
Guanidine Derivatives Containing the Chalcone Skeleton Are Potent Antiproliferative Compounds against Human Leukemia Cells
 , , and
, , and
Abstract
1. Introduction
2. Results
2.1. Synthesis
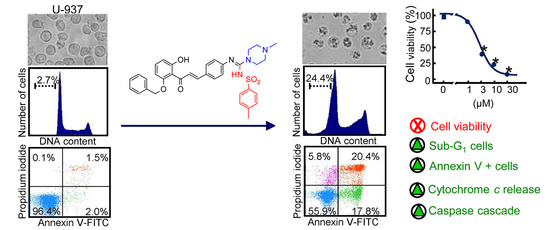
2.2. Hybrid Compounds Inhibit the Viability of Human Cancer Cells
2.3. Hybrid Compounds Induce Changes in Cell Cycle in Human U-937 Leukaemia Cells
2.4. Hybrid Compounds Induce Caspase Activation, PARP Cleavage, and Cytochrome c Release in Human U-937 Leukemia Cells
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. General Procedure for the Synthesis of Hybrid Molecules
4.2.1. Synthesis of Chalcone 3
4.2.2. General Method One-Pot Synthesis of Guanidines (6a–6k)
4.3. Cell Culture and Cytotoxicity Assays
4.4. Quantification of Sub-G1 Cells and Analysis of Cell Cycle and Apoptosis by Flow Cytometry
4.5. Assay of Caspase Activity
4.6. Western Blot Analysis
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, C.; Yu, J.; Fleck, B.A.; Hoare, S.R.; Saunders, J.; Foster, A.C. Phenylguanidines as selective nonpeptide melanocortin-5 receptor antagonists. J. Med. Chem. 2004, 47, 4083–4088. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Sood, A.K.; Goyal, K.; Singh, A.; Sharma, V.; Guliya, N.; Gulati, S.; Kumar, S. Chalcone Scaffolds as Anticancer Drugs: A Review on Molecular Insight in Action of Mechanisms and Anticancer Properties. Anticancer Agents Med. Chem. 2021, 21, 1650–1670. [Google Scholar] [CrossRef] [PubMed]
- Ashour, H.F.; Abou-Zeid, L.A.; El-Sayed, M.A.; Selim, K.B. 1,2,3-Triazole-Chalcone hybrids: Synthesis, in vitro cytotoxic activity and mechanistic investigation of apoptosis induction in multiple myeloma RPMI-8226. Eur. J. Med. Chem. 2020, 189, 112062. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, I.; Saavedra, E.; Del Rosario, H.; Perdomo, J.; Quintana, J.; Prencipe, F.; Oliva, P.; Romagnoli, R.; Estévez, F. Apoptosis pathways triggered by a potent antiproliferative hybrid chalcone on human melanoma cells. Int. J. Mol. Sci. 2021, 22, 13462. [Google Scholar] [CrossRef]
- Berlinck, R.G.S.; Bernardi, D.I.; Fill, T.; Fernandes, A.A.G.; Jurberg, I.D. The chemistry and biology of guanidine secondary metabolites. Nat. Prod. Rep. 2021, 38, 586–667. [Google Scholar] [CrossRef]
- Berlinck, R.G.; Burtoloso, A.C.; Kossuga, M.H. The chemistry and biology of organic guanidine derivatives. Nat. Prod. Rep. 2008, 25, 919–954. [Google Scholar] [CrossRef]
- Wijnen, R.; Pecoraro, C.; Carbone, D.; Fiuji, H.; Avan, A.; Peters, G.J.; Giovannetti, E.; Diana, P. Cyclin dependent kinase-1 (CDK-1) inhibition as a novel therapeutic strategy against pancreatic ductal adenocarcinoma (PDAC). Cancers 2021, 13, 4389. [Google Scholar] [CrossRef]
- Liu, W.; He, M.; Li, Y.; Peng, Z.; Wang, G. A review on synthetic chalcone derivatives as tubulin polymerisation inhibitors. J. Enzym. Inhibin. Med. Chem. 2022, 37, 9–38. [Google Scholar] [CrossRef]
