

Genome-Wide Association Analysis for Hybrid Breeding in Wheat

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
2.1. Phenotypic Characteristics of the Accessions
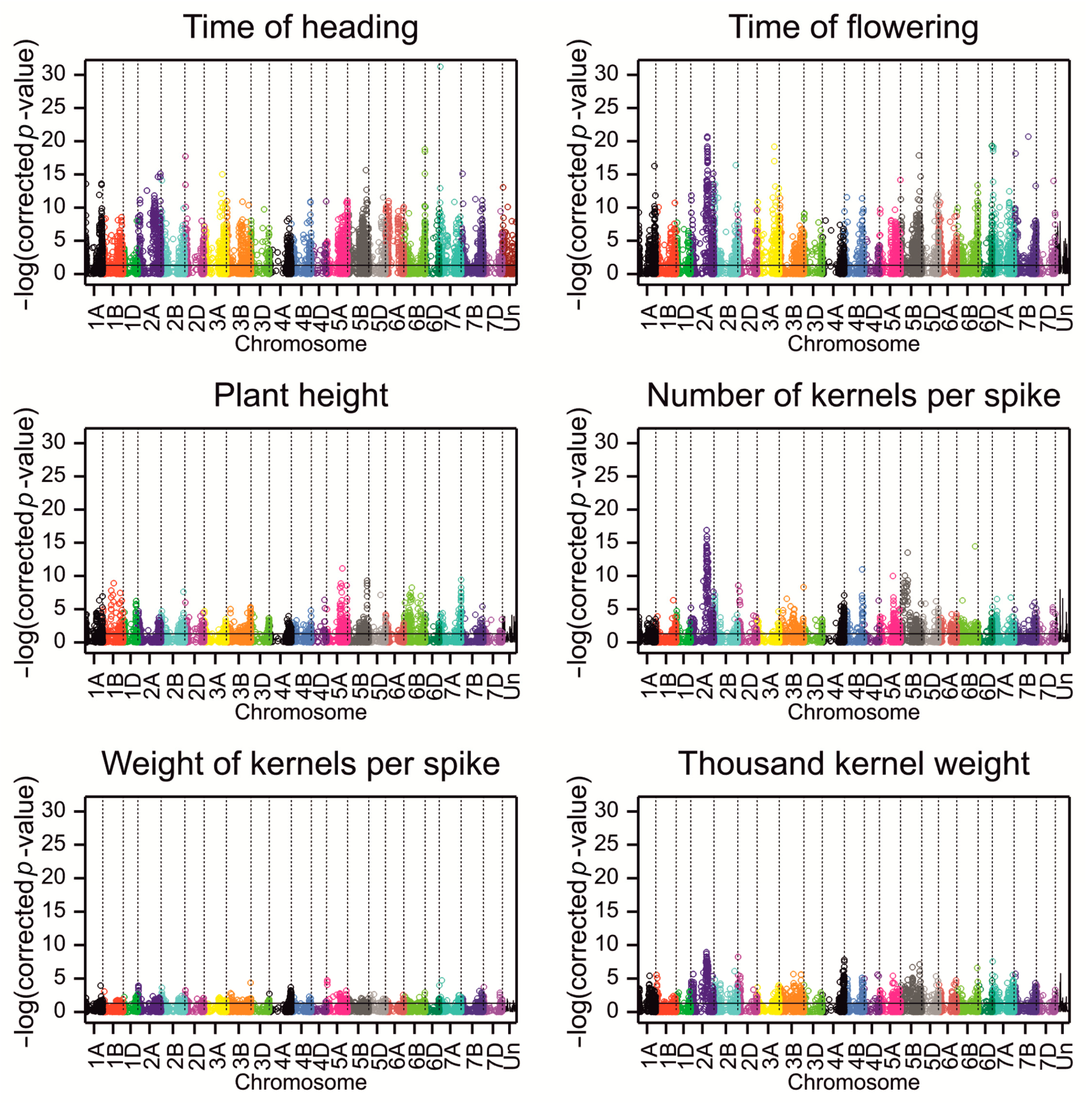
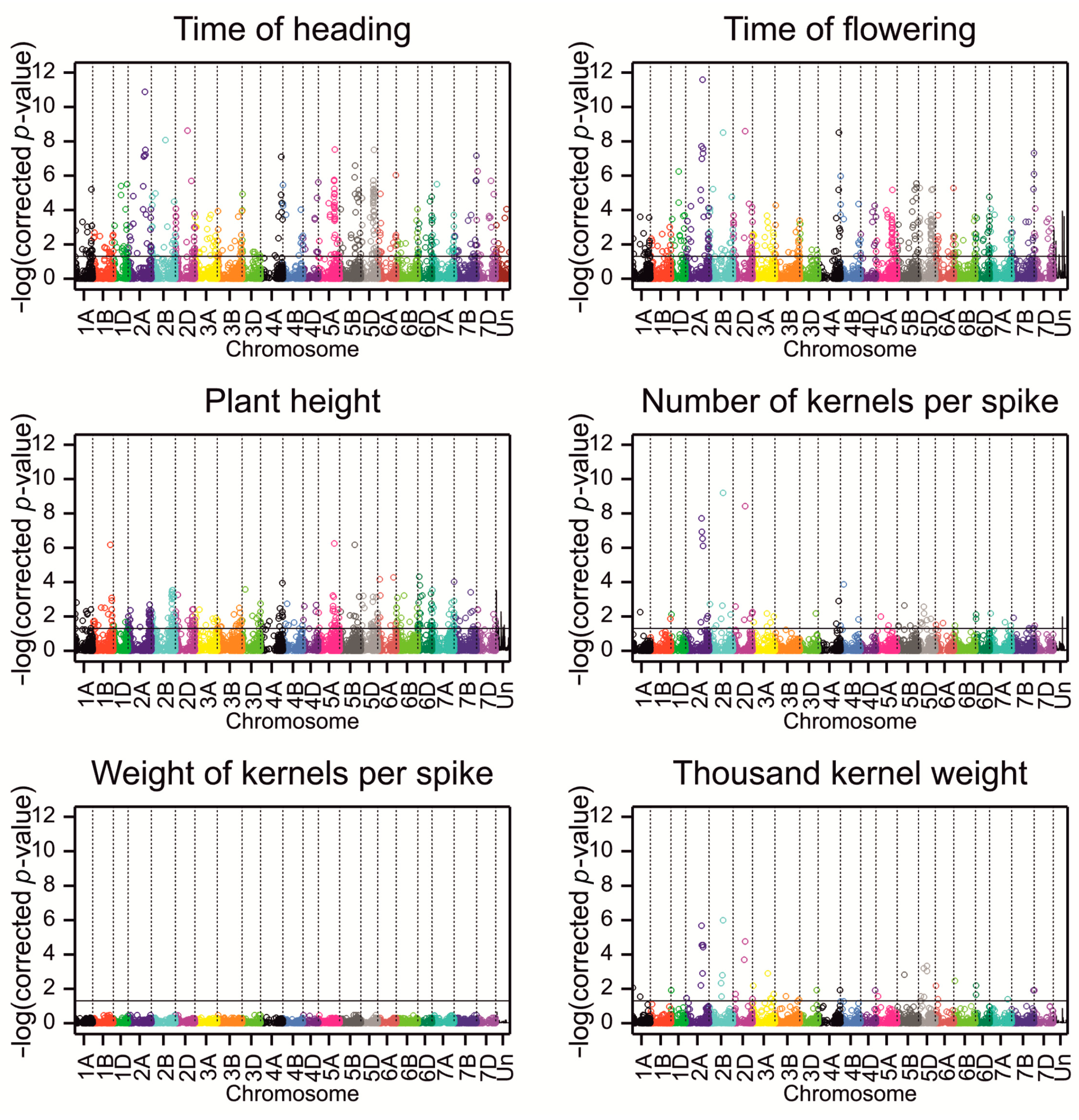
2.2. Association Analysis
2.3. Marker LD Clusters
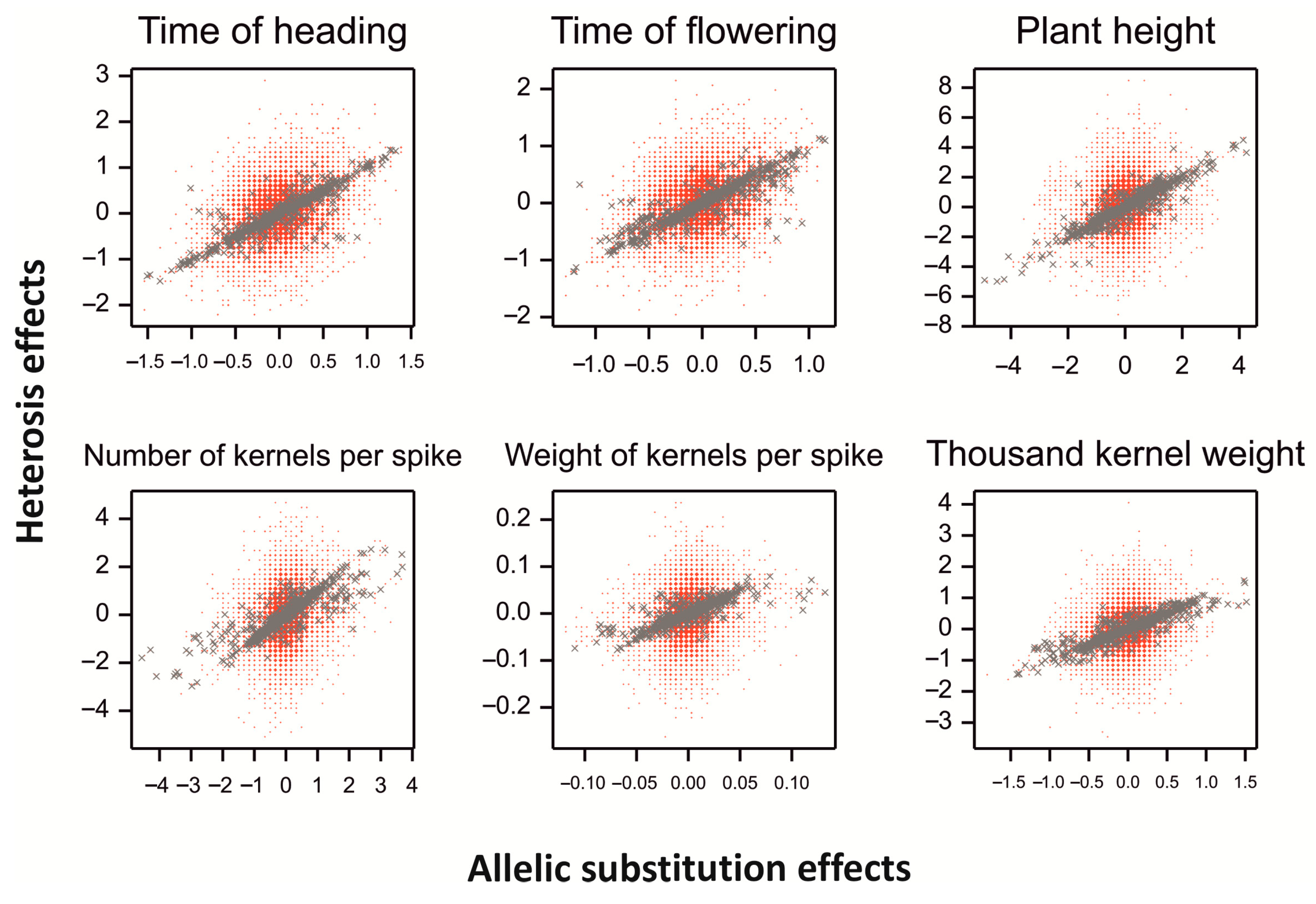
2.4. Heterosis
2.5. Allelic Substitution Effects vs. Heterosis Effects
2.6. GO Annotation of Associated Markers
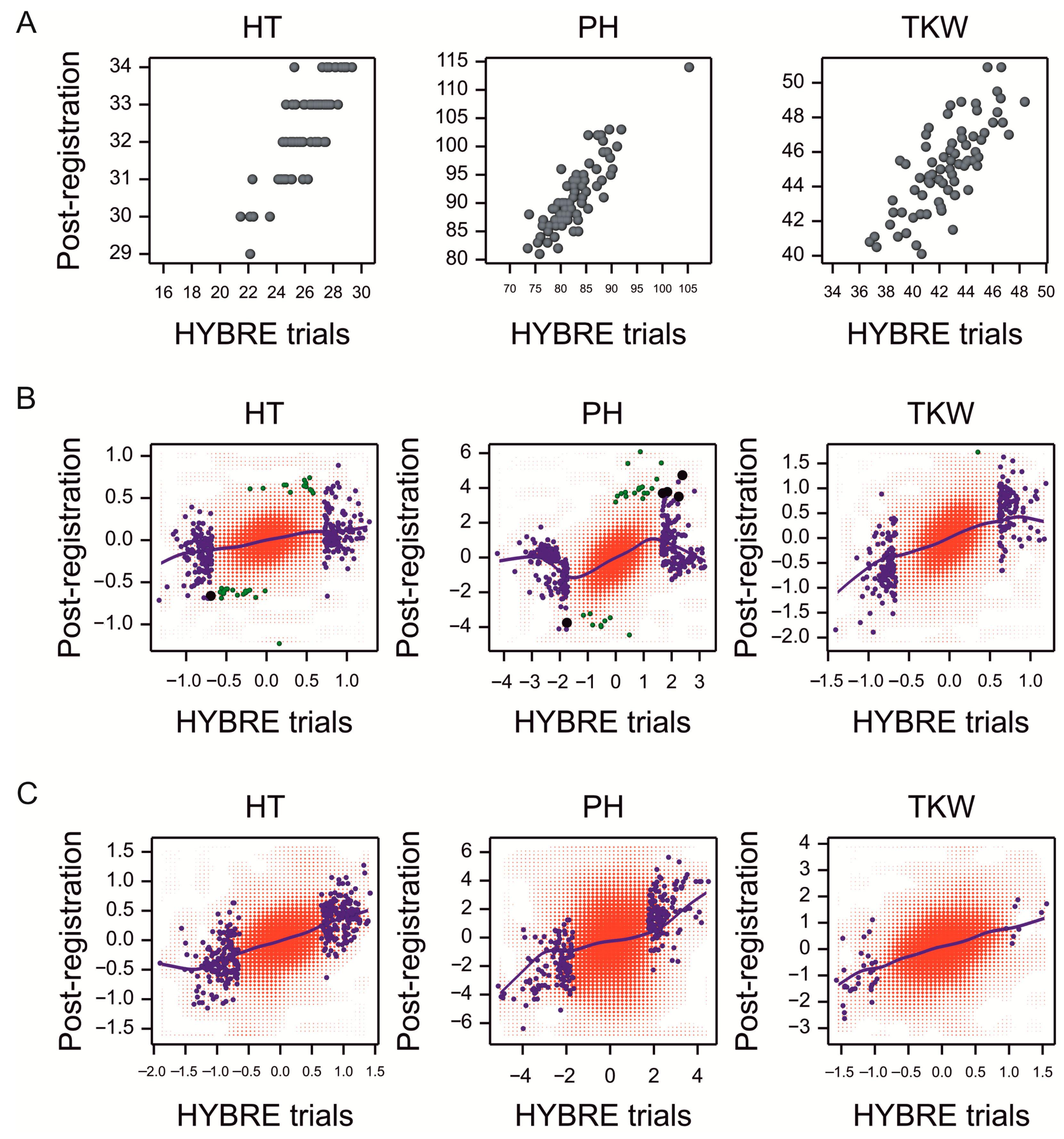
2.7. Validation of Results Using Post-Registration Trial Data
3. Discussion
3.1. Flowering-Related QTLs and Genes
3.2. Plant Height-Related QTLs and Genes
3.3. Spike Traits Related to QTLs and Genes
3.4. SNP Translation Effects
4. Materials and Methods
4.1. Germplasm Resources
4.2. Field Phenotyping
4.3. Genotyping
4.4. Data Analysis
5. Conclusions
- The successful parallel selection of homozygous parental genotypes (based on traits regulated by additive genes controlling phenology and plant height) and components revealing a high heterosis effect (the choice based on the highest values of spike-yield-related traits revealed by heterozygous genotypes) using GWAS.
- Demonstrating that the application of clustered markers for the choice of genotypes in multi-feature processes may be more efficient than classic selection based on single marker polymorphism (SNP)
