
DAMPs Released from Proinflammatory Macrophages Induce Inflammation in Cardiomyocytes via Activation of TLR4 and TNFR
 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. LPS-Stimulated Macrophages Induce a Strong Cytokine Response in Cardiomyocytes
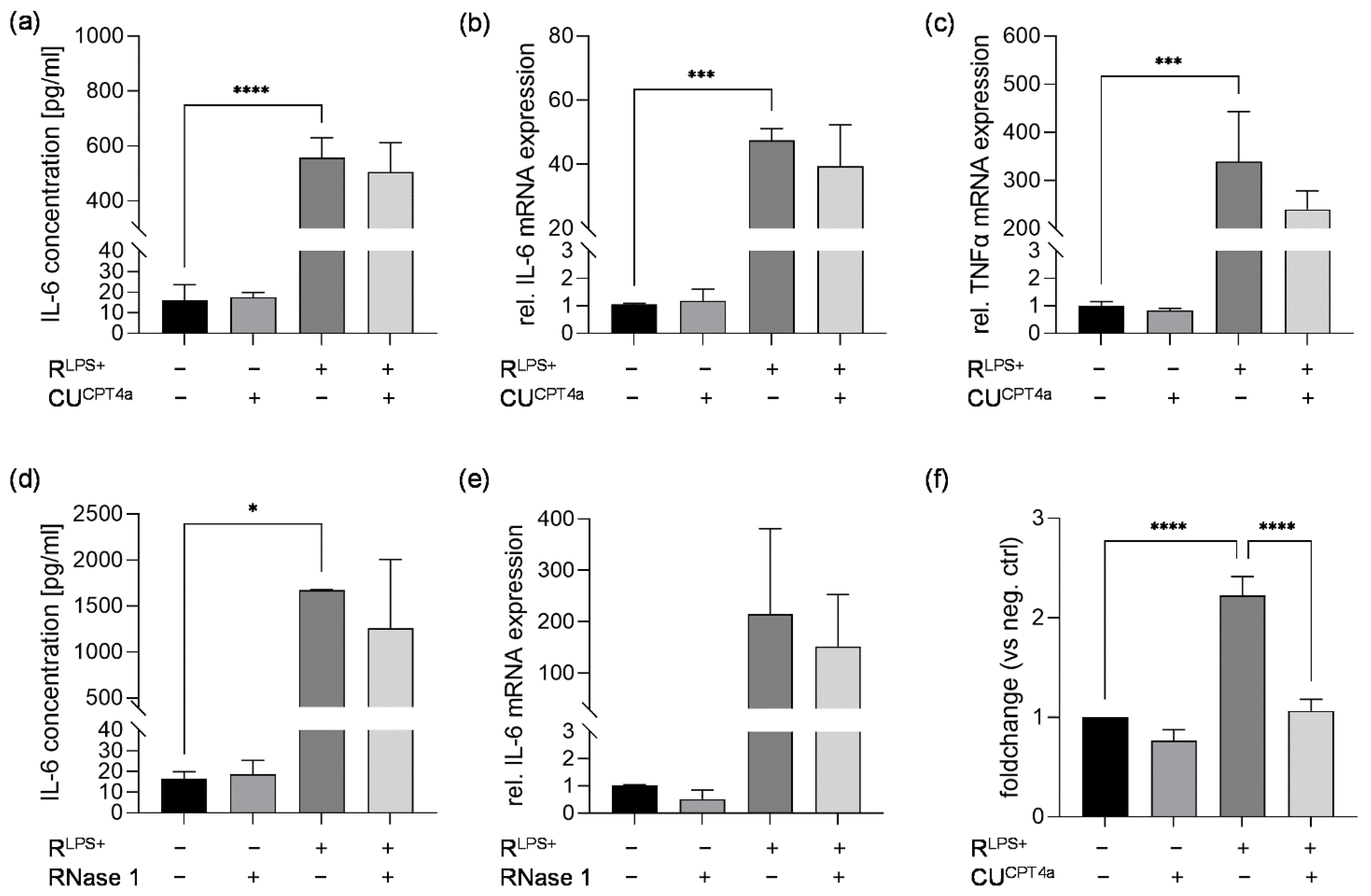
2.2. TLR3 Inhibition Reduces Cardiac Caspase-3/7 Activation without Attenuating Cardiac Inflammation and Cardiac Apoptosis Induced by LPS-Stimulated Macrophages
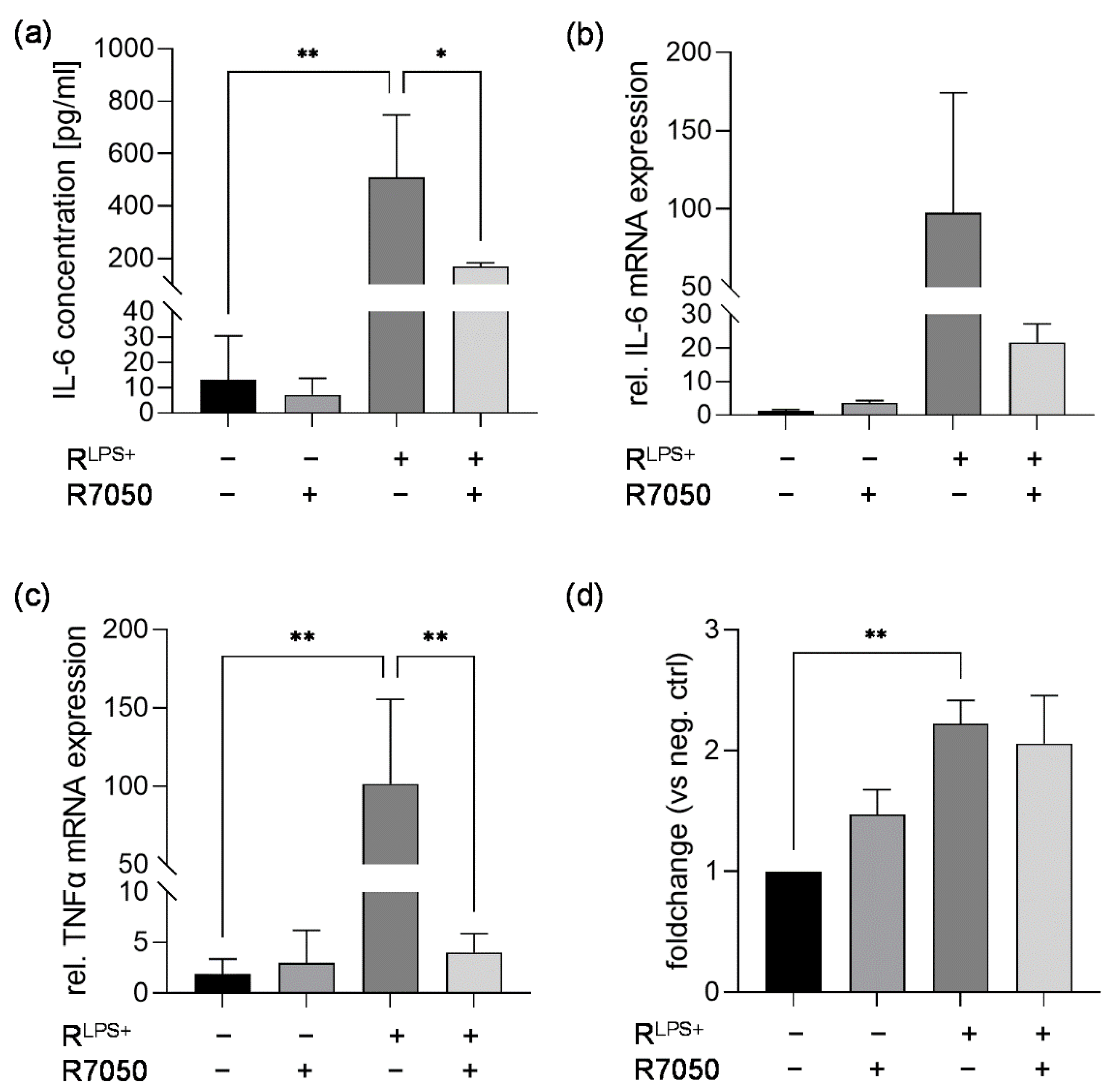
2.3. TLR4 Inhibition Reduces Cardiac Inflammation Induced by LPS-Stimulated Macrophages
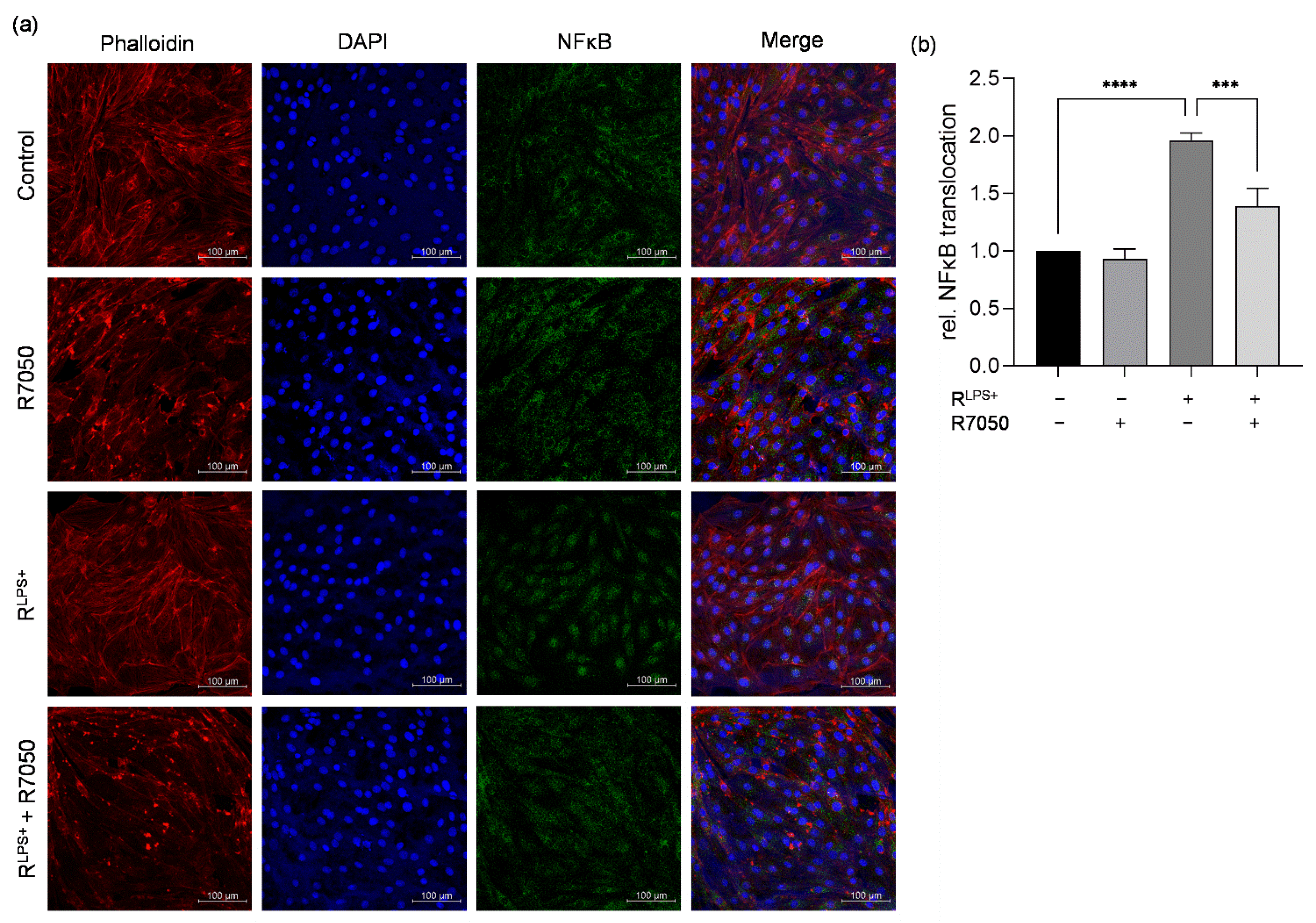
2.4. TNFR Inhibition Attenuates Cardiac Inflammation and NFκB Translocation Induced by LPS-Stimulated Macrophages
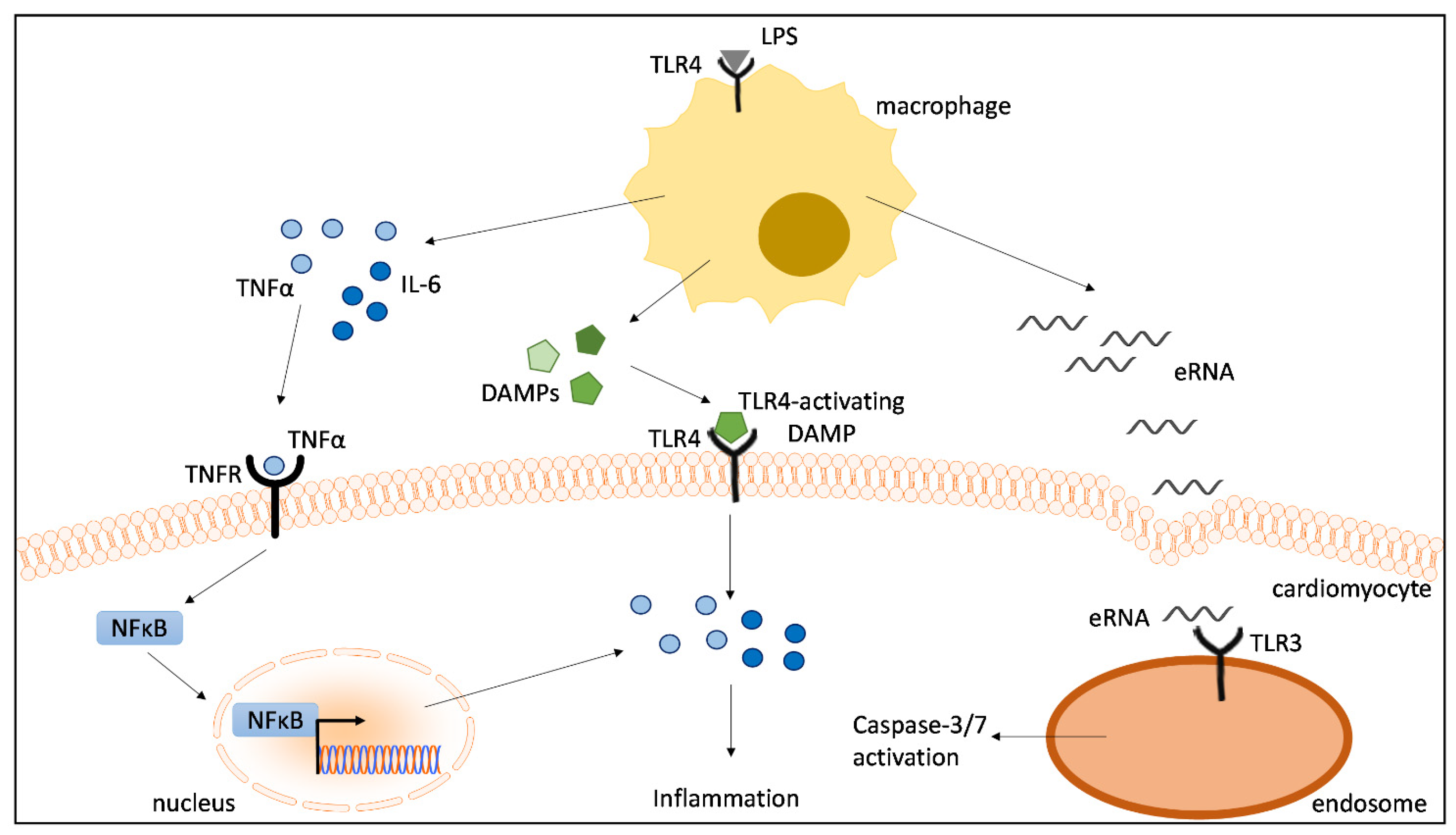
3. Discussion
4. Materials and Methods
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.-D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Geri, G.; Vignon, P.; Aubry, A.; Fedou, A.-L.; Charron, C.; Silva, S.; Repessé, X.; Vieillard-Baron, A. Cardiovascular clusters in septic shock combining clinical and echocardiographic parameters: A post hoc analysis. Intensive Care Med. 2019, 45, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.; Derwall, M.; Al Zoubi, S.; Zechendorf, E.; Reuter, D.A.; Thiemermann, C.; Schuerholz, T. The Septic Heart: Current Understanding of Molecular Mechanisms and Clinical Implications. Chest 2019, 155, 427–437. [Google Scholar] [CrossRef]
- Gabarin, R.S.; Li, M.; Zimmel, P.A.; Marshall, J.C.; Li, Y.; Zhang, H. Intracellular and Extracellular Lipopolysaccharide Signaling in Sepsis: Avenues for Novel Therapeutic Strategies. J. Innate Immun. 2021, 13, 323–332. [Google Scholar] [CrossRef]
- Luan, Y.Y.; Dong, N.; Xie, M.; Xiao, X.Z.; Yao, Y.M. The significance and regulatory mechanisms of innate immune cells in the development of sepsis. J. Interf. Cytokine Res. 2014, 34, 2–15. [Google Scholar] [CrossRef] [Green Version]
