
Shifting the pH Optima of (R)-Selective Transaminases by Protein Engineering


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
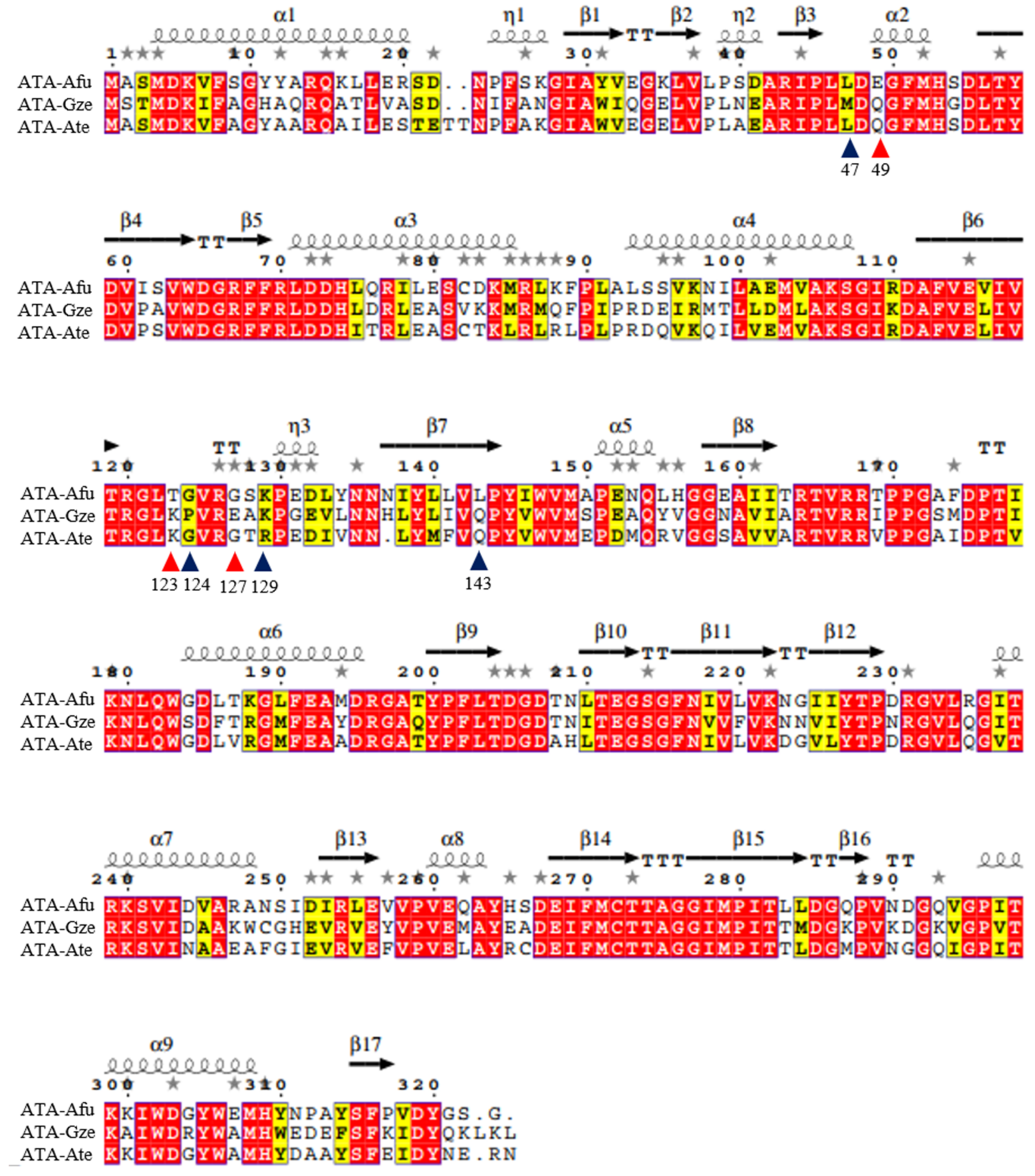
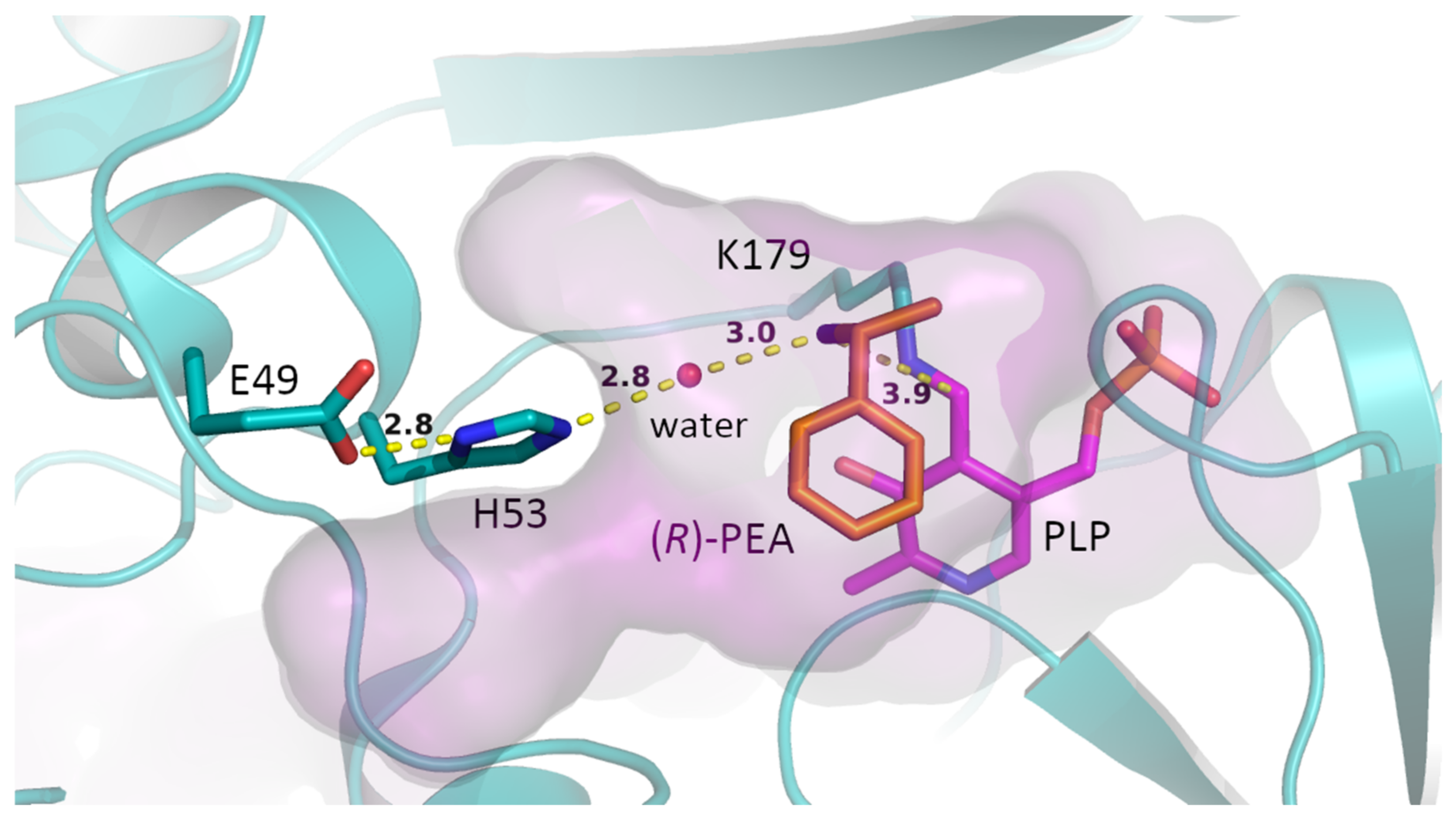
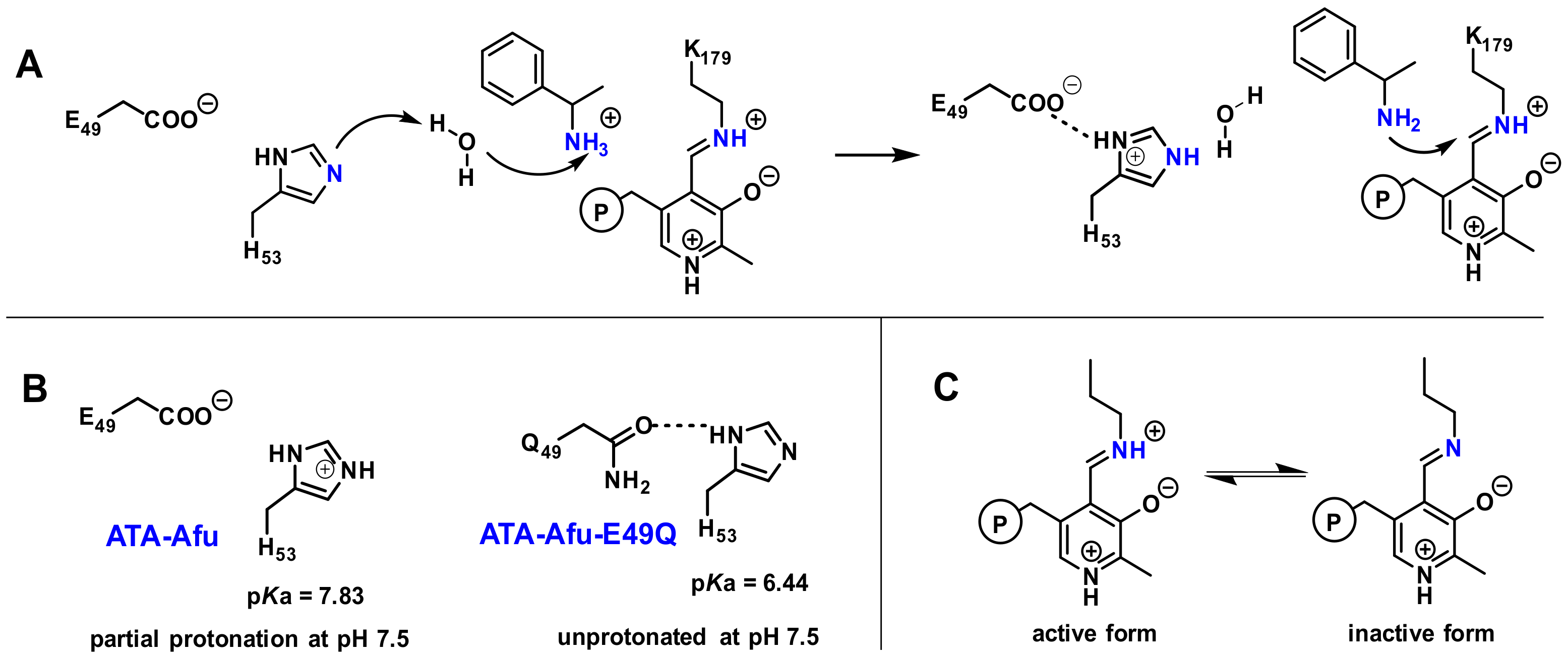
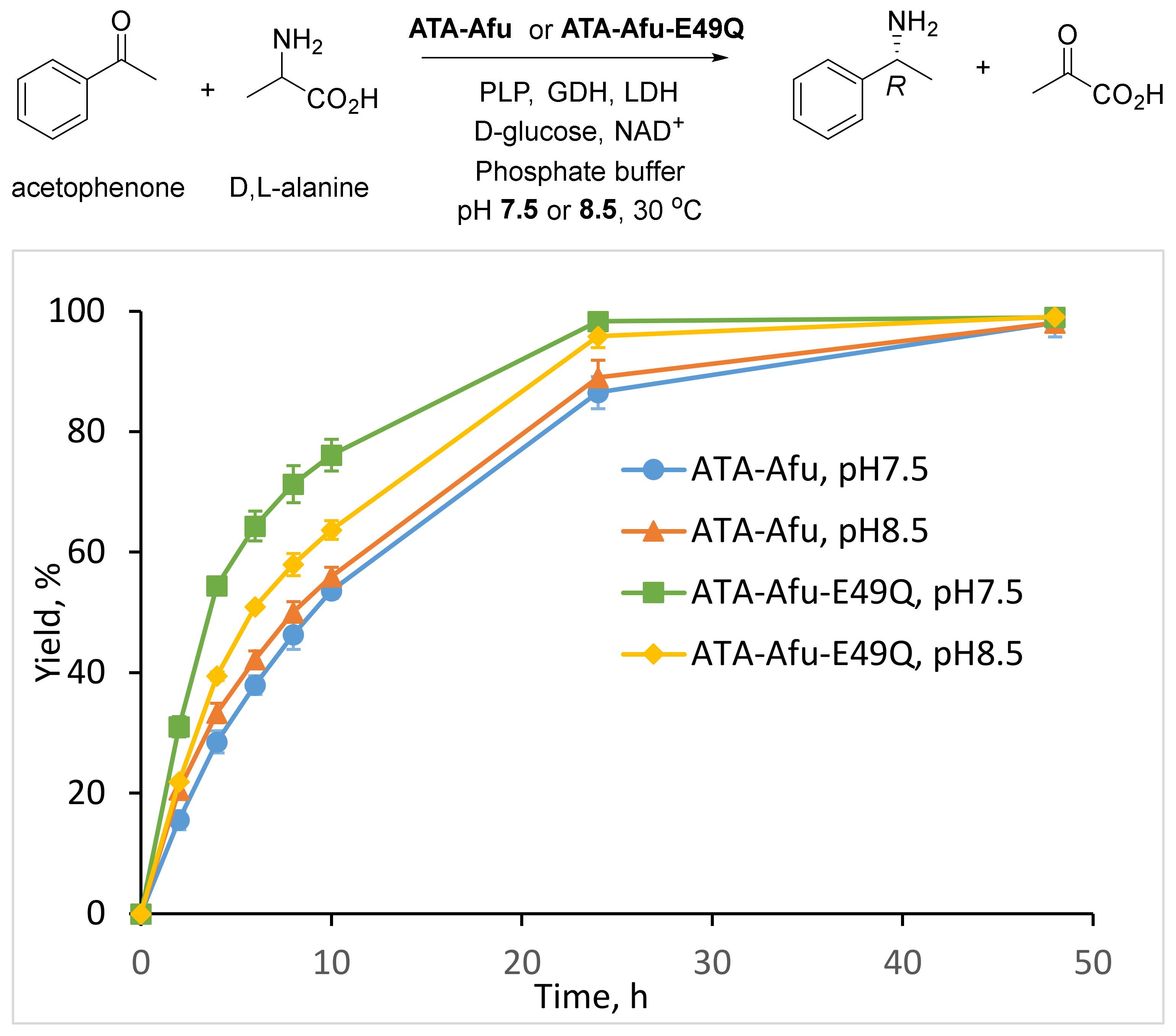
2. Results and Discussion
3. Materials and Methods
3.1. Site-Directed Mutagenesis
3.2. Gene Expression and Purification of the Enzyme Variants
3.3. Determination of Activity of Transaminases
3.4. Docking Experiments
3.5. pKa Prediction and Evaluation
3.6. Asymmetric Synthesis of (R)-1-Phenylethylamine
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lawrence, S.A. Amines: Synthesis, Properties and Applications, 1st ed.; Cambridge University: Cambridge, UK, 2004. [Google Scholar]
- Nugent, T.C. Chiral Amine Synthesis: Methods, Developments and Applications, 1st ed.; Willy-VCH: Weinheim, Germany, 2010. [Google Scholar]
- Yin, Q.; Shi, Y.; Wang, J.; Zhang, X. Direct catalytic asymmetric synthesis of α–chiral primary amines. Chem. Soc. Rev. 2020, 49, 6141–6153. [Google Scholar] [CrossRef] [PubMed]
- Höhne, M.; Bornscheuer, U.T. Biocatalytic routes to optically active amines. ChemCatChem. 2009, 1, 42–51. [Google Scholar] [CrossRef]
- Kohls, H.; Steffen-Munsberg, F.; Höhne, M. Recent achievements in developing the biocatalytic toolbox for chiral amine synthesis. Curr. Opin. Chem. Biol. 2014, 19, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Snajdrova, R.; Moore, J.C.; Baldenius, K.; Bornscheuer, U.T. Biocatalysis: Enzymatic synthesis for industrial applications. Angew. Chem. Int. Ed. 2020, 60, 88–119. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, M.; Farnberger, J.E.; Kroutil, W. The industrial age of biocatalytic transamination. Eur. J. Org. Chem. 2015, 2015, 6965–6982. [Google Scholar] [CrossRef] [Green Version]