- Validation of the GWAS results using post-registration trial data.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AMMI | Additive main-effects and multiplicative interaction |
| BLUP | Best linear unbiased prediction |
| FT | Time of flowering |
| GBS | Genotyping by Sequencing |
| GS | Growth stage (BBCH scale) |
| GWAS | Genome-wide association studies |
| HYBRE | The project acronym and the name of the consortium |
| HT | Time of heading |
| KN | Number of kernels per spike |
| KW | Weight of kernels per spike |
| PH | Plant height |
| PRT | Post-registration trials |
| SNP | Single Nucleotide Polymorphism |
| TKW | Thousand kernel weight |
| VEP | Variant Effect Predictor |
References
- Poutanen, K.S.; Kårlund, A.O.; Gómez-Gallego, C.; Johansson, D.P.; Scheers, N.M.; Marklinder, I.M.; Eriksen, A.K.; Silventoinen, P.C.; Nordlund, E.; Sozer, N.; et al. Grains—A major source of sustainable protein for health. Nutr. Rev. 2022, 80, 1648–1663. [Google Scholar] [CrossRef] [PubMed]
- FAOSTAT. Production domain. In Crops; FAO: Rome, Italy, 2019. [Google Scholar]
- El Hanafi, S.; Bendaou, N.; Kehel, Z.; Sanchez-Garcia, M.; Tadesse, W. Phenotypic evaluation of elite spring bread wheat genotypes for hybrid potential traits. Euphytica 2020, 216, 168. [Google Scholar] [CrossRef]