- Hulsmans, M.; Sam, F.; Nahrendorf, M. Monocyte and macrophage contributions to cardiac remodeling. J. Mol. Cell. Cardiol. 2016, 93, 149–155. [Google Scholar] [CrossRef] [Green Version]
- Gomez, I.; Duval, V.; Silvestre, J.S. Cardiomyocytes and Macrophages Discourse on the Method to Govern Cardiac Repair. Front. Cardiovasc. Med. 2018, 5, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavine, K.J.; Pinto, A.R.; Epelman, S.; Kopecky, B.J.; Clemente-Casares, X.; Godwin, J.; Rosenthal, N.; Kovacic, J.C. The Macrophage in Cardiac Homeostasis and Disease: JACC Macrophage in CVD Series (Part 4). J. Am. Coll. Cardiol. 2018, 72, 2213–2230. [Google Scholar] [CrossRef] [PubMed]
- Lafuse, W.P.; Wozniak, D.J.; Rajaram, M.V.S. Role of Cardiac Macrophages on Cardiac Inflammation, Fibrosis and Tissue Repair. Cells 2020, 10, 51. [Google Scholar] [CrossRef]
- Drosatos, K.; Lymperopoulos, A.; Kennel, P.J.; Pollak, N.; Schulze, P.C.; Goldberg, I.J. Pathophysiology of sepsis-related cardiac dysfunction: Driven by inflammation, energy mismanagement, or both? Curr. Heart Fail. Rep. 2015, 12, 130–140. [Google Scholar] [CrossRef]
- Habimana, R.; Choi, I.; Cho, H.J.; Kim, D.; Lee, K.; Jeong, I. Sepsis-induced cardiac dysfunction: A review of pathophysiology. Acute Crit. Care 2020, 35, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Saraf, A.; Rampoldi, A.; Chao, M.; Li, D.; Armand, L.; Hwang, H.; Liu, R.; Jha, R.; Fu, H.; Maxwell, J.T.; et al. Functional and molecular effects of TNF-α on human iPSC-derived cardiomyocytes. Stem Cell Res. 2021, 52, 102218. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Feng, Y.; Zou, L.; Wang, L.; Chen, H.H.; Cai, J.Y.; Xu, J.M.; Sosnovik, D.E.; Chao, W. Role of extracellular RNA and TLR3-Trif signaling in myocardial ischemia-reperfusion injury. J. Am. Heart Assoc. 