- Curtin, N.J.; Szabo, C. Poly(ADP-ribose) polymerase inhibition: Past, present and future. Nat. Rev. Drug. Discov. 2020, 19, 711–736. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Kalkavan, H.; Green, D.R. MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 2018, 25, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Correia, C.; Lee, S.H.; Meng, X.W.; Vincelette, N.D.; Knorr, K.L.; Ding, H.; Nowakowski, G.S.; Dai, H.; Kaufmann, S.H. Emerging understanding of Bcl-2 biology: Implications for neoplastic progression and treatment. Biochim. Biophys. Acta 2015, 1853, 1658–1671. [Google Scholar] [CrossRef] [PubMed]
- Kaloni, D.; Diepstraten, S.T.; Strasser, A.; Kelly, G.L. BCL-2 protein family: Attractive targets for cancer therapy. Apoptosis 2022. [Google Scholar] [CrossRef] [PubMed]
- Ruvolo, P.; Deng, X.; May, W. Phosphorylation of Bcl2 and regulation of apoptosis. Leukemia 2001, 15, 515–522. [Google Scholar] [CrossRef]
- Dirsch, V.M.; Stuppner, H.; Vollmar, A.M. Helenalin triggers a CD95 death receptor-independent apoptosis that is not affected by overexpression of Bcl-x(L) or Bcl-2. Cancer Res. 2001, 61, 5817–5823. [Google Scholar]
- Yoo, S.H.; Yoon, Y.G.; Lee, J.S.; Song, Y.S.; Oh, J.S.; Park, B.S.; Kwon, T.K.; Park, C.; Choi, Y.H.; Yoo, Y.H. Etoposide induces a mixed type of programmed cell death and overcomes the resistance conferred by Bcl-2 in Hep3B hepatoma cells. Int. J. Oncol. 2012, 41, 1443–1454. [Google Scholar] [CrossRef]
- Smith, J.A.; Maloney, D.J.; Hecht, S.M.; Lannigan, D.A. Structural basis for the activity of the RSK-specific inhibitor, SL0101. Bioorg. Med. Chem. 2007, 15, 5018–5034. [Google Scholar] [CrossRef]
- Duval, R.; Kolb, S.; Braud, E.; Genest, D.; Garbay, C. Rapid discovery of triazolobenzylidene-thiazolopyrimidines (TBTP) as CDC25 phosphatase inhibitors by parallel click chemistry and in situ screening. J. Comb. Chem. 2009, 11, 947–950. [Google Scholar] [CrossRef]
- Saavedra, E.; Del Rosario, H.; Brouard, I.; Quintana, J.; Estévez, F. 6′-Benzyloxy-4-bromo-2′-hydroxychalcone is cytotoxic against human leukaemia cells and induces caspase-8- and reactive oxygen species-dependent apoptosis. Chem. Biol. Interact. 2019, 298, 137–145. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Estévez-Sarmiento, F.; Hernández, E.; Brouard, I.; León, F.; García, C.; Quintana, J.; Estévez, F. 3′-Hydroxy-3,4′-dimethoxyflavone-induced cell death in human leukaemia cells is dependent on caspases and reactive oxygen species and attenuated by the inhibition of JNK/SAPK. Chem. Biol. Interact. 2018, 288, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Estévez, S.; Marrero, M.T.; Quintana, J.; Estévez, F. Eupatorin-induced cell death in human leukemia cells is dependent on caspases and activates the mitogen-activated protein kinase pathway. PLoS ONE 2014, 9, e112536. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chalcone | Isocyanate | Amine | Product |
|---|---|---|---|
 |  |  |  |
 |  | ||
 |  | ||
 |  | ||
 |  | ||
 |  | ||
 |  | ||
 |  |  | |
 |  | ||
 |  | ||
 |  |
| IC50 (µM) | ||||||
|---|---|---|---|---|---|---|
