [Google Scholar] [CrossRef]
- Sata, H.; Shimizu, M.; Iwasaki, T.; Ikeda, H.; Soejima, A.; Kozhevnikov, A.E.; Kozhevnikova, Z.V.; Im, H.T.; Jang, S.K.; Azuma, T.; et al. Phylogeography of the East Asian grassland plant, Viola orientalis (Violaceae), inferred from plastid and nuclear restriction site-associated DNA sequencing data. J. Plant Res. 2021, 134, 1181–1198. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Liu, W.; Xue, Q.; Hou, B.; Luo, J.; Ding, X. Phylogeography of the endangered orchid Dendrobium moniliforme in East Asia inferred from chloroplast DNA sequences. Mitochondrial DNA A DNA Mapp. Seq. Anal. 2017, 28, 880–891. [Google Scholar] [PubMed]
- Kokubugata, G.; Peng, C.I. Floral morphology and recognition of varieties in Conandron ramondioides (Gesneriaceae) from Japan and Taiwan. Edinb. J. Bot. 2004, 61, 21–30. [Google Scholar] [CrossRef]
- Morris, A.B.; Ickert-Bond, S.M.; Brunson, D.B.; Soltis, D.E.; Soltis, P.S. Phylogeographical structure and temporal complexity in American sweetgum (Liquidambar styraciflua; Altingiaceae). Mol. Ecol. 2008, 17, 3889–3900. [Google Scholar] [CrossRef] [PubMed]
- Schaal, B.A.; Hayworth, D.A.; Olsen, K.M.; Rauscher, J.T.; Smith, W.A. Phylogeographic studies in plants: Problems and prospects. Mol. Ecol. 1998, 7, 465–474. [Google Scholar] [CrossRef]
- Shaw, J.; Shafer, H.L.; Leonard, O.R.; Kovach, M.J.; Schorr, M.; Morris, A.B. Chloroplast DNA sequence utility for the lowest phylogenetic and phylogeographic inferences in angiosperms: The tortoise and the hare IV. Am. J. Bot. 2014, 101, 1987–2004. [Google Scholar] [CrossRef] [Green Version]
- Shaw, J.; Small, R.L. Chloroplast DNA phylogeny and phylogeography of the North American plums (Prunus subgenus Prunus section Prunocerasus, Rosaceae). Am. J. Bot. 2005, 92, 2011–2030. [Google Scholar] [CrossRef] [Green Version]
- Knowles, L.L.; Maddison, W.P. Statistical Phylogeography. Mol. Ecol. 2002, 11, 2623–2635. [Google Scholar] [CrossRef]
- Wolfe, K.H.; Li, W.H.; Sharp, P.M. Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc. Natl. Acad. Sci. USA 1987, 84, 9054–9058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, Y.X.; Sun, Y.; Zhang, X.P.; Lee, J.; Fu, C.X.; Comes, H.P. Molecular phylogeography of East Asian Kirengeshoma (Hydrangeaceae) in relation to quaternary climate change and landbridge configurations. N. Phytol. 2009, 183, 480–495. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.F.; Gong, X.; Chiang, Y.C.; Kuroda, C.; Linder, P. Phylogenetic patterns and disjunct distribution in Ligularia hodgsonii Hook. (Asteraceae). J. Biogeogr. 2013, 40, 1741–1754. [Google Scholar] [CrossRef]
- Huang, S.; Chiang, Y.C.; Schaal, B.A.; Chou, C.H.; Chiang, T.Y. Organelle DNA phylogeography of Cycas taitungensis, a relict species in Taiwan. Mol. Ecol. 2001, 10, 2669–2681. [Google Scholar] [CrossRef]
- Wolf, S.; Moritz, R.F.A. Foraging distance in Bombus terrestris L. (Hymenoptera: Apidae). Apidologie 2008, 39, 419–427. [Google Scholar] [CrossRef]
- Ma, H.W. Autonomous Selfing Provides Potential Reproductive Assurance in Conandron ramondioides (Gesneriaceae). Master’s Thesis, National Taiwan University, Taipei, Taiwan, 2017. [Google Scholar]
- Peat, J.; Goulson, D. Effects of experience and weather on foraging rate and pollen versus nectar collection in the bumblebee, Bombus terrestris. Behav. Ecol. Sociobiol. 2005, 58, 152–156. [Google Scholar] [CrossRef]