2014, 3, e000683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koczera, P.; Martin, L.; Marx, G.; Schuerholz, T. The Ribonuclease A Superfamily in Humans: Canonical RNases as the Buttress of Innate Immunity. Int. J. Mol. Sci. 2016, 17, 1278. [Google Scholar] [CrossRef] [Green Version]
- Krämer, T.J.; Hübener, P.; Pöttker, B.; Gölz, C.; Neulen, A.; Pantel, T.; Goetz, H.; Ritter, K.; Schäfer, M.K.E.; Thal, S.C. Ribonuclease-1 treatment after traumatic brain injury preserves blood–brain barrier integrity and delays secondary brain damage in mice. Sci. Rep. 2022, 12, 5731. [Google Scholar] [CrossRef] [PubMed]
- Zechendorf, E.; O’Riordan, C.E.; Stiehler, L.; Wischmeyer, N.; Chiazza, F.; Collotta, D.; Denecke, B.; Ernst, S.; Müller-Newen, G.; Coldewey, S.M.; et al. Ribonuclease 1 attenuates septic cardiomyopathy and cardiac apoptosis in a murine model of polymicrobial sepsis. JCI Insight 2021, 5, e131571. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhao, M.; Liu, S.; Guo, J.; Lu, Y.; Cheng, J.; Liu, J. Macrophage-derived extracellular vesicles: Diverse mediators of pathology and therapeutics in multiple diseases. Cell Death Dis. 2020, 11, 924. [Google Scholar] [CrossRef]
- Chen, B.; Luo, L.; Wei, X.; Gong, D.; Li, Z.; Li, S.; Tang, W.; Jin, L. M1 Bone Marrow-Derived Macrophage-Derived Extracellular Vesicles Inhibit Angiogenesis and Myocardial Regeneration Following Myocardial Infarction via the MALAT1/MicroRNA-25-3p/CDC42 Axis. Oxid. Med. Cell. Longev. 2021, 2021, 9959746. [Google Scholar] [CrossRef]
- Zheng, S.; Gong, M.; Chen, J. Extracellular vesicles enriched with miR-150 released by macrophages regulates the TP53-IGF-1 axis to alleviate myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H969–H979. [Google Scholar] [CrossRef]