- Guo, F.; Berglund, P. Transaminase biocatalysis: Optimization and application. Green Chem. 2017, 19, 333–360. [Google Scholar] [CrossRef] [Green Version]
- Slabu, I.; Galman, J.L.; Lloyd, R.C.; Turner, N.J. Discovery, engineering, and synthetic application of transaminase biocatalysts. ACS Catal. 2017, 7, 8263–8284. [Google Scholar] [CrossRef]
- Kelly, S.A.; Pohle, S.; Wharry, S.; Mix, S.; Allen, C.C.R.; Moody, T.S. Application of ω–transaminases in the pharmaceutical industry. Chem. Rev. 2018, 118, 349–367. [Google Scholar] [CrossRef]
- Savile, C.K.; Janey, J.M.; Mundorff, E.C.; Moore, J.C.; Tam, S.; Jarvis, W.R.; Colbeck, J.C.; Krebber, A.; Fleitz, F.J.; Brands, J.; et al. Biocatalytic asymmetric synthesis of chiral amines from ketones applied to sitagliptin manufacture. Science 2010, 329, 305–309. [Google Scholar] [CrossRef] [Green Version]
- Höhne, M.; Schätzle, S.; Jochens, H.; Robins, K.; Bornscheuer, U.T. Rational assignment of key motifs for function guides in silico enzyme identification. Nat. Chem. Biol. 2010, 6, 807–813. [Google Scholar] [CrossRef]
- Schätzle, S.; Steffen-Munsberg, F.; Thontowi, A.; Höhne, M.; Robins, K.; Bornscheuer, U.T. Enzymatic asymmetric synthesis of enantiomerically pure aliphatic, aromatic and arylaliphatic amines with (R)-selective amine transaminases. Adv. Synth. Catal. 2011, 353, 2439–2445. [Google Scholar] [CrossRef]
