
Genetic Deletion of AT1a Receptor or Na+/H+ Exchanger 3 Selectively in the Proximal Tubules of the Kidney Attenuates Two-Kidney, One-Clip Goldblatt Hypertension in Mice

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Basal Physiological Phenotypes and Their Responses to the Development of 2K1C Goldblatt Hypertension in All Strains of Mice
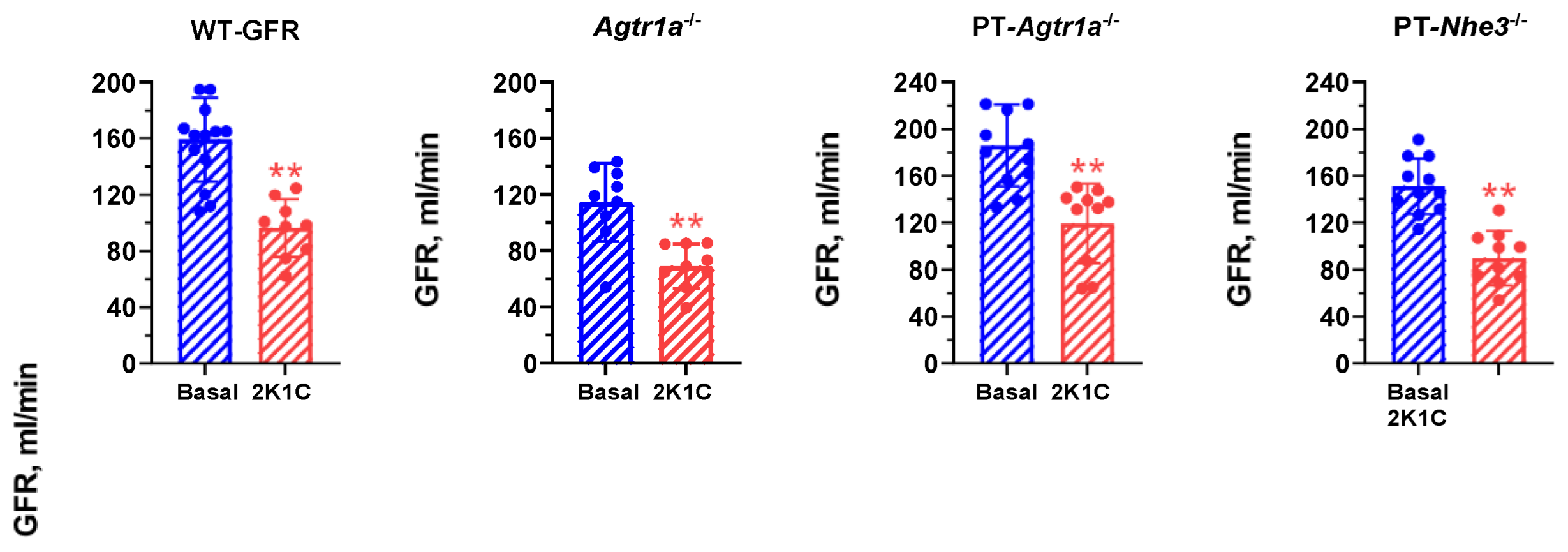
2.2. Effects of Induction of 2K1C Goldblatt Hypertension on Whole-Kidney Glomerular Filtration Rate in All Mouse Models
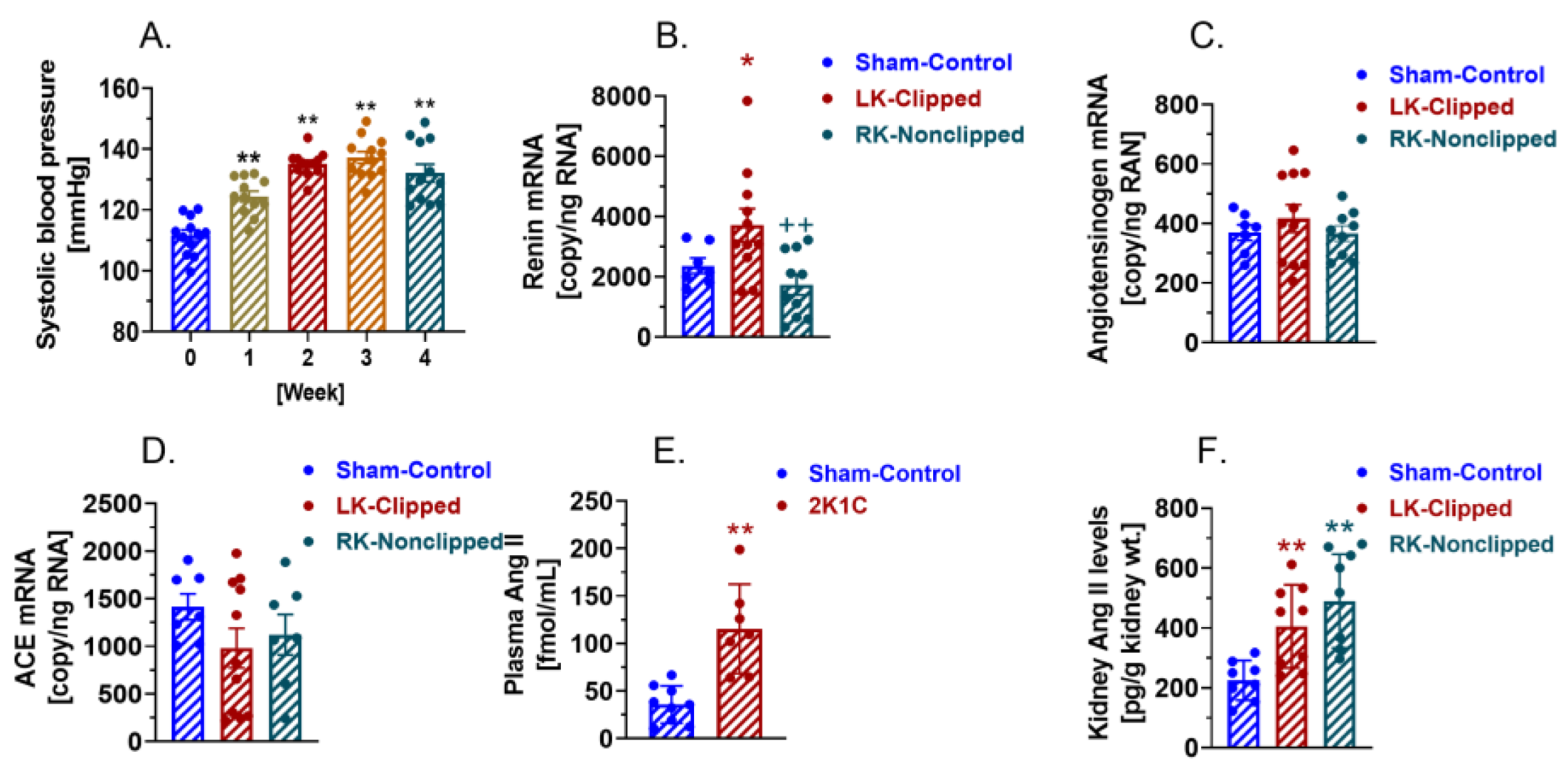
2.3. AT1 (AT1a) Receptor-Mediated, Ang II-Dependent Development of 2K1C Goldblatt Hypertension in WT Mice
2.4. Activation of Circulating and Intratubular RAS in WT Mice with 2K1C Goldblatt Hypertension
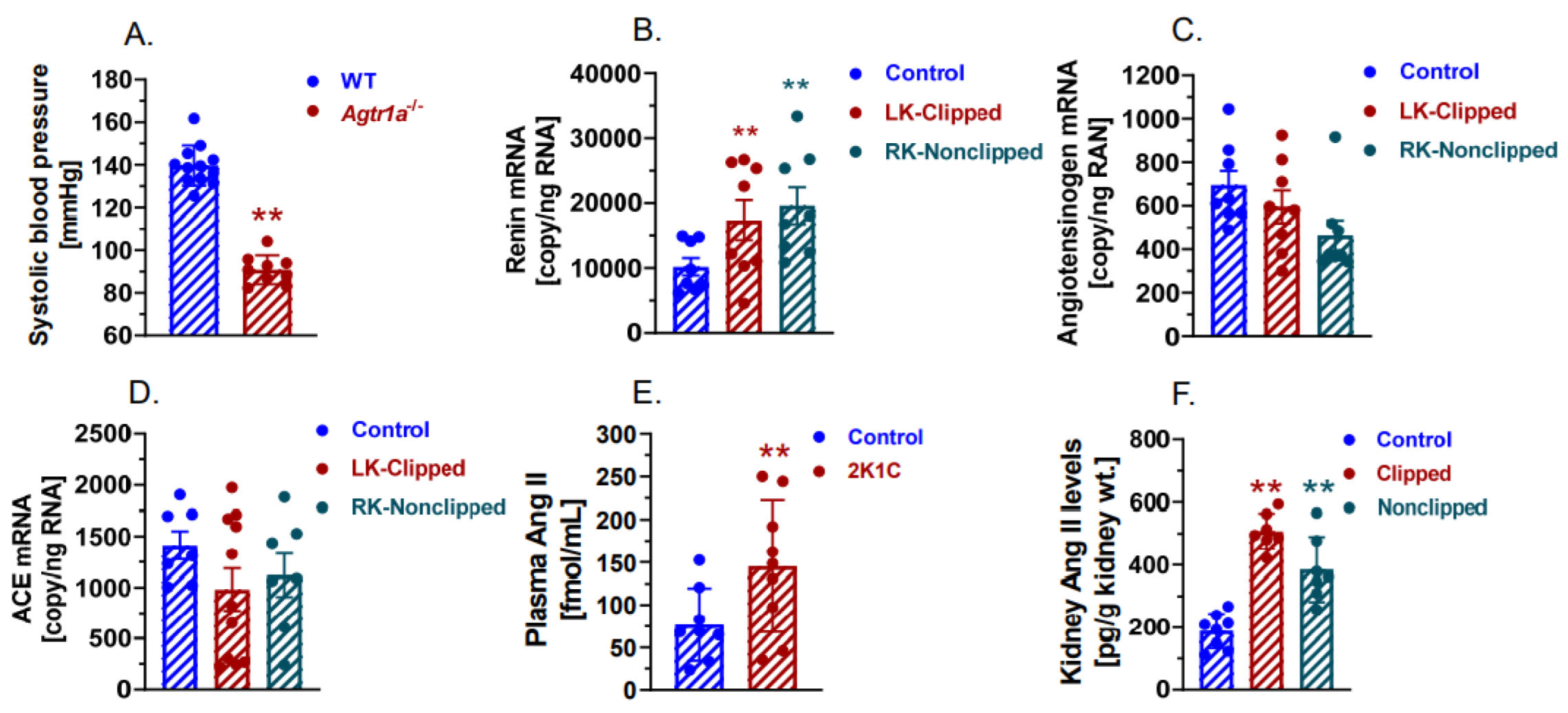
2.5. The Development of 2K1C Goldblatt Hypertension Was Completely Blocked in Whole-Body Agtr1a−/− Mice
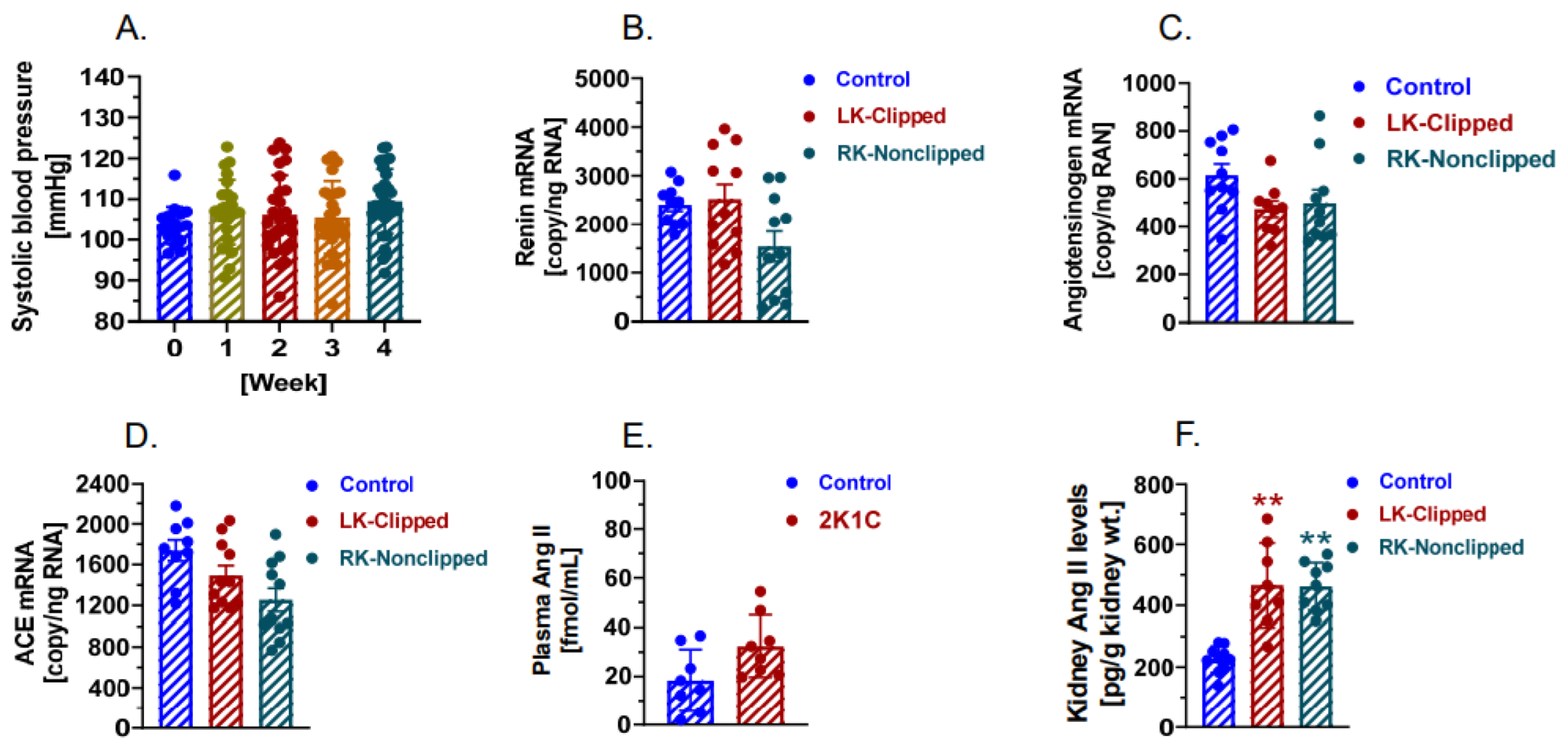
2.6. The Development of 2K1C Goldblatt Hypertension Was Significantly Attenuated in Proximal Tubule-Specific PT-Agtr1a−/− Mice
2.7. The Development of 2K1C Goldblatt Hypertension Was Significantly Attenuated in Proximal Tubule-Specific PT-Nhe3−/− Mice
2.8. Deletion of AT1 (AT1a) Receptors or NHE3 Selectively in the Proximal Tubules Promotes the Natriuretic Response in PT-Agtr1a−/− or PT-Nhe3−/− Mice during the Development of 2K1C Goldblatt Hypertension