- Gandham, S.; Su, X.; Wood, J.; Nocera, A.L.; Alli, S.C.; Milane, L.; Zimmerman, A.; Amiji, M.; Ivanov, A.R. Technologies and Standardization in Research on Extracellular Vesicles. Trends Biotechnol. 2020, 38, 1066–1098. [Google Scholar] [CrossRef]
- Burgelman, M.; Vandendriessche, C.; Vandenbroucke, R.E. Extracellular Vesicles: A Double-Edged Sword in Sepsis. Pharmaceuticals 2021, 14, 829. [Google Scholar] [CrossRef] [PubMed]
- Essandoh, K.; Yang, L.; Wang, X.; Huang, W.; Qin, D.; Hao, J.; Wang, Y.; Zingarelli, B.; Peng, T.; Fan, G.-C. Blockade of exosome generation with GW4869 dampens the sepsis-induced inflammation and cardiac dysfunction. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2015, 1852, 2362–2371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Feng, Y.; Jeyaram, A.; Jay, S.M.; Zou, L.; Chao, W. Circulating Plasma Extracellular Vesicles from Septic Mice Induce Inflammation via MicroRNA- and TLR7-Dependent Mechanisms. J. Immunol. 2018, 201, 3392–3400. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Lai, J.; Yang, L.; Ruan, G.; Chaugai, S.; Ning, Q.; Chen, C.; Wang, D.W. Trimetazidine prevents macrophage-mediated septic myocardial dysfunction via activation of the histone deacetylase sirtuin 1. Br. J. Pharmacol. 2016, 173, 545–561. [Google Scholar] [CrossRef] [Green Version]
- Ruan, W.; Ji, X.; Qin, Y.; Zhang, X.; Wan, X.; Zhu, C.; Lv, C.; Hu, C.; Zhou, J.; Lu, L.; et al. Harmine Alleviated Sepsis-Induced Cardiac Dysfunction by Modulating Macrophage Polarization via the STAT/MAPK/NF-κB Pathway. Front. Cell Dev. Biol. 2021, 9, 792257. [Google Scholar] [CrossRef]
- Jia, L.; Wang, Y.; Wang, Y.; Ma, Y.; Shen, J.; Fu, Z.; Wu, Y.; Su, S.; Zhang, Y.; Cai, Z.; et al. Heme Oxygenase-1 in Macrophages Drives Septic Cardiac Dysfunction via Suppressing Lysosomal Degradation of Inducible Nitric Oxide Synthase. Circ. Res. 2018, 122, 1532–1544. [Google Scholar] [CrossRef]
