
Effects of Apamin on MPP+-Induced Calcium Overload and Neurotoxicity by Targeting CaMKII/ERK/p65/STAT3 Signaling Pathways in Dopaminergic Neuronal Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
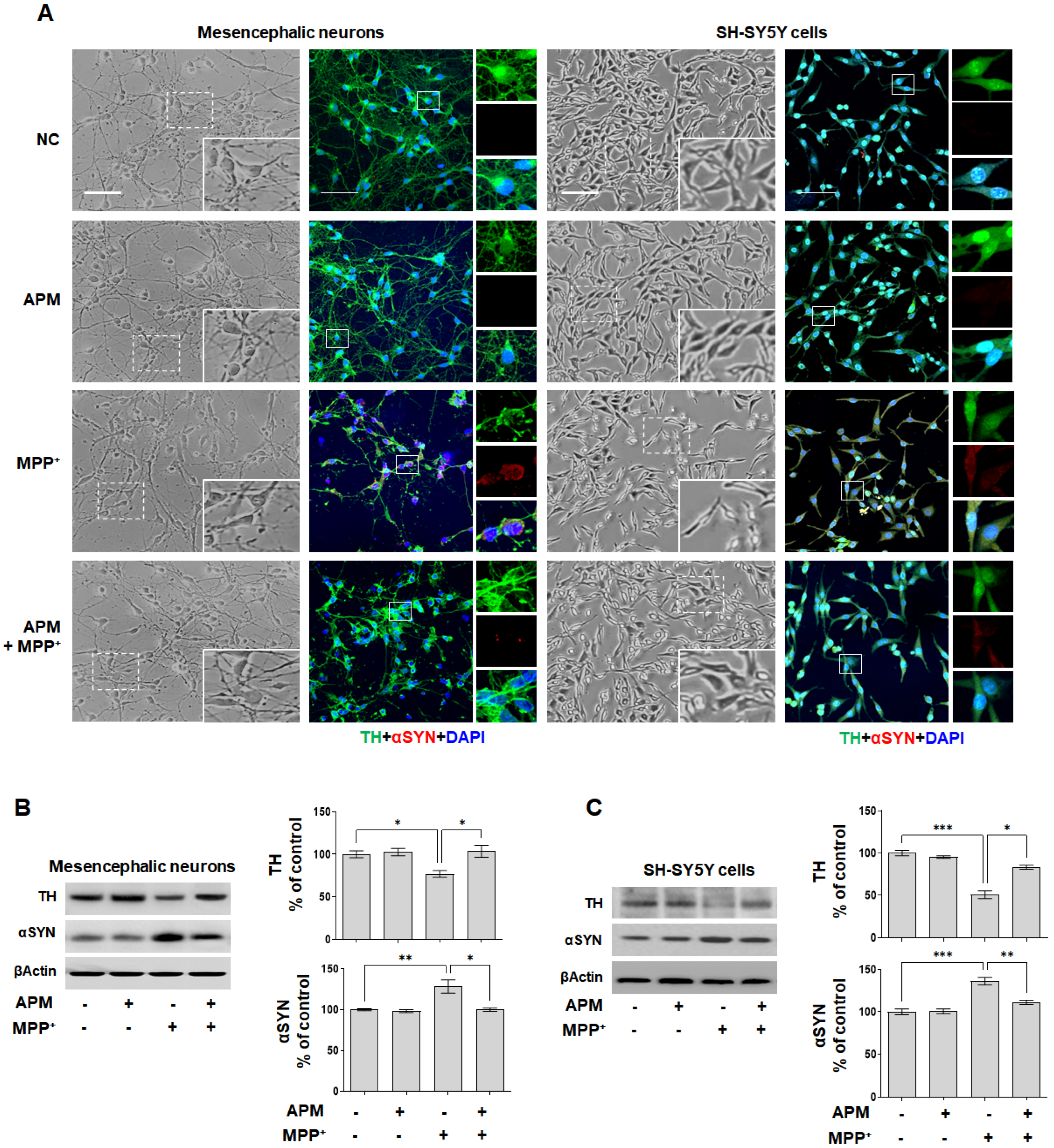
2.1. APM Protects SH-SY5Y Cells and Rat Embryo Primary Mesencephalic Neurons against MPP+-Induced Neurotoxicity
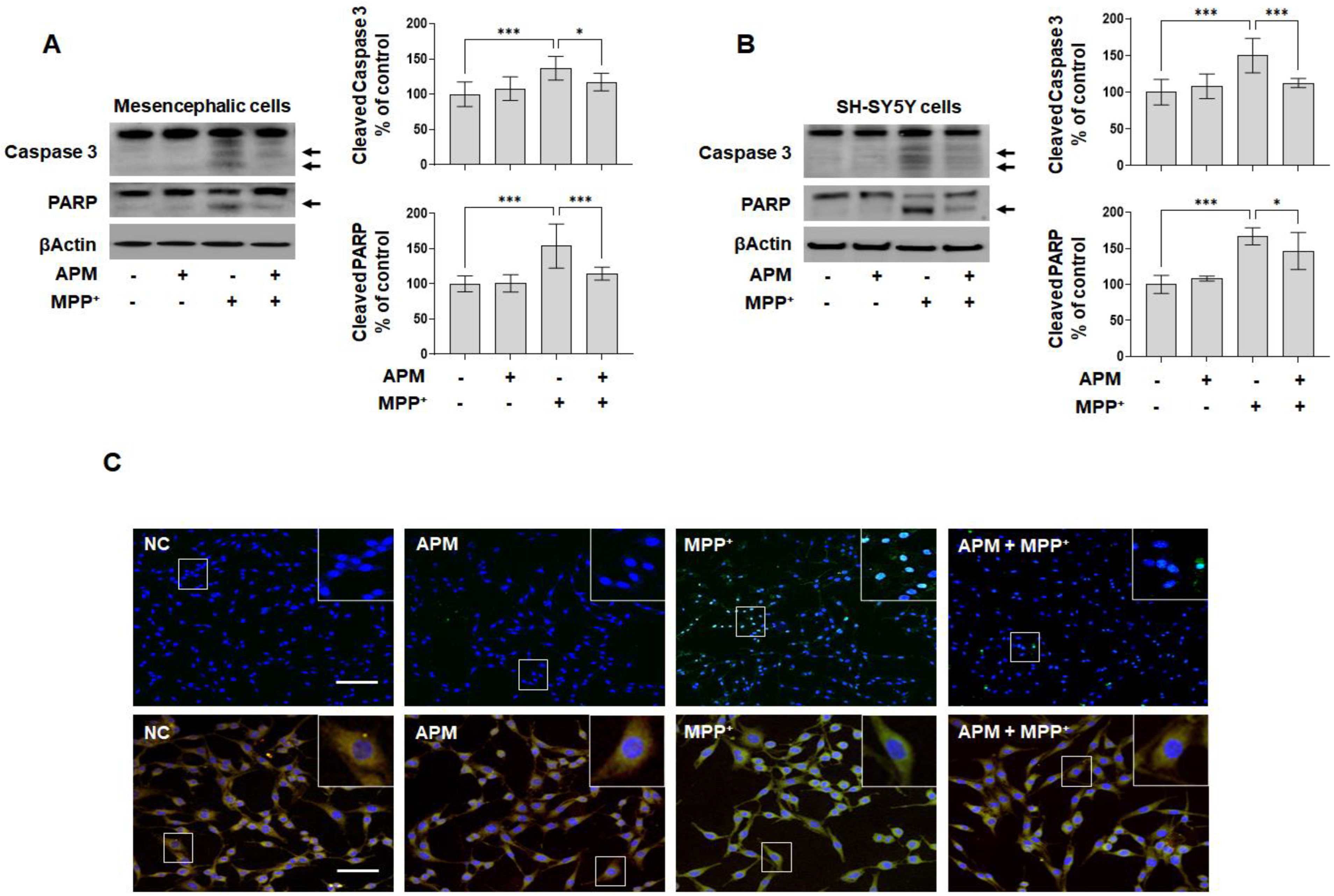
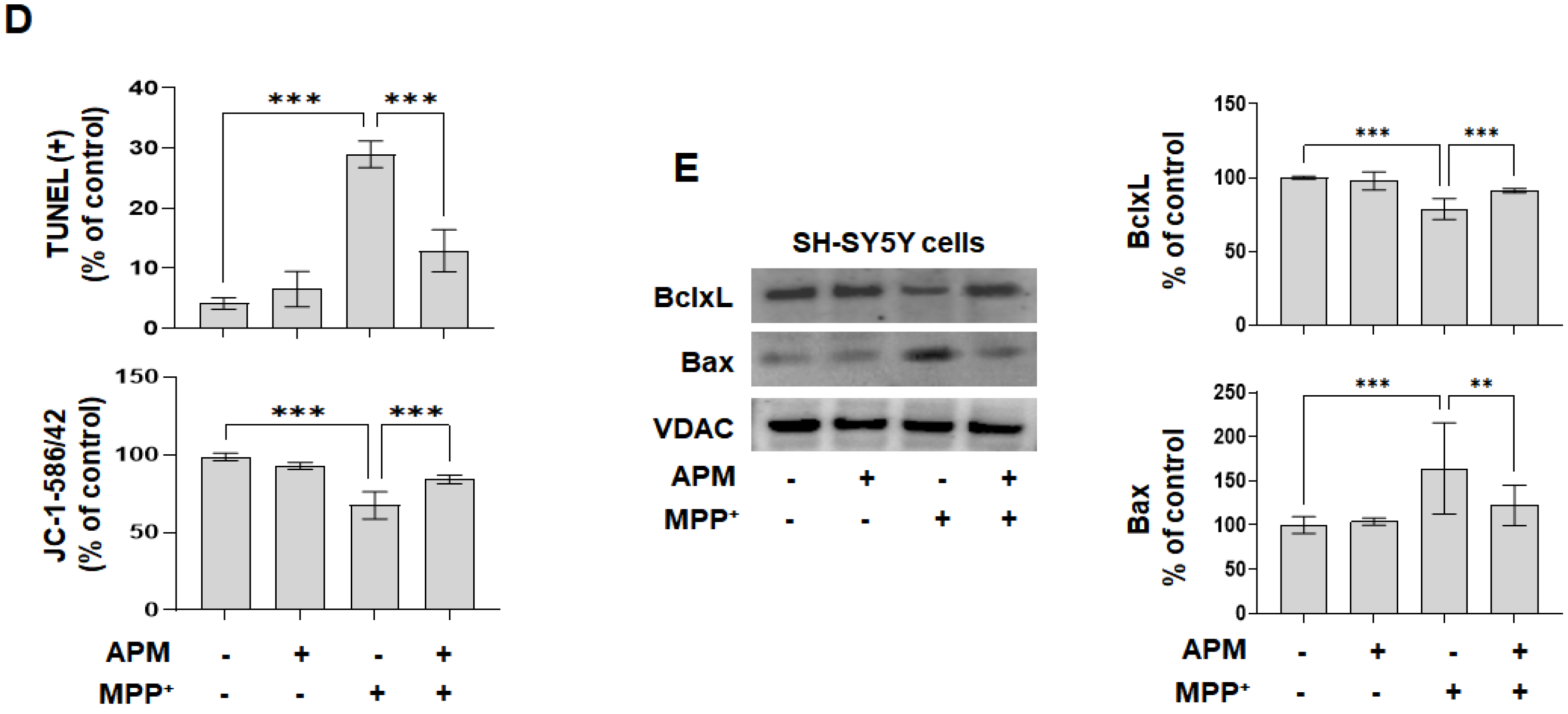
2.2. APM Ameliorates MPP+-Induced Mitochondrial-Dependent Apoptosis Pathway in SH-SY5Y and Rat Embryo Primary Mesencephalic Neurons
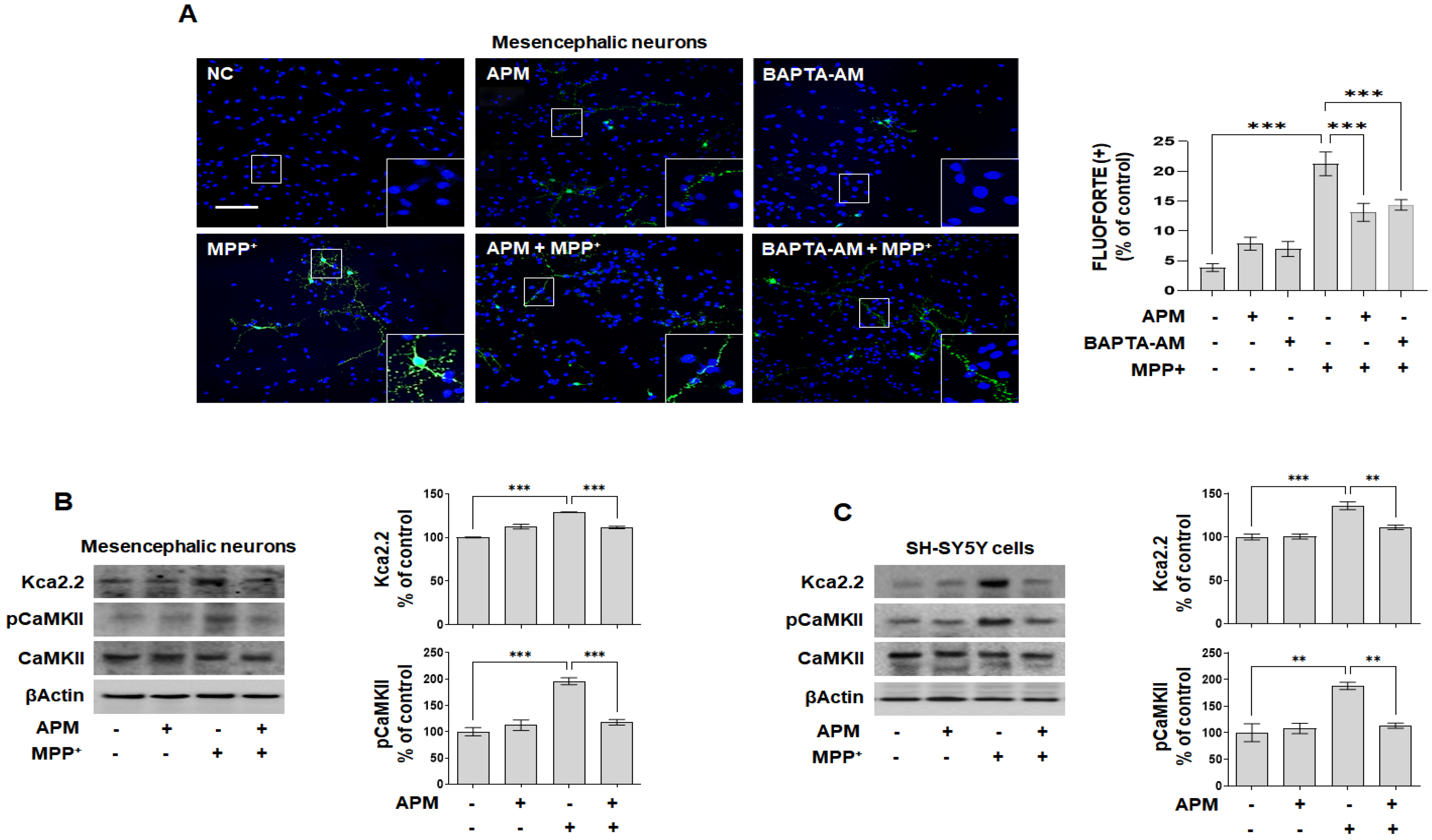
2.3. APM, a Strong Inhibiter of SK2 Channel, Protects MPP+-Induced Loss of TH-Positive Neurons and Accumulation of αSYN via Ca2+ Signaling
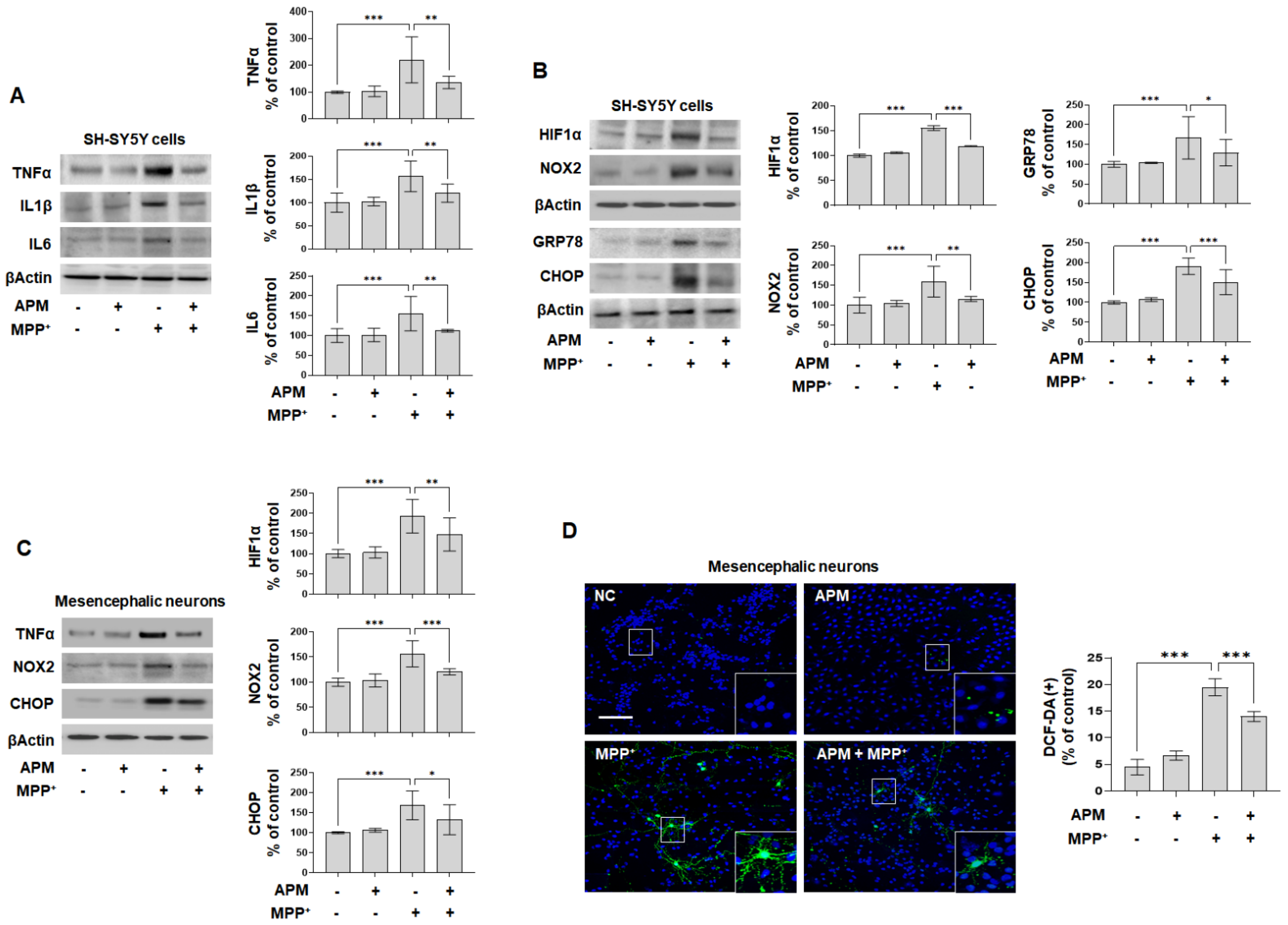
2.4. APM Alleviates MPP+-Induced Neuroinflammatory Response, Oxidative and ER Stress
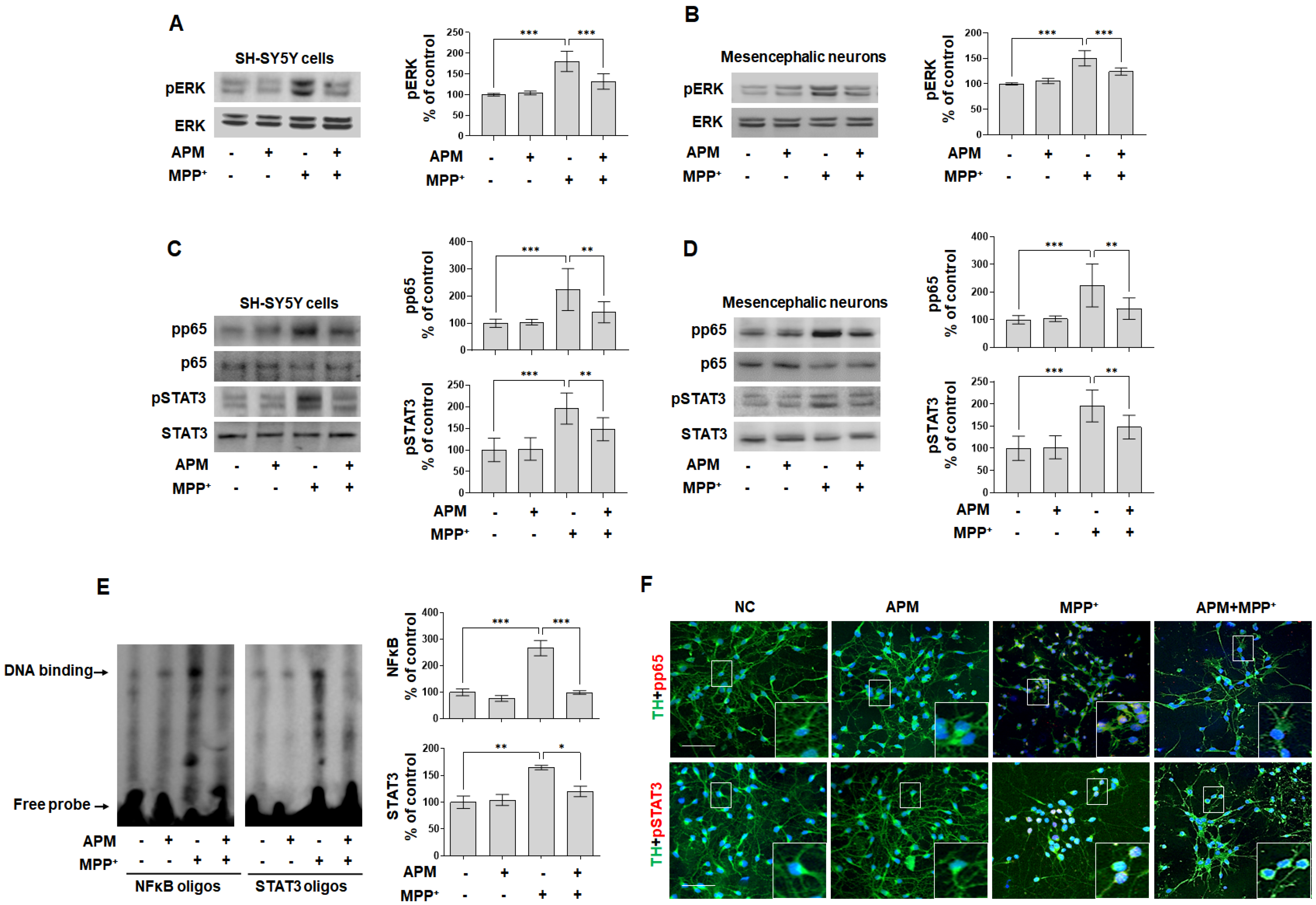
2.5. APM Ameliorates MPP+-Induced Neurotoxicity via ERK/STAT/p65 Signaling Pathway in Dopaminergic Neuronal Cells
3. Discussion
4. Materials and Methods
4.1. Cell Cultures and Reagents
4.2. Morphology Examination
4.3. Cytotoxicity Assay
4.4. Quantitative Real-Time Polymerase Chain Reaction (PCR) Analysis
4.5. Enzyme-Linked Immunosorbent Assay (ELISA)
4.6. Immunoblot Analysis
4.7. Terminal Deoxynucleotidyl-Transferase-Mediated dUTP Nick End Labelling (TUNEL) Staining
4.8. JC-1 Mitochondrial Transmembrane Potential Assay
4.9. Detection of Intracellular Ca2+ and ROS Expression
4.10. Immunofluorescent Staining
4.11. Electrophoretic Mobility Shift Analysis
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jovanovic-Tucovic, M.; Harhaji-Trajkovic, L.; Dulovic, M.; Tovilovic-Kovacevic, G.; Zogovic, N.; Jeremic, M.; Mandic, M.; Kostic, V.; Trajkovic, V.; Markovic, I. AMP-activated protein kinase inhibits MPP+-induced oxidative stress and apoptotic death of SH-SY5Y cells through sequential stimulation of Akt and autophagy. Eur. J. Pharmacol. 2019, 863, 172677. [Google Scholar] [CrossRef]
- Cui, W.; Zhang, Z.; Li, W.; Hu, S.; Mak, S.; Zhang, H.; Han, R.; Yuan, S.; Li, S.; Sa, F.; et al. The anti-cancer agent SU4312 unexpectedly protects against MPP+-induced neurotoxicity via selective and direct inhibition of neuronal NOS. Br. J. Pharmacol. 2013, 168, 1201–1214. [Google Scholar] [CrossRef] [Green Version]
- Jantas, D.; Greda, A.; Golda, S.; Korostynski, M.; Grygier, B.; Roman, A.; Pilc, A.; Lason, W. Neuroprotective effects of metabotropic glutamate receptor group II and III activators against MPP+-induced cell death in human neuroblastoma SH-SY5Y cells: The impact of cell differentiation state. Neuropharmacology 2014, 83, 36–53. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef]