- Koszelewski, D.; Lavandera, I.; Clay, D.; Rozzell, D.; Kroutil, W. Asymmetric synthesis of optically pure pharmacologically relevant amines employing ω–transaminases. Adv. Synth. Catal. 2008, 350, 2761–2766. [Google Scholar] [CrossRef]
- Höhne, M.; Kühl, S.; Robins, K.; Bornscheuer, U.T. Efficient asymmetric synthesis of chiral amines by combining transaminase and pyruvate decarboxylase. ChemBioChem 2008, 9, 363–365. [Google Scholar] [CrossRef]
- Skalden, L.; Peters, C.; Dickerhoff, J.; Nobili, A.; Joosten, H.-J.; Weisz, K.; Höhne, M.; Bornscheuer, U.T. Two subtle amino acid changes in a transaminase substantially enhance or invert enantiopreference in cascade syntheses. ChemBioChem 2015, 16, 1041–1045. [Google Scholar] [CrossRef]
- Kohls, H.; Anderson, M.; Dickerhoff, J.; Weisz, K.; Córdova, A.; Berglund, P.; Brundiek, H.; Bornscheuer, U.T.; Höhne, M. Selective access to all four diastereomers of a 1,3-amino alcohol by combination of a keto reductase- and an amine transaminase-catalysed reaction. Adv. Synth. Catal. 2015, 357, 1808–1814. [Google Scholar] [CrossRef]
- Sviatenko, O.; Ríos-Lombardía, N.; Morís, F.; González-Sabín, J.; Manideep, K.V.; Merdivan, S.; Günther, S.; Süss, P.; Höhne, M. One-pot synthesis of 4-aminocyclohexanol isomers by combining a keto reductase and an amine transaminase. ChemCatChem 2019, 11, 5794–5799. [Google Scholar] [CrossRef]
- Yi, D.; Bayer, T.; Badenhorst, C.P.S.; Wu, S.; Dörr, M.; Höhne, M.; Bornscheuer, U.T. Recent trends in biocatalysis. Chem. Soc. Rev. 2021, 50, 8003–8049. [Google Scholar] [CrossRef]
- Bornscheuer, U.T.; Huisman, G.W.; Kazlauskas, R.J.; Lutz, S.; Moore, J.C.; Robins, K. Engineering the third wave of biocatalysis. Nature 2012, 485, 185–194. [Google Scholar] [CrossRef]