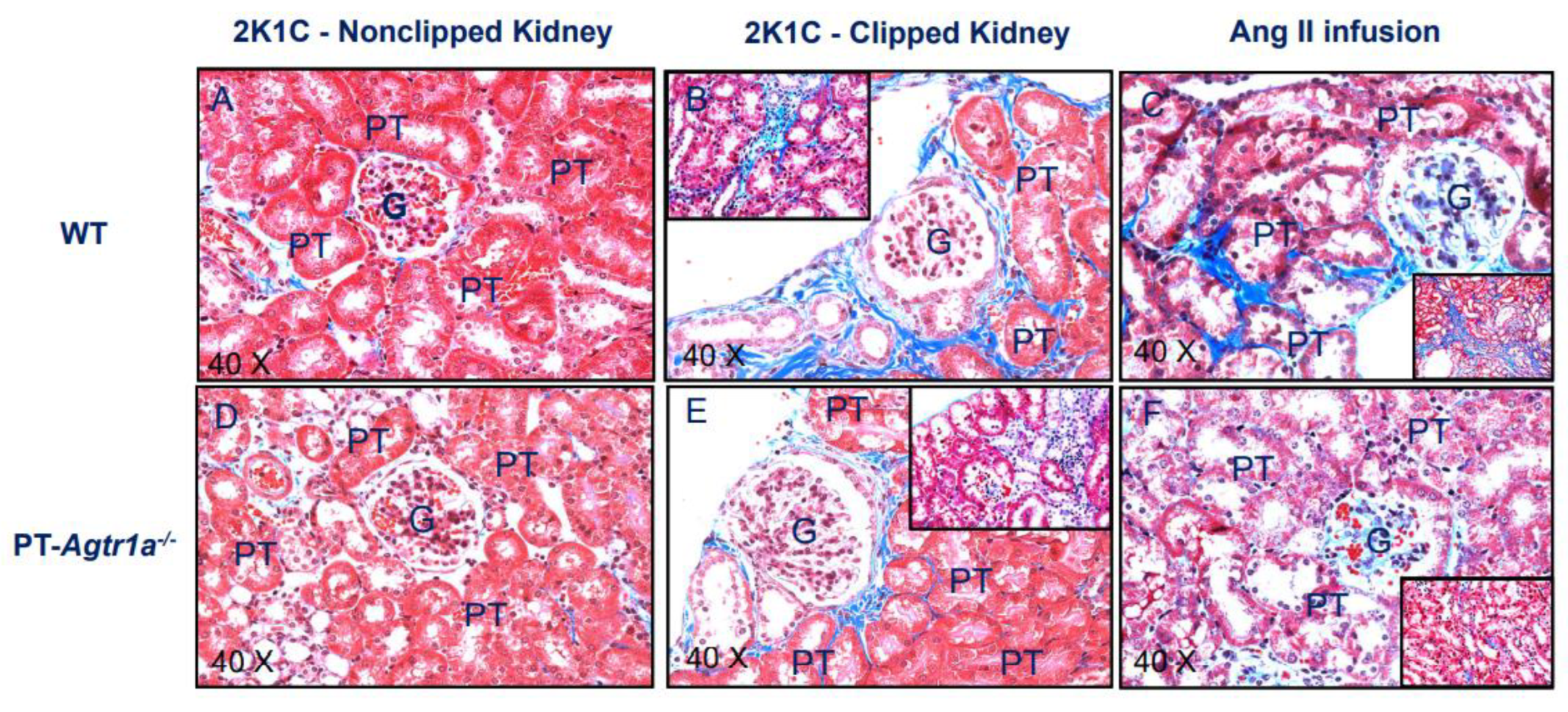
2.9. Deletion of AT1 (AT1a) Receptors Selectively in the Proximal Tubules Attenuates Tubulointerstitial Injury in PT-Agtr1a−/− Mice during the Development of 2K1C Goldblatt Hypertension
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Induction of 2K1C Goldblatt Hypertension in WT, Global Agtr1a−/−, PT-Agtr1a−/− and PT-Nhe3−/− Mice
4.3. Induction of Ang II-Induced Hypertension in WT, Global Agtr1a−/−, PT-Agtr1a−/− and PT-Nhe3−/− Mice
4.4. Implantation of Telemetry Probes for Blood Pressure Measurement in Wild-Type, Global Agtr1a−/−, PT-Agtr1a−/−, and PT-Nhe3−/− Mice
4.5. Measurement of Whole-Kidney Glomerular Filtration Rate (GFR) Using Fluorescein Labeled FITC-Sinistrin in WT, Global Agtr1a−/−, and PT-Agtr1a−/− Mice
4.6. Glomerular and Tubulointerstitial Histology in Wild-Type and Proximal Tubule-Specific PT-Agtr1a−/− Mice
4.7. Digital Droplet PCR Analysis of the Expression of Renin, Angiotensinogen, and Angiotensin-Converting Enzyme mRNAs in the Clipped and Nonclipped Kidneys of WT, Global Agtr1a−/−, and PT-Agtr1a−/− Mice
4.8. Measurement of Plasma and Kidney Ang II Levels in Wild-Type, Global Agtr1a−/−, and PT-Agtr1a−/− Mice
4.9. Measurement of Blood Volume, Hematocrit and 24 h Urinary Na+, K+, and Cl− Excretion
4.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 2K1C | two-kidney: one-clip (2K1C) Goldblatt hypertension |