- Selvaraj, S.; Sun, Y.; Watt, J.A.; Wang, S.; Lei, S.; Birnbaumer, L.; Singh, B.B. Neurotoxin-induced ER stress in mouse dopaminergic neurons involves downregulation of TRPC1 and inhibition of AKT/mTOR signaling. J. Clin. Investig. 2012, 122, 1354–1367. [Google Scholar] [CrossRef] [Green Version]
- Ricke, K.M.; Pass, T.; Kimoloi, S.; Fahrmann, K.; Jungst, C.; Schauss, A.; Baris, O.R.; Aradjanski, M.; Trifunovic, A.; Eriksson Faelker, T.M.; et al. Mitochondrial Dysfunction Combined with High Calcium Load Leads to Impaired Antioxidant Defense Underlying the Selective Loss of Nigral Dopaminergic Neurons. J. Neurosci. 2020, 40, 1975–1986. [Google Scholar] [CrossRef]
- Claros, S.; Cabrera, P.; Valverde, N.; Romero-Zerbo, S.Y.; López-González, M.V.; Shumilov, K.; Rivera, A.; Pavia, J.; Martín-Montañez, E.; Garcia-Fernandez, M. Insulin-like Growth Factor II Prevents MPP+ and Glucocorticoid Mitochondrial-Oxidative and Neuronal Damage in Dopaminergic Neurons. Antioxidants 2022, 11, 41. [Google Scholar] [CrossRef]
- Martín Giménez, V.M.; de las Heras, N.; Ferder, L.; Lahera, V.; Reiter, R.J.; Manucha, W. Potential Effects of Melatonin and Micronutrients on Mitochondrial Dysfunction during a Cytokine Storm Typical of Oxidative/Inflammatory Diseases. Diseases 2021, 9, 30. [Google Scholar] [CrossRef]
- Surmeier, D.J.; Guzman, J.N.; Sanchez-Padilla, J.; Schumacker, P.T. The role of calcium and mitochondrial oxidant stress in the loss of substantia nigra pars compacta dopaminergic neurons in Parkinson’s disease. Neuroscience 2011, 198, 221–231. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhang, J.; Rong, H.; Zhang, X.; Dong, M. Ferulic Acid Ameliorates MPP+/MPTP-Induced Oxidative Stress via ERK1/2-Dependent Nrf2 Activation: Translational Implications for Parkinson Disease Treatment. Mol. Neurobiol. 2020, 57, 2981–2995. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, Y. Cryptotanshinone ameliorates MPP+-induced oxidative stress and apoptosis of SH-SY5Y neuroblastoma cells: The role of STAT3 in Parkinson’s disease. Metab. Brain Dis. 2022, 37, 1477–1485. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Chung, Y.; Lee, Y.; Lee, Y.; Cho, J.W.; Shin, E.J.; Kim, H.C.; Oh, Y.J. Buffering of cytosolic calcium plays a neuroprotective role by preserving the autophagy-lysosome pathway during MPP+-induced neuronal death. Cell Death Discov. 2019, 5, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, K.; Eguchi, Y.; Imagawa, Y.; Akai, S.; Mochizuki, H.; Tsujimoto, Y. MPP+ induces necrostatin-1- and ferrostatin-1-sensitive necrotic death of neuronal SH-SY5Y cells. Cell Death Discov. 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sukumaran, P.; Sun, Y.; Antonson, N.; Singh, B.B. Dopaminergic neurotoxins induce cell death by attenuating NF-κB-mediated regulation of TRPC1 expression and autophagy. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2018, 32, 1640–1652. [Google Scholar] [CrossRef] [Green Version]