- Lisiecki, L.E.; Raymo, M.E. A Pliocene-Pleistocene stack of 57 globally distributed benthic δ18O records. Paleoceanography 2005, 20, PA1003. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.; Lang, X. Last Glacial Maximum East Asian Monsoon: Results of PMIP Simulations. J. Clim. 2010, 23, 5030–5038. [Google Scholar] [CrossRef]
- Mishima, M.; Kawahata, H.; Suzuki, A.; Inoue, M.; Okai, T.; Omura, A. Reconstruction of the East China Sea paleoenvironment at 16 ka by comparison of fossil and modern Faviidae corals from the Ryukyus, southwestern Japan. J. Quatenary Sci. 2009, 24, 928–936. [Google Scholar] [CrossRef]
- Ruddiman, W.F.; Raymo, M.E.; Martinson, D.G.; Clement, B.M.; Backman, J. Pleistocene evolution: Northern hemisphere ice sheets and North Atlantic Ocean. Paleoceanography 1989, 4, 353–412. [Google Scholar] [CrossRef]
- Cao, Y.N.; Comes, H.P.; Sakaguchi, S.; Chen, L.Y.; Qiu, Y.X. Evolution of East Asia’s Arcto-Tertiary relict Euptelea (Eupteleaceae) shaped by Late Neogene vicariance and Quaternary climate change. BMC Evol. Biol. 2016, 16, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, X.P.; Zhang, X.C. Phylogeography of the widespread fern Lemmaphyllum in East Asia: Species differentiation and population dynamics in response to change in climate and geography. J. Syst.Evol. 2022, 60, 411–432. [Google Scholar] [CrossRef]
- Ota, H. Geographic patterns of endemism and speciation in amphibians and reptiles of the Ryukyu Archipelago, Japan, with special reference to their paleogeographical implications. Popul. Ecol. 1998, 40, 189–204. [Google Scholar] [CrossRef]
- Hsin, K.T.; Wang, C.N. Expression shifts of floral symmetry genes correlate to flower actinomorphy in East Asia endemic Conandron ramondioides (Gesneriaceae). Bot. Stud. 2018, 59, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hileman, L.C. Trends in flower symmetry evolution revealed through phylogenetic and developmental genetic advances. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130348. [Google Scholar] [CrossRef] [Green Version]
- Hsu, H.J.; He, C.W.; Kuo, W.H.; Hsin, K.T.; Lu, J.Y.; Pan, Z.J.; Wang, C.N. Genetic Analysis of Floral Symmetry Transition in African Violet Suggests the Involvement of Trans-acting Factor for CYCLOIDEA Expression Shifts. Front. Plant Sci. 2018, 9, 1008. [Google Scholar] [CrossRef] [Green Version]
- Luo, D.; Carpenter, R.; Vincent, C.; Copsey, L.; Coen, E. Origin of floral asymmetry in Antirrhinum. Nature 1996, 383, 794–799. [Google Scholar] [CrossRef]
- Preston, J.C.; Hileman, L.C. Developmental genetics of floral symmetry evolution. Trends Plant Sci. 2009, 14, 147–154. [Google Scholar] [CrossRef]
- Alvarez, I.; Wendel, J.F. Ribosomal ITS sequences and plant phylogenetic inference. Mol. Phylogenet. Evol. 2003, 29, 417–434. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, B.G.; Sanderson, M.J.; Porter, J.M.; Wojciechowski, M.F.; Campbell, C.S.; Donoghue, M.J. The its Region of Nuclear Ribosomal DNA: A Valuable Source of Evidence on Angiosperm Phylogeny. Ann. Mo. Bot. Gard. 1995, 82, 247–277. [Google Scholar] [CrossRef]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W.; Fungal Barcoding Consotium; Fungal Barcoding Consotium Author List; Bolchacova, E.; et al. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, D. Concerted evolution: Molecular mechanism and biological implications. Am. J. Hum. Genet. 1999, 64, 24–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, B.; Zeng, X.M.; Gao, X.F.; Jin, D.-P.; Zhang, L.B. ITS non-concerted evolution and rampant hybridization in the legume genus Lespedeza (Fabaceae). Sci. Rep. 2017, 7, 40057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Sousa Queiroz, C.; de Carvalho Batista, F.R.; de Oliveira, L.O. Evolution of the 5.8S nrDNA gene and internal transcribed spacers in Carapichea ipecacuanha (Rubiaceae) within a phylogeographic context. Mol. Phylogenet. Evol. 2011, 59, 293–302. [Google Scholar] [CrossRef]