| ACE | angiotensin-converting enzyme |
| Agtr1a−/− | mutant mice with whole-body deletion of the angiotensin II AT1a receptor |
| Ang I | angiotensin I |
| Ang II | angiotensin II |
| AT1 (AT1a) | type 1 (1a) receptor for Ang II |
| GFR | glomerular filtration rate |
| iL-Sglt2-Cre | mutant mice with tissue-specific expression of Cre recombinase selectively in the proximal tubules of the kidney driven by the sodium and glucose cotransporter 2 (Sglt2) promoter |
| NHE3 | the Na+/H+ exchanger 3 |
| PT-Agtr1a−/− | mutant mice with genetic deletion of AT1 (AT1a) receptors selectively in the proximal tubules of the kidney |
| PT-Nhe3−/− | mutant mice with genetic deletion of the Na+/H+ exchanger 3 (NHE3) selectively in the proximal tubules of the kidney |
| RAS | the renin-angiotensin system |
| WT | wild-type mice |
References
- Harrison, D.G.; Coffman, T.M.; Wilcox, C.S. Pathophysiology of Hypertension: The Mosaic Theory and Beyond. Circ. Res. 2021, 128, 847–863. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, S.; Caulfield, M.; Dominiczak, A.F. Genetic and Molecular Aspects of Hypertension. Circ. Res. 2015, 116, 937–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carey, R.M.; Calhoun, D.A.; Bakris, G.L.; Brook, R.D.; Daugherty, S.L.; Dennison-Himmelfarb, C.R.; Egan, B.M.; Flack, J.M.; Gidding, S.S.; Judd, E.; et al. Resistant Hy-pertension: Detection, Evaluation, and Management: A Scientific Statement from the American Heart Association. Hypertension 2018, 72, e53–e90. [Google Scholar] [CrossRef] [PubMed]