- Trombetta-Lima, M.; Krabbendam, I.E.; Dolga, A.M. Calcium-activated potassium channels: Implications for aging and age-related neurodegeneration. Int. J. Biochem. Cell Biol. 2020, 123, 105748. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, H.; Selvaraj, S.; Sukumaran, P.; Lei, S.; Birnbaumer, L.; Singh, B.B. Inhibition of L-Type Ca2+ Channels by TRPC1-STIM1 Complex Is Essential for the Protection of Dopaminergic Neurons. J. Neurosci. 2017, 37, 3364–3377. [Google Scholar] [CrossRef] [Green Version]
- Haque, M.E.; Akther, M.; Azam, S.; Choi, D.K.; Kim, I.S. GPR4 Knockout Improves the Neurotoxin-Induced, Caspase-Dependent Mitochondrial Apoptosis of the Dopaminergic Neuronal Cell. Int. J. Mol. Sci. 2020, 21, 7517. [Google Scholar] [CrossRef]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef] [Green Version]
- Pedarzani, P.; Stocker, M. Molecular and cellular basis of small-and intermediate-conductance, calcium-activated potassium channel function in the brain. Cell Mol. Life Sci. 2008, 65, 3196–3217. [Google Scholar] [CrossRef] [Green Version]
- Willis, M.; Trieb, M.; Leitner, I.; Wietzorrek, G.; Marksteiner, J.; Knaus, H.G. Small-conductance calcium-activated potassium type 2 channels (SK2, KCa2.2) in human brain. Brain Struct. Funct. 2017, 222, 973–979. [Google Scholar] [CrossRef]
- Teng, L.; Kou, C.; Lu, C.; Xu, J.; Xie, J.; Lu, J.; Liu, Y.; Wang, Z.; Wang, D. Involvement of the ERK pathway in the protective effects of glycyrrhizic acid against the MPP+-induced apoptosis of dopaminergic neuronal cells. Int. J. Mol. Med. 2014, 34, 742–748. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Lee, J.Y.; Lim, W.; You, S.; Song, G. Butylated Hydroxyanisole Exerts Neurotoxic Effects by Promoting Cytosolic Calcium Accumulation and Endoplasmic Reticulum Stress in Astrocytes. J. Agric. Food Chem. 2019, 67, 9618–9629. [Google Scholar] [CrossRef]
- Yürekli, V.A.; Gürler, S.; Nazıroğlu, M.; Uğuz, A.C.; Koyuncuoğlu, H.R. Zonisamide Attenuates MPP+-Induced Oxidative Toxicity Through Modulation of Ca2+ Signaling and Caspase-3 Activity in Neuronal PC12 Cells. Cell. Mol. Neurobiol. 2013, 33, 205–212. [Google Scholar] [CrossRef]