- Zheng, X.; Cai, D.; Yao, L.; Teng, Y. Non-concerted ITS evolution, early origin and phylogenetic utility of ITS pseudogenes in Pyrus. Mol. Phylogenet. Evol. 2008, 48, 892–903. [Google Scholar] [CrossRef]
- Harpke, D.; Peterson, A. Non-concerted ITS evolution in Mammillaria (Cactaceae). Mol. Phylogenet. Evol. 2006, 41, 579–593. [Google Scholar] [CrossRef]
- Avise, J.C. Phylogeography: The History and Formation of Species; Harvard University Press: Cambridge, MA, USA, 2000. [Google Scholar]
- Neigel, J.; Avise, J. Evolutionary Processes and Theory; Elsevier Inc.: Amsterdam, The Netherlands, 1986. [Google Scholar]
- Palumbi, S.R.; Cipriano, F.; Hare, M.P. Predicting nuclear gene coalescence from mitochondrial data: The three-times rule. Evolution 2001, 55, 859–868. [Google Scholar] [CrossRef]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef] [Green Version]
- Taberlet, P.; Coissac, E.; Pompanon, F.; Gielly, L.; Miquel, C.; Valentini, A.; Vermat, T.; Corthier, G.; Brochmann, C.; Willerslev, E. Power and limitations of the chloroplast trnL (UAA) intron for plant DNA barcoding. Nucleic Acids Res. 2007, 35, e14. [Google Scholar] [CrossRef] [Green Version]
- Tate, J.A.; Simpson, B.B. Paraphyly of Tarasa (Malvaceae) and Diverse Origins of the Polyploid Species. Syst. Bot. 2003, 28, 723–737. [Google Scholar]
- Xiao, L.H.; Wang, Y.Z. Single nucleotide polymorphisms of Gcyc1 (Cycloidea) in Conandron ramondioides (Gesneriaceae) from Southeast China. Plant Syst. Evol. 2007, 269, 145–157. [Google Scholar] [CrossRef]
- Möller, M.; Cronk, Q.C. Phylogeny and disjunct distribution: Evolution of Saintpaulia (Gesneriaceae). Proc. Royal Soc. B 1997, 264, 1827–1836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsin, K.T.; Wang, C.N. Cheng, Y.S. Screening three single-copy nuclear markers to infer population divergence in Conandron ramondioides (Gesneriaceae). J. Nat. Taiwan Museum 2021, 74, 15–25. [Google Scholar]
- Krak, K.; Alvarez, I.; Caklová, P.; Costa, A.; Chrtek, J.; Fehrer, J. Development of novel low-copy nuclear markers for Hieraciinae (Asteraceae) and their perspective for other tribes. Am. J. Bot. 2012, 99, e74–e77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Hall, T. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [Green Version]
- Excoffier, L.; Lischer, H.E. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Excoffier, L.; Smouse, P.E.; Quattro, J.M. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 1992, 131, 479–491. [Google Scholar] [CrossRef]
- Aktas, C. R Package Haplotypes: Haplotype Inference and Statistical Analysis of Genetic Variation; R Foundation for Statistical Computing: Vienna, Austria, 2015. [Google Scholar]
- Simmons, M.P.; Ochoterena, H. Gaps as Characters in Sequence-Based Phylogenetic Analyses. Syst. Biol. 2000, 49, 369–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heled, J.; Drummond, A.J. Bayesian Inference of Species Trees from Multilocus Data. Mol. Biol. Evol. 2010, 27, 570–580. [Google Scholar] [CrossRef] [PubMed]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.P.; Yang, Z. Rate variation and estimation of divergence times using strict and relaxed clocks. BMC Evol. Biol. 2011, 11, 271. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hey, J. Isolation with Migration Models for More Than Two Populations. Mol. Biol. Evol. 2010, 27, 905–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watterson, G.A. On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 1975, 7, 256–276. [Google Scholar] [CrossRef]