- Rucker, A.J.; Rudemiller, N.P.; Crowley, S.D. Salt, Hypertension, and Immunity. Annu. Rev. Physiol. 2018, 80, 283–307. [Google Scholar] [CrossRef]
- Carey, R.M.; Sakhuja, S.; Calhoun, D.A.; Whelton, P.K.; Muntner, P. Prevalence of apparent treatment-resistant hy-pertension in the United States. Hypertension 2019, 73, 424–431. [Google Scholar] [CrossRef]
- Navar, L.G.; Zou, L.; Von Thun, A.; Tarng Wang, C.; Imig, J.D.; Mitchell, K.D. Unraveling the mystery of Goldblatt hy-pertension. News Physiol. Sci. 1998, 13, 170–176. [Google Scholar]
- Whelton, P.K.; Carey, R.M.; Aronow, W.S.; Casey, D.E., Jr.; Collins, K.J.; Dennison Himmelfarb, C.; DePalma, S.M.; Gidding, S.; Jamerson, K.A.; Jones, D.W.; et al. Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: A Report of the Ameri-can College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension 2018, 71, e13–e115. [Google Scholar]
- Treadway, K.K.; Slater, E.E. Renovascular hypertension. Annu. Rev. Med. 1984, 35, 665–692. [Google Scholar] [CrossRef]
- Koratala, A.; Chamarthi, G.; Touyz, R.M.; Dominiczak, A.F.; Elijovich, F.; Spence, J.D.; Grim, C.E.; Taler, S.J.; Mohandas, R. Renovascular hypertension: One size does not fit all: Challenges in diagnosis and management. Hypertension 2021, 77, 1022–1028. [Google Scholar] [CrossRef]