- Miller, D.C.; Athavale, S.V.; Arnold, F.H. Combining chemistry and protein engineering for new-to-nature biocatalysis. Nat. Synth. 2022, 1, 18–23. [Google Scholar] [CrossRef]
- Thomas, P.G.; Russell, A.J.; Fersht, A.R. Tailoring the pH dependence of enzyme catalysis using protein engineering. Nature 1985, 318, 375–376. [Google Scholar] [CrossRef]
- Russell, A.J.; Fersht, A.R. Rational modification of enzyme catalysis by engineering surface charge. Nature 1987, 328, 496–500. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, G.; Jones, J.B. Chemical modification at a single site can induce significant shifts in the pH profiles of a serine protease. J. Am. Chem. Soc. 1998, 120, 8582–8586. [Google Scholar] [CrossRef]
- Pokhrel, S.; Joo, J.C.; Yoo, Y.J. Shifting the optimum pH of Bacillus circulans xylanase towards acidic side by introducing arginine. Biotechnol. Bioprocess Eng. 2013, 18, 35–42. [Google Scholar] [CrossRef]
- Xu, H.; Zhang, F.; Shang, H.; Li, X.; Wang, J.; Qiao, D.; Cao, Y. Alkalophilic adaptation of XynB endoxylanase from Aspergillus niger via rational design of pKa of catalytic residues. J. Biosci. Bioeng. 2013, 115, 618–622. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Xie, Y.; Luo, M.; Wang, S.; Hu, Y.; Liu, Y.; Feng, Y.; Yang, G.-Y. Sequence homolog-based molecular engineering for shifting the enzymatic pH optimum. Synth. Syst. Biotechnol. 2016, 1, 195–206. [Google Scholar] [CrossRef] [Green Version]
- Qiu, J.; Han, H.; Sun, B.; Chen, L.; Yu, C.; Peng, R.; Yao, Q. Residue mutations of xylanase in Aspergillus kawachii alter its optimum pH. Microbiol. Res. 2016, 182, 1–7. [Google Scholar] [CrossRef]