- Briggle, L.W.; Reitz, L.P. Classification of Triticum Species and of Wheat Varieties Grown in the United States; U.S. Department of Agriculture: Washington, DC, USA, 1963. [Google Scholar]
- Fabrizius, M.A.; Busch, R.H.; Khan, K.; Huckle, L. Genetic diversity and heterosis of spring wheat crosses. Crop Sci. 1998, 38, 1108–1112. [Google Scholar] [CrossRef]
- Krystkowiak, K.; Adamski, T.; Surma, M.; Kaczmarek, Z. Relationship between phenotypic and genetic diversity of parental genotypes and the specific combining ability and heterosis effects in wheat (Triticum aestivum L.). Euphytica 2009, 165, 419–434. [Google Scholar] [CrossRef]
- Whitford, R.; Fleury, D.; Reif, J.C.; Garcia, M.; Okada, T.; Korzun, V.; Langridge, P. Hybrid breeding in wheat: Technologies to improve hybrid wheat seed production. J. Exp. Bot. 2013, 64, 5411–5428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mühleisen, J.; Piepho, H.-P.; Maurer, H.P.; Longin, C.F.; Reif, J.C. Yield stability of hybrids versus lines in wheat, barley, and triticale. Theor. Appl. Genet. 2014, 127, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Creech, C. Making the Case for Certified Wheat Seed. CropWatch. 2017. Available online: https://cropwatch.unl.edu/2017/making-case-certified-wheat-seed (accessed on 28 November 2022).