- Moreno, M.; Giralt, E. Three valuable peptides from bee and wasp venoms for therapeutic and biotechnological use: Melittin, apamin and mastoparan. Toxins 2015, 7, 1126–1150. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.-H.; An, H.-J.; Kim, J.-Y.; Gwon, M.-G.; Gu, H.; Lee, S.-J.; Park, J.Y.; Park, K.-D.; Han, S.-M.; Kim, M.-K.; et al. Apamin inhibits TNF-α- and IFN-γ-induced inflammatory cytokines and chemokines via suppressions of NF-κB signaling pathway and STAT in human keratinocytes. Pharmacol. Rep. 2017, 69, 1030–1035. [Google Scholar] [CrossRef]
- Cho, H.J.; Jeong, Y.J.; Park, K.K.; Park, Y.Y.; Chung, I.K.; Lee, K.G.; Yeo, J.H.; Han, S.M.; Bae, Y.S.; Chang, Y.C. Bee venom suppresses PMA-mediated MMP-9 gene activation via JNK/p38 and NF-kappaB-dependent mechanisms. J. Ethnopharmacol. 2010, 127, 662–668. [Google Scholar] [CrossRef]
- Lee, W.R.; Pak, S.C.; Park, K.K. The protective effect of bee venom on fibrosis causing inflammatory diseases. Toxins 2015, 7, 4758–4772. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; An, H.J.; Kim, W.H.; Park, Y.Y.; Park, K.D.; Park, K.K. Apamin suppresses biliary fibrosis and activation of hepatic stellate cells. Int. J. Mol. Med. 2017, 39, 1188–1194. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Park, J.H.; Kim, K.H.; Lee, W.R.; An, H.J.; Min, B.K.; Han, S.M.; Kim, K.S.; Park, K.K. Apamin inhibits THP-1-derived macrophage apoptosis via mitochondria-related apoptotic pathway. Exp. Mol. Pathol. 2012, 93, 129–134. [Google Scholar] [CrossRef]
- Gu, H.; Han, S.M.; Park, K.K. Therapeutic Effects of Apamin as a Bee Venom Component for Non-Neoplastic Disease. Toxins 2020, 12, 195. [Google Scholar] [CrossRef]
- Alvarez-Fischer, D.; Noelker, C.; Vulinović, F.; Grünewald, A.; Chevarin, C.; Klein, C.; Oertel, W.H.; Hirsch, E.C.; Michel, P.P.; Hartmann, A. Bee venom and its component apamin as neuroprotective agents in a Parkinson disease mouse model. PLoS ONE 2013, 8, e61700. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.Y.; Lee, Y.E.; Doo, K.H.; Lee, J.H.; Jung, W.S.; Moon, S.K.; Park, J.M.; Ko, C.N.; Kim, H.; Rhee, H.Y.; et al. Efficacy of Combined Treatment with Acupuncture and Bee Venom Acupuncture as an Adjunctive Treatment for Parkinson’s Disease. J. Altern. Complement. Med. 2018, 24, 25–32. [Google Scholar] [CrossRef]
- Kirkley, K.S.; Popichak, K.A.; Afzali, M.F.; Legare, M.E.; Tjalkens, R.B. Microglia amplify inflammatory activation of astrocytes in manganese neurotoxicity. J. Neuroinflamm. 2017, 14, 99. [Google Scholar] [CrossRef] [Green Version]
- Khalil, W.K.; Assaf, N.; ElShebiney, S.A.; Salem, N.A. Neuroprotective effects of bee venom acupuncture therapy against rotenone-induced oxidative stress and apoptosis. Neurochem. Int. 2015, 80, 79–86. [Google Scholar] [CrossRef]
- Cho, H.J.; Park, J.H.; Nam, J.H.; Chang, Y.C.; Park, B.; Hoe, H.S. Ascochlorin Suppresses MMP-2-Mediated Migration and Invasion by Targeting FAK and JAK-STAT Signaling Cascades. J. Cell Biochem. 2018, 119, 300–313. [Google Scholar] [CrossRef]