- Hey, J. The divergence of chimpanzee species and subspecies as revealed in multipopulation isolation-with-migration analyses. Mol. Biol. Evol. 2010, 27, 921–933. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, R.; Wakeley, J. Distinguishing migration from isolation: A Markov chain Monte Carlo approach. Genetics 2001, 158, 885–896. [Google Scholar] [CrossRef] [PubMed]
- Tajima, F. Evolutionary relationship of DNA sequences in finite populations. Genetics 1983, 105, 437–460. [Google Scholar] [CrossRef]
- Fu, Y.X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997, 147, 915–925. [Google Scholar] [CrossRef]
- McDonald, J.H.; Kreitman, M. Adaptive protein evolution at the Adh locus in Drosophila. Nature 1991, 351, 652–654. [Google Scholar] [CrossRef] [PubMed]
- Egea, R.; Casillas, S.; Barbadilla, A. Standard and generalized McDonald-Kreitman test: A website to detect selection by comparing different classes of DNA sites. Nucleic Acids Res. 2008, 36, W157–W162. [Google Scholar] [CrossRef] [Green Version]
- Heled, J.; Drummond, A.J. Bayesian inference of population size history from multiple loci. BMC Evol. Biol. 2008, 8, 289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elith, J.; Graham, C.H.; Anderson, R.P.; Dudík, M.; Ferrier, S.; Guisan, A.; Hijmans, R.J.; Huettmann, F.; Leathwick, J.R.; Lehmann, A.; et al. Novel methods improve prediction of species’ distributions from occurrence data. Ecography 2006, 29, 129–151. [Google Scholar] [CrossRef] [Green Version]
- Phillips, S.J.; Anderson, R.P.; Schapire, R.E. Maximum entropy modeling of species geographic distributions. Ecol. Modell. 2006, 190, 231–259. [Google Scholar] [CrossRef] [Green Version]
- Hijmans, R.J.; Cameron, S.E.; Parra, J.L.; Jones, P.G.; Jarvis, A. Very high resolution interpolated climate surfaces for global land areas. Int. J. Climatol. 2005, 25, 1965–1978. [Google Scholar] [CrossRef]
- van Etten, J. R Package gdistance: Distances and Routes on Geographical Grids. J. Stat. Softw. 2017, 76, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Perret, M.; Chautems, A.; De Araujo, A.O.; Salamin, N. Temporal and spatial origin of Gesneriaceae in the New World inferred from plastid DNA sequences. Bot. J. Linn. 2012, 171, 61–79. [Google Scholar] [CrossRef] [Green Version]
- Möller, M.; Pfosser, M.; Jang, C.G.; Mayer, V.; Clark, A.; Hollingsworth, M.L.; Barfuss, M.H.J.; Wang, Y.Z.; Kiehn, M.; Weber, A. A preliminary phylogeny of the ‘didymocarpoid Gesneriaceae’ based on three molecular data sets: Incongruence with available tribal classifications. Am. J. Bot. 2009, 96, 989–1010. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.; Middleton, D.J.; Forrestet, A.; Kiew, R.; Lim, C.L.; Rafidah, A.R.; Sontag, S.; Triboun, P.; Wei, Y.G.; Yao, T.L.; et al. Molecular systematics and remodelling of Chirita and associated genera (Gesneriaceae). Taxon 2011, 60, 767–790. [Google Scholar] [CrossRef]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cyrtandra * | Hemiboea * | Oreocharis * | ||||
|---|---|---|---|---|---|---|
| Polymorphism | Divergence | Polymorphism | Divergence | Polymorphism | Divergence | |
| Synonymous (dS) | 5 | 24 | 9 | 20 | 8 | 11 |
| Non-synonymous (dN) | 20 | 24 | 41 | 30 | 16 | 21 |