- Gupta, P.K.; Mir, R.R.; Mohan, A.; Kumar, J. Wheat Genomics: Present Status and Future Prospects. Int. J. Plant Genom. 2008, 896451. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.C.; Wong, D.; Forrest, K.; Allen, A.; Chao, S.; Huang, B.E.; Maccaferri, M.; Salvi, S.; Milner, S.G.; Cattivelli, L.; et al. Characterization of polyploid wheat genomic diversity using a high-density 90000 single nucleotide polymorphism array. Plant Biotechnol. 2014, 12, 787–796. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.D.; He, Z.H.; Rasheed, A.; Wen, W.E.; Yan, J.; Zhang, P.Z.; Wan, Y.X.; Zhang, Y.; Xie, C.J.; Xia, X.C. Genome-wide association mapping of black point reaction in common wheat (Triticum aestivum L.). BMC Plant Biol. 2017, 17, 220. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Xu, Z.; Fan, X.; Zhou, Q.; Cao, J.; Wang, F.; Ji, G.; Yang, L.; Feng, B.; Wang, T.A. Genome-Wide Association Study of Wheat Spike Related Traits in China. Front. Plant Sci. 2018, 9, 1584. [Google Scholar] [CrossRef]
- Li, F.J.; Wen, W.E.; He, Z.H.; Liu, J.D.; Jin, H.; Cao, S.H.; Geng, H.W.; Yan, J.; Zhang, P.Z.; Wan, Y.X.; et al. Genome-wide linkage mapping of yield related traits in three Chinese bread wheat populations using high-density SNP markers. Theor. Appl. Genet. 2018, 131, 1903–1924. [Google Scholar] [CrossRef] [PubMed]
- Muqaddasi, Q.H.; Lohwasser, U.; Nagel, M.; Börner, A.; Pillen, K.; Röder, M.S. Genome-Wide Association Mapping of Anther Extrusion in Hexaploid Spring Wheat. PLoS ONE 2016, 11, e0155494. [Google Scholar] [CrossRef] [Green Version]
- Adhikari, A.; Basnet, B.R.; Crossa, J.; Dreisigacker, S.; Camarillo, F.; Bhati, P.K.; Jarquin, D.; Manes, Y.; Ibrahim, A.M.H. Genome-Wide Association Mapping and Genomic Prediction of Anther Extrusion in CIMMYT Hybrid Wheat Breeding Program via Modeling Pedigree, Genomic Relationship, and Interaction With the Environment. Front. Genet. 2020, 11, 586687. [Google Scholar] [CrossRef] [PubMed]
- Gahlaut, V.; Jaiswal, V.; Singh, S.; Balyan, H.S.; Gupta, P.K. Multi-Locus Genome Wide Association Mapping for Yield and Its Contributing Traits in Hexaploid Wheat under Different Water Regimes. Sci. Rep. 2019, 9, 19486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamil, M.; Ali, A.; Gul, A.; Ghafoor, A.; Napar, A.A.; Ibrahim, A.M.H.; Naveed, N.H.; Yasin, N.A.; Mujeeb-Kazi, A. Genome-wide association studies of seven agronomic traits under two sowing conditions in bread wheat. BMC Plant Biol. 2019, 19, 149. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Wen, W.; Liu, J.; Zhang, Y.; Cao, S.; He, Z.; Rasheed, A.; Jin, H.; Zhang, C.; Yan, J.; et al. Genetic architecture of grain yield in bread wheat based on genome-wide association studies. BMC Plant Biol. 2019, 19, 168. [Google Scholar] [CrossRef]
- Tsai, H.-Y.; Janss, L.L.; Andersen, J.R.; Orabi, J.; Jensen, J.D.; Jahoor, A.; Jensen, J. Genomic prediction and GWAS of yield, quality and disease-related traits in spring barley and winter wheat. Sci. Rep. 2020, 10, 3347. [Google Scholar] [CrossRef] [Green Version]
- De Vries, A.P. Flowering biology of wheat, particularly in view of hybrid seed production—A review. Euphytica 1971, 20, 152–170. [Google Scholar] [CrossRef]
- Longin, F.; Mühleisen, J.; Maurer, H.P.; Zhang, H.; Gowda, M.; Reif, J. Hybrid breeding in autogamous cereals. Theor. Appl. Genet. 2012, 125, 1087–1096. [Google Scholar] [CrossRef]
- Langer, S.M.; Longin, C.F.H.; Würschum, T. Phenotypic evaluation of floral and flowering traits with relevance for hybrid breeding in wheat (Triticum aestivum L.). Plant Breed. 2014, 133, 433–441. [Google Scholar] [CrossRef]
- Okada, T.; Ridma, J.E.A.; Jayasinghe, M.; Nansamba, M.; Baes, M.; Warner, P.; Kouidri, A.; Correia, D.; Nguyen, V.; Whitford, R.; et al. Unfertilized ovary pushes wheat flower for cross-pollination. J. Exp. Bot. 2018, 69, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Selva, C.; Riboni, M.; Baumann, U.; Würschum, T.; Whitford, R.; Tucker, M.R. Hybrid breeding in wheat: How shaping floral biology can offer new perspectives. Funct. Plant Biol. 2020, 47, 675–694. [Google Scholar] [CrossRef] [PubMed]
- Zajączkowska, U.; Denisow, B.; Łotocka, B.; Dołkin-Lewko, A.; Rakoczy-Trojanowska, M. Spikelet movements, anther extrusion and pollen production in wheat cultivars with contrasting tendencies to cleistogamy. BMC Plant Biol. 2021, 21, 136. [Google Scholar] [CrossRef]
- Hayes, J.D. Hybrid Wheat—Results and Problems, by A. A. Pickett. 259 pp. Berlin, Hamburg: Paul Parey. DM 96.00 (paperback). ISBN 3 489 53510 3. J. Agric. Sci. 1993, 121, 294. [Google Scholar] [CrossRef]
- Tyrka, M.; Mokrzycka, M.; Bakera, B.; Tyrka, D.; Szeliga, M.; Stojałowski, S.; Matysik, P.; Rokicki, M.; Rakoczy-Trojanowska, M.; Krajewski, P. Evaluation of genetic structure in European wheat cultivars and advanced breeding lines using high-density genotyping-by sequencing approach. BMC Genom. 2021, 22, 81. [Google Scholar] [CrossRef] [PubMed]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [Green Version]
- Gupta, D.R.; Avila, C.S.R.; Win, J.; Soanes, D.M.; Ryder, L.S.; Croll, D.; Bhattacharjee, P.; Hossain, M.S.; Mahmud, N.U.; Mehebub, M.S.; et al. Cautionary Notes on Use of the MoT3 Diagnostic Assay for Magnaporthe oryzae Wheat and Rice Blast Isolates. Phytopathology 2019, 109, 504–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waines, J.G.; Hegde, S.G. Intraspecific Gene Flow in Bread Wheat as Affected by Reproductive Biology and Pollination Ecology of Wheat Flowers. Crop Sci. 2003, 43, 451–463. [Google Scholar] [CrossRef]