- Weaver, J.D.; Mullaney, E.J.; Lei, X.G. Altering the substrate specificity site of Aspergillus niger PhyB shifts the pH optimum to pH 3.2. Appl. Microbiol. Biotechnol. 2007, 76, 117–122. [Google Scholar] [CrossRef]
- Ushasree, M.V.; Vidya, J.; Pandey, A. Replacement P212H altered the pH-temperature profile of phytase from Aspergillus niger NII 08121. Appl. Microbiol. Biotechnol. 2015, 175, 3084–3092. [Google Scholar]
- Yasuda, T.; Takeshita, H.; Iida, R.; Ueki, M.; Nakajima, T.; Kaneko, Y.; Mogi, K.; Kominato, Y.; Kishi, K. A Single amino acid substitution can shift the optimum pH of DNase I for enzyme activity: Biochemical and molecular analysis of the piscine DNase I family. Biochim. Biophys. Acta 2004, 1672, 174–183. [Google Scholar] [CrossRef]
- Qiu, S.; Lai, L. Tailoring the pH dependence of human non-pancreatic secretory phospholipase A2 by engineering surface charges. Appl. Biochem. Biotechnol. 2013, 171, 1454–1464. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, M.; Yang, T.; Zhang, X.; Rao, Z. Surface charge-based rational design of aspartase modifies the optimal pH for efficient β–aminobutyric acid production. Int. J. Biol. Macromol. 2020, 164, 4165–4172. [Google Scholar] [CrossRef]
- Sugino, M.; Kajita, S.; Banno, K.; Shirai, T.; Yamane, T.; Kato, M.; Kobayashi, T.; Tsukagoshi, N. Upward shift of the pH optimum of Acremonium ascorbate oxidase. Biochim. Biophys. Acta 2002, 1596, 36–46. [Google Scholar] [CrossRef]