- Uhlén, M.; Karlsson, M.J.; Hober, A.; Svensson, A.-S.; Scheffel, J.; Kotol, D.; Zhong, W.; Tebani, A.; Strandberg, L.; Edfors, F.; et al. The human secretome. Sci. Signal. 2019, 12, eaaz0274. [Google Scholar] [CrossRef] [Green Version]
- Biemmi, V.; Milano, G.; Ciullo, A.; Cervio, E.; Burrello, J.; Dei Cas, M.; Paroni, R.; Tallone, T.; Moccetti, T.; Pedrazzini, G.; et al. Inflammatory extracellular vesicles prompt heart dysfunction via TRL4-dependent NF-κB activation. Theranostics 2020, 10, 2773–2790. [Google Scholar] [CrossRef]
- Arango Duque, G.; Descoteaux, A. Macrophage cytokines: Involvement in immunity and infectious diseases. Front. Immunol. 2014, 5, 491. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Lin, L.; Zhang, Z.; Zhang, H.; Hu, H. Targeting NF-κB pathway for the therapy of diseases: Mechanism and clinical study. Signal Transduct. Target. Ther. 2020, 5, 209. [Google Scholar] [CrossRef]
- Kakihana, Y.; Ito, T.; Nakahara, M.; Yamaguchi, K.; Yasuda, T. Sepsis-induced myocardial dysfunction: Pathophysiology and management. J. Intensive Care 2016, 4, 22. [Google Scholar] [CrossRef] [Green Version]
- Sato, R.; Kuriyama, A.; Takada, T.; Nasu, M.; Luthe, S.K. Prevalence and risk factors of sepsis-induced cardiomyopathy: A retrospective cohort study. Medicine 2016, 95, e5031. [Google Scholar] [CrossRef] [PubMed]
- Salari, H.; Walker, M.J. Cardiac dysfunction caused by factors released from endotoxin-activated macrophages. Circ. Shock 1989, 27, 263–272. [Google Scholar]
- Yang, D.; Jiang, Y.; Qian, H.; Liu, X.; Mi, L. Silencing Cardiac Troponin I-Interacting Kinase Reduces Lipopolysaccharide-Induced Sepsis-Induced Myocardial Dysfunction in Rat by Regulating Apoptosis-Related Proteins. BioMed Res. Int. 2021, 2021, 5520051. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Y.; Zhang, Y.Q. Isoflurane reduces endotoxin-induced oxidative, inflammatory, and apoptotic responses in H9c2 cardiomyocytes. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 3976–3987. [Google Scholar] [PubMed]