- Racedo, J.; Gutiérrez, L.; Perera, M.F.; Ostengo, S.; Pardo, E.M.; Cuenya, M.I.; Welin, B.; Castagnaro, A.P. Genome-wide association mapping of quantitative traits in a breeding population of sugarcane. BMC Plant Biol. 2016, 16, 142. [Google Scholar] [CrossRef] [Green Version]
- Mwadzingeni, L.; Shimelis, H.; Rees, D.J.G.; Tsilo, T.J. Genome-wide association analysis of agronomic traits in wheat under drought-stressed and non-stressed conditions. PLoS ONE 2017, 12, e0171692. [Google Scholar] [CrossRef]
- Mourad, A.M.I.; Sallam, A.; Belamkar, V.; Wegulo, S.; Bowden, R.; Jin, Y.; Mahdy, E.; Bakheit, B.; El-Wafaa, A.A.; Poland, J.; et al. Genome-Wide Association Study for Identification and Validation of Novel SNP Markers for Sr6 Stem Rust Resistance Gene in Bread Wheat. Front. Plant Sci. 2018, 9, 380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malosetti, M.; Ribaut, J.M.; van Eeuwijk, F.A. The statistical analysis of multi-environment data: Modeling genotype-by-environment interaction and its genetic basis. Front. Physiol. 2013, 4, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denisow, B.; Masierowska, M.; Winiarczyk, K.; Rakoczy-Trojanowska, M. The pollen dispersal ability for cross-pollination in winter wheat (Triticum aestivum L.) is related to anther extrusion capability rather than to pollen output. S. Afr. J. Bot. 2022, 148, 283–292. [Google Scholar] [CrossRef]
- Allan, R.E. Wheat. In Hybridization of Crop Plants; Agronomy Books. 3; Fehr, W.R., Hadley, H.H., Eds.; Iova State University: Ames, IA, USA, 1980; Available online: https://lib.dr.iastate.edu/agron_books/3 (accessed on 28 November 2022).
- Wilkinson, P.A.; Winfield, M.O.; Barker, G.L.A.; Allen, A.M.; Burridge, A.; Coghill, J.A.; Burridge, A.; Edwards, K.J. CerealsDB 2.0: An integrated resource for plant breeders and scientists. BMC Bioinform. 2013, 13, 219. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; She, M.; Yang, R.; Jiang, Y.; Qin, Y.; Zhai, S.; Balotf, S.; Zhao, Y.; Anwar, M.; Alhabbar, Z.; et al. Yield-Related QTL Clusters and the Potential Candidate Genes in Two Wheat DH Populations. Int. J. Mol. Sci. 2021, 22, 11934. [Google Scholar] [CrossRef]
- Arjona, J.; Royo, C.; Dreisigacker, S.; Ammar, K.; Villegas, D. Effect of Ppd-A1 and Ppd-B1 Allelic Variants on Grain Number and Thousand Kernel Weight of Durum Wheat and Their Impact on Final Grain Yield. Front. Plant Sci. 2018, 9, 888. [Google Scholar] [CrossRef] [Green Version]
- Zikhali, M.; Griffiths, S. The effect of Earliness per se (Eps) genes on flowering time in bread wheat. In Advances in Wheat Genetics: From Genome to Field, Proceedings of the 12th International Wheat Genetics Symposium, Yokohama, Japan, 8–14 September 2013; Yasunari, O., Shigeo, T., Hirokazu, H., Eds.; Springer: Tokyo, Japan, 2015; pp. 339–345. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wen, W.; Hanif, M.; Xia, X.; Wang, H.; Liu, S.; Liu, J.; Yang, L.; Cao, S.; He, Z. TaELF3-1DL, a homolog of ELF3, is associated with heading date in bread wheat. Mol. Breed. 2016, 36, 161. [Google Scholar] [CrossRef]
- Peng, F.Y.; Hu, Z.; Yang, R.-C. Genome-Wide Comparative Analysis of Flowering-Related Genes in Arabidopsis, Wheat, and Barley. Int. J. Plant Genom. 2015, 874361. [Google Scholar] [CrossRef] [Green Version]
- Lu, Q.; Lillemo, M.; Skinnes, H.; He, X.; Shi, J.; Ji, F.; Dong, Y.; Bjørnstad, A. Anther extrusion and plant height are associated with Type I resistance to Fusarium head blight in bread wheat line ‘Shanghai-3/Catbird’. Theor. Appl. Genet. 2013, 126, 317–334. [Google Scholar] [CrossRef]
- Boeven, P.H.G.; Longin, C.F.H.; Leiser, W.L.; Kollers, S.; Ebmeyer, E.; Würschum, T. Genetic architecture of male floral traits required for hybrid wheat breeding. Theor. Appl. Genet. 2016, 129, 2343–2357. [Google Scholar] [CrossRef]
- Würschum, T.; Liu, G.; Boeven, P.H.G.; Longin, C.F.H.; Mirdita, V.; Kazman, E.; Zhao, Y.; Reif, J.C. Exploiting the Rht portfolio for hybrid wheat breeding. Theor. Appl. Genet. 2018, 131, 1433–1442. [Google Scholar] [CrossRef] [PubMed]
- McIntosch, R.A.; Yamazaki, Y.; Devos, K.M.; Dubcovsky, J.; Rogers, W.J.; Appels, R. Catalogue of gene symbols for wheat. In Proceedings of the 10th International Wheat Genetics Symposium, Paestrum, Italy, 1–6 September 2003; Pogna, N.E., Romano, N., Pogna, E.A., Galterio, G., Eds.; Instituto Sperimentale per la Cerealicoltura: Rome, Italy, 2003; pp. 1–34. [Google Scholar]
- Tian, X.; Wen, W.; Xie, L.; Fu, L.; Xu, D.; Fu, C.; Wang, D.; Chen, X.; Xia, X.; Chen, Q.; et al. Molecular Mapping of Reduced Plant Height Gene Rht24 in Bread Wheat. Front. Plant Sci. 2017, 8, 998. [Google Scholar] [CrossRef] [Green Version]
- Würschum, T.; Langer, S.M.; Longin, C.F.H.; Tucker, M.R.; Leiser, W.L. A modern Green Revolution gene for reduced height in wheat. Plant J. 2017, 92, 892–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanke, C.D.; Ling, J.; Plieske, J.; Kollers, S.; Ebmeyer, E.; Korzun, V.; Argillier, O.; Stiewe, G.; Hinze, M.; Neumann, K.; et al. Whole Genome Association Mapping of Plant Height in Winter Wheat (Triticum aestivum L.). PLoS ONE 2014, 9, e113287. [Google Scholar] [CrossRef] [Green Version]
- Korzun, V.; Röder, M.; Ganal, M.W.; Worland, A.J.; Law, C.N. Genetic analysis of the dwarfing gene (Rht8) in wheat. Part I. Molecular mapping of Rht8 on the short arm of chromosome 2D of bread wheat (Triticum aestivum L.). Theor. Appl. Genet. 1998, 96, 1104–1109. [Google Scholar] [CrossRef]
- Ellis, M.H.; Rebetzke, G.J.; Azanza, F.; Richards, R.A.; Spielmeyer, W. Molecular mapping of gibberellin-responsive dwarfing genes in bread wheat. Theor. Appl. Genet. 2005, 111, 423–430. [Google Scholar] [CrossRef]