- Han, S.; Lee, K.; Yeo, J.; Kweon, H.; Woo, S.; Lee, M.; Baek, H.; Kim, S.; Park, K. Effect of honey bee venom on microglial cells nitric oxide and tumor necrosis factor-alpha production stimulated by LPS. J. Ethnopharmacol. 2007, 111, 176–181. [Google Scholar] [CrossRef]
- Park, J.; Jang, K.M.; Park, K.K. Apamin Suppresses LPS-Induced Neuroinflammatory Responses by Regulating SK Channels and TLR4-Mediated Signaling Pathways. Int. J. Mol. Sci. 2020, 21, 4319. [Google Scholar] [CrossRef]
- Li, Y.; Guo, Y.; Tang, J.; Jiang, J.; Chen, Z. New insights into the roles of CHOP-induced apoptosis in ER stress. Acta Biochim. Biophys. Sin. 2015, 47, 146–147. [Google Scholar] [CrossRef] [Green Version]
- Niedzielska, E.; Smaga, I.; Gawlik, M.; Moniczewski, A.; Stankowicz, P.; Pera, J.; Filip, M. Oxidative Stress in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 4094–4125. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Kim, K.H.; Lee, W.R.; An, H.J.; Lee, S.J.; Han, S.M.; Lee, K.G.; Park, Y.Y.; Kim, K.S.; Lee, Y.S.; et al. Apamin inhibits PDGF-BB-induced vascular smooth muscle cell proliferation and migration through suppressions of activated Akt and Erk signaling pathway. Vasc. Pharm. 2015, 70, 8–14. [Google Scholar] [CrossRef]
- Nabavi, S.M.; Ahmed, T.; Nawaz, M.; Devi, K.P.; Balan, D.J.; Pittalà, V.; Argüelles-Castilla, S.; Testai, L.; Khan, H.; Sureda, A.; et al. Targeting STATs in neuroinflammation: The road less traveled! Pharmacol. Res. 2019, 141, 73–84. [Google Scholar] [CrossRef]
- Chen, T.S.; Koutsilieri, E.; Rausch, W.D. MPP+ selectively affects calcium homeostasis in mesencephalic cell cultures from embryonal C57/B16 mice. J. Neural Transm./Gen. Sect. JNT 1995, 100, 153–163. [Google Scholar] [CrossRef]
- Lieberman, O.J.; Choi, S.J.; Kanter, E.; Saverchenko, A.; Frier, M.D.; Fiore, G.M.; Wu, M.; Kondapalli, J.; Zampese, E.; Surmeier, D.J.; et al. α-Synuclein-Dependent Calcium Entry Underlies Differential Sensitivity of Cultured SN and VTA Dopaminergic Neurons to a Parkinsonian Neurotoxin. eNeuro 2017, 4. [Google Scholar] [CrossRef] [Green Version]
- Egunlusi, A.O.; Malan, S.F.; Omoruyi, S.I.; Ekpo, O.E.; Palchykov, V.A.; Joubert, J. Open and rearranged norbornane derived polycyclic cage molecules as potential neuroprotective agents through attenuation of MPP+- and calcium overload-induced excitotoxicity in neuroblastoma SH-SY5Y cells. Eur. J. Med. Chem. 2020, 204, 112617. [Google Scholar] [CrossRef]
- Schapira, A.H.; Olanow, C.W.; Greenamyre, J.T.; Bezard, E. Slowing of neurodegeneration in Parkinson’s disease and Huntington’s disease: Future therapeutic perspectives. Lancet 2014, 384, 545–555. [Google Scholar] [CrossRef]