- Guan, S.; Fox, J.; Mitchell, K.D.; Navar, L.G. Angiotensin and angiotensin converting enzyme tissue levels in two-kidney, one clip hypertensive rats. Hypertension 1992, 20, 763–767. [Google Scholar] [CrossRef] [Green Version]
- Wilcox, C.S.; Cardozo, J.; Welch, W.J. AT1 and TxA2/PGH2 receptors maintain hypertension throughout 2K,1C Goldblatt hypertension in the rat. Am J Physiol. 1996, 271 Pt 2, R891–R896. [Google Scholar] [CrossRef] [PubMed]
- Abdi, A.; Johns, E.J. The effect of angiotensin II receptor antagonists on kidney function in two-kidney, two-clip Goldblatt hypertensive rats. Eur. J. Pharmacol. 1997, 331, 185–192. [Google Scholar] [CrossRef]
- Cervenka, L.; Navar, L.G. Renal responses of the nonclipped kidney of two-kidney/one-clip Goldblatt hyperten-sive rats to type 1 angiotensin II receptor blockade with candesartan. J. Am. Soc. Nephrol. 1999, 10 (Suppl. 11), S197–S201. [Google Scholar] [PubMed]
- Kagiyama, S.; Varela, A.; Phillips, M.I.; Galli, S.M. Antisense Inhibition of Brain Renin-Angiotensin System Decreased Blood Pressure in Chronic 2-Kidney, 1 Clip Hypertensive Rats. Hypertension 2001, 37 Pt 2, 371–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Červenka, L.; Horáček, V.; Vaněčková, I.; Hubáček, J.A.; Oliverio, M.I.; Coffman, T.M.; Navar, L.G. Essential Role of AT1A Receptor in the Development of 2K1C Hypertension. Hypertension 2002, 40, 735–741. [Google Scholar] [CrossRef] [Green Version]