- Katare, P.B.; Nizami, H.L.; Paramesha, B.; Dinda, A.K.; Banerjee, S.K. Activation of toll like receptor 4 (TLR4) promotes cardiomyocyte apoptosis through SIRT2 dependent p53 deacetylation. Sci. Rep. 2020, 10, 19232. [Google Scholar] [CrossRef] [PubMed]
- Van Deventer, S.H.; Ten Gate, J.; Buller, H.; Sturk, A.; Pauw, W. Endotoxaemia: An early predictor of septicaemia in febrile patients. Lancet 1988, 331, 605–609. [Google Scholar] [CrossRef]
- Shenep, J.L.; Flynn, P.M.; Barrett, F.F.; Stidham, G.L.; Westenkirchner, D.F. Serial Quantitation of Endotoxemia and Bacteremia during Therapy for Gram-Negative Bacterial Sepsis. J. Infect. Dis. 1988, 157, 565–568. [Google Scholar] [CrossRef]
- Cai, J.; Wen, J.; Bauer, E.; Zhong, H.; Yuan, H.; Chen, A.F. The Role of HMGB1 in Cardiovascular Biology: Danger Signals. Antioxid. Redox Signal. 2015, 23, 1351–1369. [Google Scholar] [CrossRef]
- Li, L.; Lu, Y.-Q. The Regulatory Role of High-Mobility Group Protein 1 in Sepsis-Related Immunity. Front. Immunol. 2021, 11, 601815. [Google Scholar] [CrossRef]
- Wang, G.; Jin, S.; Huang, W.; Li, Y.; Wang, J.; Ling, X.; Huang, Y.; Hu, Y.; Li, C.; Meng, Y.; et al. LPS-induced macrophage HMGB1-loaded extracellular vesicles trigger hepatocyte pyroptosis by activating the NLRP3 inflammasome. Cell Death Discov. 2021, 7, 337. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wan, D.; Luo, X.; Song, T.; Wang, Y.; Yu, Q.; Jiang, L.; Liao, R.; Zhao, W.; Su, B. Circulating Histones in Sepsis: Potential Outcome Predictors and Therapeutic Targets. Front. Immunol. 2021, 12, 650184. [Google Scholar] [CrossRef] [PubMed]