- Yun, Y.S.; Lee, T.-H.; Nam, G.H.; Jang, D.S.; Shin, S.; Oh, B.-H.; Choi, K.Y. Origin of the different pH activity profile in two homologous ketosteroid isomerases. J. Biol. Chem. 2003, 278, 28229–28236. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Huang, N.; Zhou, L.; Cui, W.; Liu, Z.; Zhu, L.; Liu, Y.; Zhou, Z. Modulating the pH activity profiles of phenylalanine ammonia lyase from Anabaena variabilis by modification of center-near surface residues. Appl. Biochem. Biotechnol. 2017, 183, 699–711. [Google Scholar] [CrossRef]
- Hu, H.-J.; Wang, Q.-Q.; Wang, D.-X.; Ao, Y.-F. Enantioselective biocatalytic desymmetrization for synthesis of enantiopure cis-3,4-disubstituted pyrrolidines. Green Synth. Catal. 2021, 2, 324–327. [Google Scholar] [CrossRef]
- Pennacchietti, E.; Lammens, T.M.; Capitani, G.; Franssen, M.C.R.; John, R.A.; Bossa, F.; Biase, D.D. Mutation of His465 alters the pH-dependent spectroscopic properties of Escherichia coli glutamate decarboxylase and broadens the range of its activity toward more alkaline pH. J. Biol. Chem. 2009, 284, 31587–31596. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, H.; Kagamiyama, H. Transient-state kinetics of the reaction of aspartate aminotransferase with aspartate at low pH reveals dual routes in the enzyme-substrate association process. Biochemistry 1997, 36, 13558–13569. [Google Scholar] [CrossRef]
- Hayashi, H.; Mizuguchi, H.; Kagamiyama, H. The imine-pyridine torsion of the pyridoxal 5’-phosphate Schiff base of aspartate aminotransferase lowers its pKa in the unliganded enzyme and is crucial for the successive increase in the pKa during catalysis. Biochemistry 1998, 37, 15076–15085. [Google Scholar] [CrossRef]