- Peng, Z.S.; Li, X.; Yang, Z.J.; Liao, M.L. A new reduced height gene found in the tetraploid semi-dwarf wheat landrace Aiganfanmai. Genet. Mol. Res. 2011, 10, 2349–2357. [Google Scholar] [CrossRef]
- Ellis, H.; Spielmeyer, W.; Gale, R.; Rebetzke, J.; Richards, A. "Perfect" markers for the Rht-B1b and Rht-D1b dwarfing genes in wheat. Theor. Appl. Genet. 2002, 105, 1038–1042. [Google Scholar] [CrossRef]
- Pearce, S.; Saville, R.; Vaughan, S.P.; Chandler, P.M.; Wilhelm, E.P.; Sparks, C.A.; Al-Kaff, N.; Korolev, A.; Boulton, M.I.; Phillips, A.L.; et al. Molecular characterization of Rht-1 dwarfing genes in hexaploid wheat. Plant Physiol. 2011, 157, 1820–1831. [Google Scholar] [CrossRef]
- Xu, T.; Bian, N.; Wen, M.; Xiao, J.; Yuan, C.; Cao, A.; Zhang, S.; Wang, X.; Wang, H. Characterization of a common wheat (Triticum aestivum L.) high-tillering dwarf mutant. Theor. Appl. Genet. 2017, 130, 483–494. [Google Scholar] [CrossRef]
- Alemu, A.; Suliman, S.; Hagras, A.; Thabet, S.; Al-Abdallad, A.; Abdelmula, A.A.; Tadesse, W. Multi-model genome-wide association and genomic prediction analysis of 16 agronomic, physiological and quality related traits in ICARDA spring wheat. Euphytica 2021, 217, 205. [Google Scholar] [CrossRef]
- Yan, X.; Zhao, L.; Ren, Y.; Dong, Z.; Cui, D.; Chen, F. Genome-wide association study revealed that the TaGW8 gene was associated with kernel size in Chinese bread wheat. Sci. Rep. 2019, 9, 2702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Zhou, Y.; Wu, Q.; Chen, Y.; Zhang, P.; Zhang, Y.; Cao, T. Molecular characterization of a novel TaGL3–5A allele and its association with grain length in wheat (Triticum aestivum L.). Theor. Appl. Genet. 2019, 132, 1799–1814. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhou, Y.; Zhang, Y.; Hu, W.; Wu, Q.; Chen, Y.; Zhao, H. Cloning, characterization of TaGS3 and identifcation of allelic variation associated with kernel traits in wheat (Triticum aestivum L.). BMC Genet. 2019, 20, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Zhao, D.; Meng, Z.; Xu, K.; Yan, J.; Xia, X.; Zhang, Y. QTL mapping for grain yield-related traits in bread wheat via SNP-based selective genotyping. Theor. Appl. Genet. 2019, 133, 857–872. [Google Scholar] [CrossRef] [PubMed]
- Daba, S.D.; Tyagi, P.; Brown-Guedira, G.; Mohammadi, M. Genome-wide association studies to identify loci and candidate genes controlling kernel weight and length in a historical United States wheat population. Front. Plant Sci. 2018, 9, 1045. [Google Scholar] [CrossRef] [Green Version]
- Hadfield, K.A.; Bennett, A.B. Polygalacturonases: Many genes in search of a function. Plant Physiol. 1998, 117, 337–343. [Google Scholar] [CrossRef] [Green Version]
- Babu, Y.; Bayer, M. Plant Polygalacturonases Involved in Cell Elongation and Separation-The Same but Different? Plants 2014, 3, 613–623. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Yu, Y.; Liang, Y.; Anderson, C.T.; Cao, J. A Profusion of Molecular Scissors for Pectins: Classification, Expression, and Functions of Plant Polygalacturonases. Front. Plant Sci. 2018, 9, 1208. [Google Scholar] [CrossRef]
- Chateigner-Boutin, A.L.; Suliman, M.; Bouchet, B.; Alvarado, C.; Lollier, V.; Rogniaux, H.; Guillon, F.; Larré, C. Endomembrane proteomics reveals putative enzymes involved in cell wall metabolism in wheat grain outer layers. J. Exp. Bot. 2015, 66, 2649–2658. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Li, J.; Cheng, Y.; Yao, F.; Long, L.; Wang, Y.; Wu, Y.; Li, J.; Wang, J.; Jiang, Q.; et al. Genome-wide association study reveals new loci for yield-related traits in Sichuan wheat germplasm under stripe rust stress. BMC Genom. 2019, 20, 640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, K.; Yang, W. E3 Ubiquitin Ligases: Ubiquitous Actors in Plant Development and Abiotic Stress Responses. Plant Cell Physiol. 2017, 58, 1461–1476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, A.K.; Lu, Y.; Zinta, G.; Lang, Z.; Zhu, J.K. UTR-Dependent Control of Gene Expression in Plants. Trends Plant Sci. 2018, 23, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Remy, E.; Cabrito, T.R.; Batista, R.A.; Hussein, M.A.M.; Teixeira, M.C.; Athanasiadis, A.; Sá-Correia, I.; Duque, P. Intron Retention in the 5′UTR of the Novel ZIF2 Transporter Enhances Translation to Promote Zinc Tolerance in Arabidopsis. PLoS Genet. 2014, 10, e1004375. [Google Scholar] [CrossRef] [Green Version]
- Chung, B.Y.; Simons, C.; Firth, A.E.; Brown, C.M.; Hellens, R.P. Effect of 5’UTR introns on gene expression in Arabidopsis thaliana. BMC Genom. 2006, 7, 120. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Liu, B.; Wu, K.; Ye, Y.; Huang, S.; Wang, S.; Wang, Y.; Han, R.; Liu, Q.; Fu, X.; et al. Regulation of OsmiR156h through Alternative Polyadenylation Improves Grain Yield in Rice. PLoS ONE 2015, 10, e0126154. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.; Muller, A.E. Flowering time control and applications in plant breeding. Trends Plant Sci. 2009, 14, 563–573. [Google Scholar] [CrossRef]
- Gaudinier, A.; Blackman, B.K. Evolutionary processes from the perspective of flowering time diversity. New Phytol. 2020, 225, 1883–1898. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-C.; Wei, F.-J.; Chiou, W.-Y.; Tsai, Y.-C.; Wu, H.-P.; Gotarkar, D.; Wei, Z.H.; Lai, M.H.; Hsing, Y.I.C. Studies of rice Hd1 haplotypes worldwide reveal adaptation of flowering time to different environments. PLoS ONE 2020, 15, e0239028. [Google Scholar] [CrossRef]
- Research Centre for Cultivar Testing. Available online: http://www.coboru.pl (accessed on 23 June 2019).