| Total | 25 | 48 | 50 | 50 | 24 | 32 |
| χ2 value | 6.11 | 5.876 | 0.904 | |||
| p-value | 0.013 | 0.015 | 0.34 | |||
| Tajima’s D | Fu’s Fs | |
|---|---|---|
| cpDNA | 2.9 (p < 0.01) | 7.86 (p < 0.01) |
| ATG2 intron 1 | −0.15 (p > 0.1) | 0.05 (p > 0.1) |
| GroES intron 1 | 1.63 (p > 0.1) | 2.04 (p > 0.1) |
| LFY intron 1 | 1.3 (p > 0.1) | 3.34 (p > 0.1) |
| Group | Honshu+Shikoku | Taiwan+Iriomote | |
|---|---|---|---|
| Honshu+Shikoku | - | ||
| (a) ATG2 intron 1 | Taiwan+Iriomote | 0.571 *** | - |
| Southeast China | 0.748 *** | 0.691 *** | |
| Honshu+Shikoku | - | ||
| (b) GroES intron 1 | Taiwan+Iriomote | 0.449 *** | - |
| Southeast China | 0.298 *** | 0.513 *** | |
| Honshu+Shikoku | - | ||
| (c) LFY intron 1 | Taiwan+Iriomote | 0.472 *** | - |
| Southeast China | 0.674 *** | 0.848 *** | |
| Honshu+Shikoku | - | ||
| (d) cpDNA | Taiwan+Iriomote | 0.295 *** | - |
| SE-China | 0.32 *** | 0.269 *** |
| ATG2 intron 1 | ||||||
| 3 groups | ||||||
| Source of variation | d.f. | Sum of squares | Variance components | Percentage variation | Fixation indices | p |
| Among groups | 2 | 100.635 | 0.771 | 39.61 | ΦCT = 0.4 | 0.003 |
| Among populations within groups | 7 | 75.023 | 0.721 | 37.04 | ΦSC = 0.61 | <0.0001 |
| Within populations | 144 | 65.443 | 0.454 | 23.34 | ΦST = 0.767 | <0.0001 |
| Total | 153 | 153.000 | 1.947 | |||
| GroES intron 1 | ||||||
| Source of variation | d.f. | Sum of squares | Variance components | Percentage variation | Fixation indices | p |
| Among groups | 2 | 154.583 | 0.844 | 36.750 | ΦCT = 0.367 | <0.0001 |
| Among populations within groups | 12 | 109.228 | 0.541 | 23.550 | ΦSC = 0.372 | <0.0001 |
| Within populations | 226 | 205.067 | 0.911 | 39.700 | ΦST = 0.601 | <0.0001 |
| Total | 239 | 468.367 | 2.290 | |||
| LFY intron 1 | ||||||
| Source of variation | d.f. | Sum of squares | Variance components | Percentage variation | Fixation indices | p |
| Among groups | 2 | 77.365 | 0.839 | 47.810 | ΦCT = 0.478 | 0.002 |
| Among populations within groups | 7 | 64.485 | 0.657 | 37.410 | ΦSC = 0.717 | <0.0001 |
| Within populations | 124 | 32.166 | 0.259 | 14.780 | ΦST = 0.852 | <0.0001 |
| Total | 133 | 172.769 | 1.741 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsin, K.-T.; Kuo, H.-C.; Kokubugata, G.; Möller, M.; Wang, C.-N.; Cheng, Y.-S. Allopatric Lineage Divergence of the East Asian Endemic Herb Conandron ramondioides Inferred from Low-Copy Nuclear and Plastid Markers. Int. J. Mol. Sci. 2022, 23, 14932. https://doi.org/10.3390/ijms232314932
Hsin K-T, Kuo H-C, Kokubugata G, Möller M, Wang C-N, Cheng Y-S. Allopatric Lineage Divergence of the East Asian Endemic Herb Conandron ramondioides Inferred from Low-Copy Nuclear and Plastid Markers. International Journal of Molecular Sciences. 2022; 23(23):14932. https://doi.org/10.3390/ijms232314932
Chicago/Turabian StyleHsin, Kuan-Ting, Hao-Chih Kuo, Goro Kokubugata, Michael Möller, Chun-Neng Wang, and Yi-Sheng Cheng. 2022. "Allopatric Lineage Divergence of the East Asian Endemic Herb Conandron ramondioides Inferred from Low-Copy Nuclear and Plastid Markers" International Journal of Molecular Sciences 23, no. 23: 14932. https://doi.org/10.3390/ijms232314932
APA StyleHsin, K.-T., Kuo, H.-C., Kokubugata, G., Möller, M., Wang, C.-N., & Cheng, Y.-S. (2022). Allopatric Lineage Divergence of the East Asian Endemic Herb Conandron ramondioides Inferred from Low-Copy Nuclear and Plastid Markers. International Journal of Molecular Sciences, 23(23), 14932. https://doi.org/10.3390/ijms232314932