- Swart, T.; Hurley, M.J. Calcium channel antagonists as disease-modifying therapy for Parkinson’s disease: Therapeutic rationale and current status. Cns Drugs 2016, 30, 1127–1135. [Google Scholar]
- Song, Q.; Peng, S.; Zhu, X. Baicalein protects against MPP+/MPTP-induced neurotoxicity by ameliorating oxidative stress in SH-SY5Y cells and mouse model of Parkinson’s disease. NeuroToxicology 2021, 87, 188–194. [Google Scholar] [CrossRef]
- Segura-Aguilar, J. On the Role of Aminochrome in Mitochondrial Dysfunction and Endoplasmic Reticulum Stress in Parkinson’s Disease. Front. Neurosci. 2019, 13, 271. [Google Scholar] [CrossRef]
- Wu, L.-K.; Agarwal, S.; Kuo, C.-H.; Kung, Y.-L.; Day, C.H.; Lin, P.-Y.; Lin, S.-Z.; Hsieh, D.J.-Y.; Huang, C.-Y.; Chiang, C.-Y. Artemisia Leaf Extract protects against neuron toxicity by TRPML1 activation and promoting autophagy/mitophagy clearance in both in vitro and in vivo models of MPP+/MPTP-induced Parkinson’s disease. Phytomedicine 2022, 104, 154250. [Google Scholar] [CrossRef]
- Wang, X.; Cao, G.; Ding, D.; Li, F.; Zhao, X.; Wang, J.; Yang, Y. Ferruginol prevents degeneration of dopaminergic neurons by enhancing clearance of α-synuclein in neuronal cells. Fitoterapia 2022, 156, 105066. [Google Scholar] [CrossRef]
- Xu, F.; Xu, J.; Xiong, X.; Deng, Y. Salidroside inhibits MAPK, NF-κB, and STAT3 pathways in psoriasis-associated oxidative stress via SIRT1 activation. Redox Rep. 2019, 24, 70–74. [Google Scholar] [CrossRef]
- Ju, D.-T.; Sivalingam, K.; Kuo, W.-W.; Ho, T.-J.; Chang, R.-L.; Chung, L.-C.; Day, C.H.; Viswanadha, V.P.; Liao, P.-H.; Huang, C.-Y. Effect of Vasicinone against Paraquat-Induced MAPK/p53-Mediated Apoptosis via the IGF-1R/PI3K/AKT Pathway in a Parkinson’s Disease-Associated SH-SY5Y Cell Model. Nutrients 2019, 11, 1655. [Google Scholar] [CrossRef] [Green Version]
- Sang, Q.; Liu, X.; Wang, L.; Qi, L.; Sun, W.; Wang, W.; Sun, Y.; Zhang, H. Curcumin protects an SH-SY5Y cell model of Parkinson’s disease against toxic injury by regulating HSP90. Cell. Physiol. Biochem. 2018, 51, 681–691. [Google Scholar] [CrossRef]
- Furukawa, K.; Matsuzaki-Kobayashi, M.; Hasegawa, T.; Kikuchi, A.; Sugeno, N.; Itoyama, Y.; Wang, Y.; Yao, P.J.; Bushlin, I.; Takeda, A. Plasma membrane ion permeability induced by mutant α-synuclein contributes to the degeneration of neural cells. J. Neurochem. 2006, 97, 1071–1077. [Google Scholar] [CrossRef]
- Kim, D.H.; Kim, J.-H.; Hwangbo, H.; Kim, S.Y.; Ji, S.Y.; Kim, M.Y.; Cha, H.-J.; Park, C.; Hong, S.H.; Kim, G.-Y.; et al. Spermidine Attenuates Oxidative Stress-Induced Apoptosis via Blocking Ca2+ Overload in Retinal Pigment Epithelial Cells Independently of ROS. Int. J. Mol. Sci. 2021, 22, 1361. [Google Scholar] [CrossRef]