- Cullis, B.R.; Smith, A.B.; Coombes, N.E. On the design of early generation variety trials with correlated data. J. Agric. Biol. Environ. Stat. 2006, 11, 381–393. [Google Scholar] [CrossRef]
- Gauch, H.G. Statistical Analysis of Regional Yield Trials: AMMI Analysis of Factorial Designs; Elsevier: Amsterdam, The Netherlands, 1992. [Google Scholar]
- van Eeuwijk, F.A.; Bink, M.C.A.M.; Chenu, K.; Chapman, S.C. Detection and use of QTL for complex traits in multiple environments. Curr. Opin. Plant Biol. 2010, 13, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Vidotti, M.S.; Lyra, D.H.; Morosini, J.S.; Granato, Í.S.C.; Quecine, M.C.; Azevedo, J.L.D.; Fritsche-Neto, R. Additive and heterozygous (dis)advantage GWAS models reveal candidate genes involved in the genotypic variation of maize hybrids to Azospirillum brasilense. PLoS ONE 2019, 14, e0222788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VSN International. Genstat for Windows. 19th ed. Hemel Hempstead. Web Page. 2017. Available online: Genstat.co.uk (accessed on 28 November 2022).
- Kashif, M.; Khaliq, I. Heritability, correlation and path coefficient analysis for some metric traits in wheat. Int. J. Agric. Biol. 2004, 6, 138–142. [Google Scholar]
- Nukasani, V.; Potdukhe, N.R.; Bharad, S.; Deshmukh, S.; Shinde, S.M. Genetic variability, correlation and path analysis in wheat. J. Wheat Res. 2013, 5, 48–51. [Google Scholar]
- Guo, Z.; Zhao, Y.; Röder, M.S.; Reif, J.C.; Ganal, M.W.; Chen, D.; Schnurbusch, T. Manipulation and prediction of spike morphology traits for the improvement of grain yield in wheat. Sci. Rep. 2018, 8, 14435. [Google Scholar] [CrossRef] [Green Version]
- Mangini, G.; Gadaleta, A.; Colasuonno, P.; Marcotuli, I.; Signorile, A.M.; Simeone, R.; De Vita, P.; Mastrangelo, A.M.; Laidò, G.; Pecchioni, N.; et al. Genetic dissection of the relationships between grain yield components by genome-wide association mapping in a collection of tetraploid wheats. PLoS ONE 2018, 13, e0190162. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Mean | Minimum | Maximum | CV | Variance Component ± s.e. | Heritability (%) | |||
|---|---|---|---|---|---|---|---|---|---|
| G | G × Y | G × L | Error | ||||||
| HT | 25.45 | 10.5 | 34.44 | 13.8 | 6.06 ± 0.42 | 0.91 ± 0.07 | 0.36 ± 0.03 | 1.56 ± 0.03 | 90.41 |
| FT | 31.67 | 17.87 | 39.95 | 11.74 | 4.04 ± 0.32 | 1.39 ± 0.11 | 0.46 ± 0.04 | 1.38 ± 0.03 | 80.77 |
| PH | 81.5 | 42.66 | 127.73 | 17.42 | 32.52 ± 2.37 | 5.92 ± 0.55 | 2.41 ± 0.31 | 22.15 ± 0.42 | 86.79 |
| KN | 62.24 | 27.69 | 103.9 | 15.88 | 14.05 ± 1.73 | 9.68 ± 1.12 | 11.44 ± 1.03 | 63.5 ± 1.2 | 54.60 |
| KW | 2.53 | 1.03 | 4.33 | 19.49 | 0.018 ± 0.003 | 0.028 ± 0.003 | 0.023 ± 0.002 | 0.14 ± 0.003 | 38.79 |
| TKW | 41.87 | 23.86 | 55.96 | 12.03 | 4.36 ± 0.56 | 5.41 ± 0.42 | 2.25 ± 0.18 | 9.72 ± 0.18 | 52.97 |
| Trait | Number of Associations | Negative Significant Effects | Positive Significant Effects | 2th Percentile | |||
|---|---|---|---|---|---|---|---|
| Min | Max | Min | Max | Lower | Upper | ||
| HT | 4206 | −1.57 | −0.0024 | 0.0003 | 1.49 | −0.68 | 0.72 |
| FT | 4108 | −1.30 | −0.0027 | 0.0014 | 1.13 | −0.61 | 0.64 |
| PH | 1999 | −4.24 | −0.0006 | 0.0013 | 3.18 | −1.72 | 1.63 |
| KN | 1858 | −4.29 | −0.0015 | 0.0113 | 3.27 | −1.36 | 1.54 |
| KW | 738 | −0.09 | −0.0001 | 0.0002 | 0.12 | −0.05 | 0.05 |
| TKW | 1908 | −1.41 | −0.0002 | 0.0001 | 1.19 | −0.66 | 0.61 |
| Trait | Number of Associations | Number of SNPs with Mean LD <0.01 | Number of SNP Clusters | With GE Interaction (% out of Significant) | % in Subgenome | % in Genes | Number of SNPs Affecting Protein Translation | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | B | D | High | Low | Moderate | Modifier * | ||||||
| HT | 444 | 71 | 220 | 66.0 | 47.1 | 36.9 | 13.3 | 55.4 | 0 | 57 | 45 | 342 (144) |
| FT | 460 | 78 | 238 | 66.1 | 43.7 | 34.8 | 18.7 | 56.7 | 1 | 64 | 38 | 357 (158) |
| PH | 391 | 21 | 123 | 38.1 | 20.2 | 63.2 | 13.3 | 59.6 | 0 | 52 | 66 | 273 (115) |
| KN | 170 | 25 | 94 | 31.2 | 44.1 | 32.4 | 21.8 | 55.9 | 0 | 18 | 20 | 132 (57) |