- Nair, R.R.; Mazza, D.; Brambilla, F.; Gorzanelli, A.; Agresti, A.; Bianchi, M.E. LPS-Challenged Macrophages Release Microvesicles Coated With Histones. Front. Immunol. 2018, 9, 1463. [Google Scholar] [CrossRef]
- Shah, M.; He, Z.; Rauf, A.; Beikoghli Kalkhoran, S.; Heiestad, C.M.; Stensløkken, K.-O.; Parish, C.R.; Soehnlein, O.; Arjun, S.; Davidson, S.M.; et al. Extracellular histones are a target in myocardial ischaemia–reperfusion injury. Cardiovasc. Res. 2021, 118, 1115–1125. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Liang, Y.; Shao, F.; Wang, X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3–mediated pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 20054–20059. [Google Scholar] [CrossRef] [Green Version]
- Virca, G.D.; Kim, S.Y.; Glaser, K.B.; Ulevitch, R.J. Lipopolysaccharide Induces Hyporesponsiveness to Its Own Action in RAW 264.7 Cells*. J. Biol. Chem. 1989, 264, 21951–21956. [Google Scholar] [CrossRef] [PubMed]
- Rolski, F.; Błyszczuk, P. Complexity of TNF-α Signaling in Heart Disease. J. Clin. Med. 2020, 9, 3267. [Google Scholar] [CrossRef]
- Moe, K.T.; Khairunnisa, K.; Yin, N.O.; Chin-Dusting, J.; Wong, P.; Wong, M.C. Tumor necrosis factor-α-induced nuclear factor-kappaB activation in human cardiomyocytes is mediated by NADPH oxidase. J. Physiol. Biochem. 2014, 70, 769–779. [Google Scholar] [CrossRef]
- Linkermann, A.; Green, D.R. Necroptosis. N. Engl. J. Med. 2014, 370, 455–465. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Kos, R.; Garssen, J.; Redegeld, F. Molecular Insights into the Mechanism of Necroptosis: The Necrosome as a Potential Therapeutic Target. Cells 2019, 8, 1486. [Google Scholar] [CrossRef] [Green Version]
- Bertheloot, D.; Latz, E.; Franklin, B.S. Necroptosis, pyroptosis and apoptosis: An intricate game of cell death. Cell. Mol. Immunol. 2021, 18, 1106–1121. [Google Scholar] [CrossRef] [PubMed]