| Compound | U-937 | HL-60 | MOLT-3 | NALM-6 | U-937/Bcl-2 | SK-MEL-1 |
| 3 | - | - | 7.4 ± 0.4 | 11.6 ± 1.6 | - | - |
| 6a | 11.4 ± 6.5 | 3.7 ± 1.6 | 2.9 ± 1.1 | 2.9 ± 0.5 | 2.9 ± 0.2 | - |
| 6b | 18.5 ± 9.6 | 20.5 ± 9.5 | 2.0 ± 0.7 | 27.0 ± 0.8 | 11.7 ± 7.0 | - |
| 6c | 9.5 ± 5.3 | 4.2 ± 0.5 | 2.5 ± 0.2 | 9.9 ± 3.1 | 4.2 ± 1.2 | - |
| 6d | - | 28.5 ± 14.3 | 3.3 ± 0.6 | 6.6 ± 1.2 | 24.6 ± 5.4 | - |
| 6e | 5.4 ± 2.1 | 3.8 ± 1.1 | 3.2 ± 1.5 | 3.1 ± 0.5 | 2.3 ± 0.9 | 14.3 ± 5.7 |
| 6f | 1.6 ± 0.6 | 1.6 ± 1.0 | 1.5 ± 0.0 | 2.7 ± 0.0 | 2.2 ± 0.8 | 4.5 ± 0.4 |
| 6g | - | - | - | - | - | - |
| 6h | 10.6 ± 2.6 | 5.2 ± 0.9 | 4.3 ± 0.5 | 5.4 ± 0.2 | 14.0 ± 6.6 | 5.2 ± 1.0 |
| 6i | 3.5 ± 1.1 | 3.2 ± 0.6 | 7.1 ± 1.3 | 10.8 ± 0.5 | 5.1 ± 1.8 | 13.8 ± 1.2 |
| 6j | 8.6 ± 1.1 | 6.2 ± 1.7 | 1.8 ± 0.5 | 4.1 ± 0.5 | 11.7 ± 3.4 | 5.4 ± 0.8 |
| 6k | 14.0 ± 1.1 | 11.4 ± 5.5 | 21.7 ± 6.5 | 24.8 ± 3.3 | 14.0 ± 5.1 | - |
| Etoposide | 1.8 ± 0.2 | 0.6 ± 0.2 | 0.2 ± 0.1 | ND | ND | 9.0 ± 3.0 |
| Doxorubicin | 0.1 ± 0.0 | ND | ND | ND | ND | 0.3 ± 0.1 |
| %Sub-G1 | %G1 | %S | %G2-M | |
|---|---|---|---|---|
| Control | 2.4 ± 0.4 | 54.4 ± 0.4 | 29.0 ± 0.7 | 14.2 ± 0.2 |
| 6c | 23.5 ± 1.2 * | 37.4 ± 0.3 * | 27.0 ± 0.7 | 11.4 ± 0.4 |
| 6e | 11.9 ± 0.2 * | 36.5 ± 2.3 * | 23.6 ± 0.5 * | 27.3 ± 2.0 * |
| 6f | 25.1 ± 2.6 * | 35.2 ± 3.8 * | 28.3 ± 0.2 | 10.8 ± 1.6 |
| 6h | 7.2 ± 1.1 * | 53.7 ± 0.4 | 29.5 ± 0.3 | 9.2 ± 0.5 * |
| 6i | 31.6 ± 4.5 * | 35.9 ± 2.1 * | 23.6 ± 2.4 * | 8.4 ± 0.1 * |
| 6j | 4.7 ± 0.6 | 51.3 ± 1.1 | 32.0 ± 0.4 | 11.5 ± 0.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Estévez-Sarmiento, F.; Saavedra, E.; Brouard, I.; Peyrac, J.; Hernández-Garcés, J.; García, C.; Quintana, J.; Estévez, F. Guanidine Derivatives Containing the Chalcone Skeleton Are Potent Antiproliferative Compounds against Human Leukemia Cells. Int. J. Mol. Sci. 2022, 23, 15518. https://doi.org/10.3390/ijms232415518
Estévez-Sarmiento F, Saavedra E, Brouard I, Peyrac J, Hernández-Garcés J, García C, Quintana J, Estévez F. Guanidine Derivatives Containing the Chalcone Skeleton Are Potent Antiproliferative Compounds against Human Leukemia Cells. International Journal of Molecular Sciences. 2022; 23(24):15518. https://doi.org/10.3390/ijms232415518
Chicago/Turabian StyleEstévez-Sarmiento, Francisco, Ester Saavedra, Ignacio Brouard, Jesús Peyrac, Judith Hernández-Garcés, Celina García, José Quintana, and Francisco Estévez. 2022. "Guanidine Derivatives Containing the Chalcone Skeleton Are Potent Antiproliferative Compounds against Human Leukemia Cells" International Journal of Molecular Sciences 23, no. 24: 15518. https://doi.org/10.3390/ijms232415518
APA StyleEstévez-Sarmiento, F., Saavedra, E., Brouard, I., Peyrac, J., Hernández-Garcés, J., García, C., Quintana, J., & Estévez, F. (2022). Guanidine Derivatives Containing the Chalcone Skeleton Are Potent Antiproliferative Compounds against Human Leukemia Cells. International Journal of Molecular Sciences, 23(24), 15518. https://doi.org/10.3390/ijms232415518