| KW | 51 | 10 | 36 | 68.6 | 41.2 | 39.2 | 19.6 | 54.9 | 0 | 7 | 6 | 38 (15) |
| TKW | 272 | 22 | 115 | 61.4 | 55.1 | 36.4 | 5.9 | 58.5 | 1 | 23 | 29 | 219 (106) |
| Marker | Gene | Effect | Interpro Description |
|---|---|---|---|
| 2253029|F|0-10|CT (negative) | TraesCS2A02G482200 | MFI | NAD-dependent epimerase/dehydratase; NAD(P)-binding domain superfamily |
| 3938110|F|0-10|CG | TraesCS2B02G045100 | LOW | NB-ARC;P-loop containing nucleoside triphosphate hydrolase; Leucine-rich repeat domain superfamily |
| 1237800|F|0-13|CG (positive) | TraesCS2B02G164700 | MFI | F-box-like domain superfamily |
| 4910338|F|0-15|GC | TraesCS2B02G475700 | MDR | Zf-FLZ domain; Zf-FLZ domain |
| 2322929|F|0-10|AT | TraesCS2B02G490600 | MFI | Ribonuclease H-like superfamily; Exonuclease, RNase T/DNA polymerase III; Ribonuclease H superfamily |
| 2322355|F|0-40|CG | TraesCS2B02G521100 | LOW | Glycosyl transferase;1,3-beta-glucan synthase subunit FKS1-like, domain-1 |
| 1238701|F|0-16|GA | TraesCS2D02G127300 | LOW | F-box domain;F-box-like domain superfamily |
| 1675478|F|0-15|TG | TraesCS3A02G506600 | LOW | NAD(P)-binding domain superfamily; NAD(P)-binding domain superfamily |
| 7350269|F|0-8|TC | TraesCS3A02G517700 | LOW | |
| 1049114|F|0-20|AG | TraesCS3D02G511900 | MFI | Ubiquitin-like domain; Ubiquitin domain; Ubiquitin-like domain superfamily |
| 1675534|F|0-35|AC | TraesCS3D02G513900 | MDR | UDP-glucuronosyl/UDP-glucosyltransferase |
| 2252787|F|0-19|CT | TraesCS3D02G529700 | MFI | Coenzyme Q-binding protein COQ10, START domain; START-like domain superfamily |
| 7352843|F|0-43|AT | TraesCS3D02G531000 | MDR | Transcription initiation factor IIA, gamma subunit; Transcription factor IIA, helical; Transcription factor IIA, beta-barrel; Transcription initiation factor IIA, gamma subunit, C-terminal |
| 7352096|F|0-35|GC | TraesCS3D02G541900 | LOW | Uncharacterised conserved protein UCP015417, vWA |
| 7353078|F|0-15|CA | TraesCS3D02G542800 | MDR | Oxoglutarate/iron-dependent dioxygenase; Non-haem dioxygenase N-terminal domain; Isopenicillin N synthase-like |
| 3024403|F|0-23|CT | TraesCS3D02G543100 | LOW | DnaJ domain; Tetratricopeptide-like helical domain superfamily; Tetratricopeptide repeat-containing domain |
| 1009915|F|0-65|CG | TraesCSU02G059500 | LOW | Leucine-rich repeat, cysteine-containing subtype; SKP1/BTB/POZ domain superfamily; BTB/Kelch-associated; Leucine-rich repeat domain superfamily |
| 1056528|F|0-9|TG (negative) * | - | MFI | - |
| Trait | Number of Significant Effects | % in Genome | % in Genes | Number SNPs with Protein Translation Effect (in Genes) | ||||
|---|---|---|---|---|---|---|---|---|
| A | B | D | Low | Moderate | Modifier * | |||
| HT | 437 | 33.9 | 28.6 | 35.5 | 57.7 | 106 | 52 | 275 (94) |
| FT | 393 | 35.4 | 28.0 | 35.4 | 57.0 | 96 | 45 | 249 (83) |
| PH | 324 | 35.5 | 31.8 | 28.7 | 52.8 | 52 | 41 | 229 (78) |
| KN | 62 | 37.1 | 32.3 | 29.0 | 62.9 | 18 | 3 | 41 (18) |
| TKW | 45 | 42.2 | 20.0 | 37.8 | 68.9 | 13 | 2 | 30 (16) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mokrzycka, M.; Stojałowski, S.; Tyrka, M.; Matysik, P.; Żmijewska, B.; Marcinkowski, R.; Woźna-Pawlak, U.; Martofel, R.; Rokicki, M.; Rakoczy-Trojanowska, M.; et al. Genome-Wide Association Analysis for Hybrid Breeding in Wheat. Int. J. Mol. Sci. 2022, 23, 15321. https://doi.org/10.3390/ijms232315321
Mokrzycka M, Stojałowski S, Tyrka M, Matysik P, Żmijewska B, Marcinkowski R, Woźna-Pawlak U, Martofel R, Rokicki M, Rakoczy-Trojanowska M, et al. Genome-Wide Association Analysis for Hybrid Breeding in Wheat. International Journal of Molecular Sciences. 2022; 23(23):15321. https://doi.org/10.3390/ijms232315321
Chicago/Turabian StyleMokrzycka, Monika, Stefan Stojałowski, Mirosław Tyrka, Przemysław Matysik, Barbara Żmijewska, Rafał Marcinkowski, Urszula Woźna-Pawlak, Róża Martofel, Michał Rokicki, Monika Rakoczy-Trojanowska, and et al. 2022. "Genome-Wide Association Analysis for Hybrid Breeding in Wheat" International Journal of Molecular Sciences 23, no. 23: 15321. https://doi.org/10.3390/ijms232315321