- Mizuguchi, H.; Hayashi, H.; Okada, K.; Miyahara, I.; Hirotsu, K.; Kagamiyama, H. Strain is more important than electrostatic interaction in controlling the pKa of the catalytic group in aspartate aminotransferase. Biochemistry 2001, 40, 353–360. [Google Scholar] [CrossRef]
- Yano, T.; Mizuno, T.; Kagamiyama, H. A hydrogen-bonding network modulating enzyme function: Asparagine-194 and tyrosine-225 of Escherichia coli aspartate aminotransferase. Biochemistry 1993, 32, 1810–1815. [Google Scholar] [CrossRef]
- Cassimjee, K.E.; Humble, M.S.; Land, H.; Abedi, V.; Berglund, P. Chromobacterium violaceum ω–transaminase variant Trp60Cys shows increased specificity for (S)-1-phenylethylamine and 4’-substituted acetophenones, and follows Swain-Lupton parameterization. Org. Biomol. Chem. 2012, 10, 5466–5470. [Google Scholar] [CrossRef] [PubMed]
- Bezsudnova, E.Y.; Popov, V.O.; Boyko, K.M. Structural insight into the substrate specificity of PLP fold type IV transaminases. Appl. Microbiol. Biotechnol. 2020, 104, 2343–2357. [Google Scholar] [CrossRef] [PubMed]
- Available online: http://www.yasara.org/ (accessed on 1 October 2022).
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016, 44, W344–W350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: https://www.ncbi.nlm.nih.gov/tools/cobalt/ (accessed on 1 October 2022).
- Available online: https://espript.ibcp.fr/ESPript/cgi-bin/ESPript.cgi (accessed on 1 October 2022).
- The PyMOL Molecular Graphics System; Version 2.3.0; Schrödinger, LLC.: New York, NY, USA, 2019.
- Davies, M.T. A universal buffer solution for use in ultra-violet spectrophotometry. Analyst 1959, 84, 248–251. [Google Scholar] [CrossRef]