- Cervenka, L.; Vanecková, I.; Husková, Z.; Vanourková, Z.; Erbanová, M.; Thumová, M.; Skaroupková, P.; Opocenský, M.; Malý, J.; Chábová, V.C.; et al. Pivotal role of angiotensin II receptor subtype 1A in the development of two-kidney, one-clip hypertension: Study in angiotensin II receptor subtype 1A knockout mice. J. Hypertens. 2008, 26, 1379–1389. [Google Scholar] [CrossRef] [Green Version]
- Prieto-Carrasquero, M.C.; Botros, F.T.; Pagan, J.; Kobori, H.; Seth, D.M.; Casarini, D.E.; Navar, L.G. Collecting Duct Renin Is Upregulated in Both Kidneys of 2-Kidney, 1-Clip Goldblatt Hypertensive Rats. Hypertension 2008, 51, 1590–1596. [Google Scholar] [CrossRef] [Green Version]
- Schricker, K.; Holmer, S.; Krämer, B.K.; Riegger, G.; Kurtz, A. Control of renin gene expression in 2 kidney-1 clip rats. Kidney Int. 1994, 46, 1539–1541. [Google Scholar] [CrossRef] [Green Version]
- Modrall, J.; Quinones, M.J.; Frankhouse, J.H.; Hsueh, W.A.; Weaver, F.A.; Kedes, L. Upregulation of Angiotensin II Type 1 Receptor Gene Expression in Chronic Renovascular Hypertension. J. Surg. Res. 1995, 59, 135–140. [Google Scholar] [CrossRef]