- Lin, J.-W.; Lin, Y.-C.; Liu, J.-M.; Liu, S.-H.; Fang, K.-M.; Hsu, R.-J.; Huang, C.-F.; Chang, K.-Y.; Lee, K.-I.; Chang, K.-C.; et al. Norketamine, the Main Metabolite of Ketamine, Induces Mitochondria-Dependent and ER Stress-Triggered Apoptotic Death in Urothelial Cells via a Ca2+-Regulated ERK1/2-Activating Pathway. Int. J. Mol. Sci. 2022, 23, 4666. [Google Scholar] [CrossRef]
- Follett, J.; Darlow, B.; Wong, M.B.; Goodwin, J.; Pountney, D.L. Potassium depolarization and raised calcium induces α-synuclein aggregates. Neurotox. Res. 2013, 23, 378–392. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, L.; Zhou, X.; Yu, Y.; Li, Z.; Zuo, D.; Wu, Y. Silver nanoparticles induce protective autophagy via Ca2+/CaMKKbeta/AMPK/mTOR pathway in SH-SY5Y cells and rat brains. Nanotoxicology 2019, 13, 369–391. [Google Scholar] [CrossRef]
- Li, J.; Zhang, S.; Liu, X.; Han, D.; Xu, J.; Ma, Y. Neuroprotective effects of leonurine against oxygen–glucose deprivation by targeting Cx36/CaMKII in PC12 cells. PLoS ONE 2018, 13, e0200705. [Google Scholar] [CrossRef]
- Wang, J.; Xu, X.; Jia, W.; Zhao, D.; Boczek, T.; Gao, Q.; Wang, Q.; Fu, Y.; He, M.; Shi, R.; et al. Calcium-/Calmodulin-Dependent Protein Kinase II (CaMKII) Inhibition Induces Learning and Memory Impairment and Apoptosis. Oxidative Med. Cell. Longev. 2021, 2021, 4635054. [Google Scholar] [CrossRef]
- Jiang, H.; Ashraf, G.M.; Liu, M.; Zhao, K.; Wang, Y.; Wang, L.; Xing, J.; Alghamdi, B.S.; Li, Z.; Liu, R. Tilianin Ameliorates Cognitive Dysfunction and Neuronal Damage in Rats with Vascular Dementia via p-CaMKII/ERK/CREB and ox-CaMKII-Dependent MAPK/NF-κB Pathways. Oxidative Med. Cell. Longev. 2021, 2021, 6673967. [Google Scholar] [CrossRef]
- Jia, W.; Kawahata, I.; Cheng, A.; Fukunaga, K. The Role of CaMKII and ERK Signaling in Addiction. Int. J. Mol. Sci. 2021, 22, 3189. [Google Scholar] [CrossRef]
- Wang, N.; Wang, J.; Zhang, Y.; Zeng, Y.; Hu, S.; Bai, H.; Hou, Y.; Wang, C.; He, H.; He, L. Imperatorin ameliorates mast cell-mediated allergic airway inflammation by inhibiting MRGPRX2 and CamKII/ERK signaling pathway. Biochem. Pharmacol. 2021, 184, 114401. [Google Scholar] [CrossRef]
- Chen, S.-P.; Sun, J.; Zhou, Y.-Q.; Cao, F.; Braun, C.; Luo, F.; Ye, D.-W.; Tian, Y.-K. Sinomenine attenuates cancer-induced bone pain via suppressing microglial JAK2/STAT3 and neuronal CAMKII/CREB cascades in rat models. Mol. Pain 2018, 14, 1744806918793232. [Google Scholar] [CrossRef] [Green Version]