- Schätzle, S.; Höhne, M.; Redestad, E.; Robins, K.; Bornscheuer, U.T. Rapid and sensitive kinetic assay for characterization of ω–transaminases. Anal. Chem. 2009, 81, 8244–8248. [Google Scholar] [CrossRef]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Dissanayake, T.; Swails, J.M.; Harris, M.E.; Roitberg, A.E.; York, D.M. Interpretation of pH-activity profiles for acid-base catalysis from molecular simulations. Biochemistry 2015, 54, 1307–1313. [Google Scholar] [CrossRef] [Green Version]
- Regnault, M. Relationships between GDH and LDH activity in their respective NADH-dependent reactions in the cheliped muscle of the crab, Cancer Pagurus. Comp. Biochem. Physiol. B 1992, 101, 367–373. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTool4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiang, C.; Ao, Y.-F.; Höhne, M.; Bornscheuer, U.T. Shifting the pH Optima of (R)-Selective Transaminases by Protein Engineering. Int. J. Mol. Sci. 2022, 23, 15347. https://doi.org/10.3390/ijms232315347
Xiang C, Ao Y-F, Höhne M, Bornscheuer UT. Shifting the pH Optima of (R)-Selective Transaminases by Protein Engineering. International Journal of Molecular Sciences. 2022; 23(23):15347. https://doi.org/10.3390/ijms232315347
Chicago/Turabian StyleXiang, Chao, Yu-Fei Ao, Matthias Höhne, and Uwe T. Bornscheuer. 2022. "Shifting the pH Optima of (R)-Selective Transaminases by Protein Engineering" International Journal of Molecular Sciences 23, no. 23: 15347. https://doi.org/10.3390/ijms232315347
APA StyleXiang, C., Ao, Y.-F., Höhne, M., & Bornscheuer, U. T. (2022). Shifting the pH Optima of (R)-Selective Transaminases by Protein Engineering. International Journal of Molecular Sciences, 23(23), 15347. https://doi.org/10.3390/ijms232315347