- Branco, A.F.; Pereira, S.P.; Gonzalez, S.; Gusev, O.; Rizvanov, A.A.; Oliveira, P.J. Gene Expression Profiling of H9c2 Myoblast Differentiation towards a Cardiac-Like Phenotype. PLoS ONE 2015, 10, e0129303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zechendorf, E.; Vaßen, P.; Zhang, J.; Hallawa, A.; Martincuks, A.; Krenkel, O.; Müller-Newen, G.; Schuerholz, T.; Simon, T.P.; Marx, G.; et al. Heparan Sulfate Induces Necroptosis in Murine Cardiomyocytes: A Medical-In silico Approach Combining In vitro Experiments and Machine Learning. Front. Immunol. 2018, 9, 393. [Google Scholar] [CrossRef]
- Borosch, S.; Dahmen, E.; Beckers, C.; Stoppe, C.; Buhl, E.M.; Denecke, B.; Goetzenich, A.; Kraemer, S. Characterization of extracellular vesicles derived from cardiac cells in an in vitro model of preconditioning. J. Extracell. Vesicles 2017, 6, 1390391. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neu, C.; Thiele, Y.; Horr, F.; Beckers, C.; Frank, N.; Marx, G.; Martin, L.; Kraemer, S.; Zechendorf, E. DAMPs Released from Proinflammatory Macrophages Induce Inflammation in Cardiomyocytes via Activation of TLR4 and TNFR. Int. J. Mol. Sci. 2022, 23, 15522. https://doi.org/10.3390/ijms232415522
Neu C, Thiele Y, Horr F, Beckers C, Frank N, Marx G, Martin L, Kraemer S, Zechendorf E. DAMPs Released from Proinflammatory Macrophages Induce Inflammation in Cardiomyocytes via Activation of TLR4 and TNFR. International Journal of Molecular Sciences. 2022; 23(24):15522. https://doi.org/10.3390/ijms232415522
Chicago/Turabian StyleNeu, Carolina, Yvonne Thiele, Fabienne Horr, Christian Beckers, Nadine Frank, Gernot Marx, Lukas Martin, Sandra Kraemer, and Elisabeth Zechendorf. 2022. "DAMPs Released from Proinflammatory Macrophages Induce Inflammation in Cardiomyocytes via Activation of TLR4 and TNFR" International Journal of Molecular Sciences 23, no. 24: 15522. https://doi.org/10.3390/ijms232415522