- Venkatesan, T.; Choi, Y.W.; Lee, J.; Kim, Y.K. Pinus densiflora needle supercritical fluid extract suppresses the expression of pro-inflammatory mediators iNOS, IL-6 and IL-1beta, and activation of inflammatory STAT1 and STAT3 signaling proteins in bacterial lipopolysaccharide-challenged murine macrophages. Daru 2017, 25, 18. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.I.; Cheng, C.I.; Kang, Y.F.; Chang, P.C.; Lin, I.P.; Kuo, Y.H.; Jhou, A.J.; Lin, M.Y.; Chen, C.Y.; Lee, C.H. Hispidulin Inhibits Neuroinflammation in Lipopolysaccharide-Activated BV2 Microglia and Attenuates the Activation of Akt, NF-kappaB, and STAT3 Pathway. Neurotox. Res. 2020, 38, 163–174. [Google Scholar] [CrossRef]
- Do, H.T.T.; Bui, B.P.; Sim, S.; Jung, J.K.; Lee, H.; Cho, J. Anti-Inflammatory and Anti-Migratory Activities of Isoquinoline-1-Carboxamide Derivatives in LPS-Treated BV2 Microglial Cells via Inhibition of MAPKs/NF-κB Pathway. Int. J. Mol. Sci. 2020, 21, 2319. [Google Scholar] [CrossRef] [Green Version]
- Ryu, K.Y.; Lee, H.J.; Woo, H.; Kang, R.J.; Han, K.M.; Park, H.; Lee, S.M.; Lee, J.Y.; Jeong, Y.J.; Nam, H.W.; et al. Dasatinib regulates LPS-induced microglial and astrocytic neuroinflammatory responses by inhibiting AKT/STAT3 signaling. J. Neuroinflamm. 2019, 16, 190. [Google Scholar] [CrossRef]
- Xu, J.; Yuan, C.; Wang, G.; Luo, J.; Ma, H.; Xu, L.; Mu, Y.; Li, Y.; Seeram, N.P.; Huang, X.; et al. Urolithins Attenuate LPS-Induced Neuroinflammation in BV2Microglia via MAPK, Akt, and NF-kappaB Signaling Pathways. J. Agric. Food Chem. 2018, 66, 571–580. [Google Scholar] [CrossRef]
- Park, J.H.; Seo, Y.H.; Jang, J.H.; Jeong, C.H.; Lee, S.; Park, B. Asiatic acid attenuates methamphetamine-induced neuroinflammation and neurotoxicity through blocking of NF-kB/STAT3/ERK and mitochondria-mediated apoptosis pathway. J. Neuroinflamm. 2017, 14, 240. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, J.; Jang, K.M.; Park, K.-K. Effects of Apamin on MPP+-Induced Calcium Overload and Neurotoxicity by Targeting CaMKII/ERK/p65/STAT3 Signaling Pathways in Dopaminergic Neuronal Cells. Int. J. Mol. Sci. 2022, 23, 15255. https://doi.org/10.3390/ijms232315255
Park J, Jang KM, Park K-K. Effects of Apamin on MPP+-Induced Calcium Overload and Neurotoxicity by Targeting CaMKII/ERK/p65/STAT3 Signaling Pathways in Dopaminergic Neuronal Cells. International Journal of Molecular Sciences. 2022; 23(23):15255. https://doi.org/10.3390/ijms232315255
Chicago/Turabian StylePark, Jihyun, Kyung Mi Jang, and Kwan-Kyu Park. 2022. "Effects of Apamin on MPP+-Induced Calcium Overload and Neurotoxicity by Targeting CaMKII/ERK/p65/STAT3 Signaling Pathways in Dopaminergic Neuronal Cells" International Journal of Molecular Sciences 23, no. 23: 15255. https://doi.org/10.3390/ijms232315255