- Mai, M.; Hilgers, K.F.; Wagner, J.; Mann, J.F.; Geiger, H. Expression of angiotensin-converting enzyme in renovascu-lar hypertensive rat kidney. Hypertension 1995, 25 Pt 2, 674–678. [Google Scholar] [CrossRef]
- Della Bruna, R.; Bernhard, I.; Gess, B.; Schricker, K.; Kurtz, A. Renin gene and angiotensin II AT1 receptor gene ex-pression in the kidneys of normal and of two-kidney/one-clip rats. Pflug. Arch. 1995, 430, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Cervenka, L.; Wang, C.T.; Mitchell, K.D.; Navar, L.G. Proximal tubular angiotensin II levels and renal functional re-sponses to AT1 receptor blockade in nonclipped kidneys of Goldblatt hypertensive rats. Hypertension 1999, 33, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Tokuyama, H.; Hayashi, K.; Matsuda, H.; Kubota, E.; Honda, M.; Okubo, K.; Takamatsu, I.; Tatematsu, S.; Ozawa, Y.; Wakino, S.; et al. Differential Regulation of Elevated Renal Angiotensin II in Chronic Renal Ischemia. Hypertension 2002, 40, 34–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadjadi, J.; Puttaparthi, K.; Welborn, M.B., 3rd; Rogers, T.E.; Moe, O.; Clagett, G.P.; Turnage, R.H.; Levi, M.; Modrall, J.G. Upregulation of autocrine-paracrine renin-angiotensin systems in chronic renovascular hypertension. J. Vasc. Surg. 2002, 36, 386–392. [Google Scholar] [CrossRef] [Green Version]
- Shao, W.; Miyata, K.; Katsurada, A.; Satou, R.; Seth, D.M.; Rosales, C.B.; Prieto, M.C.; Mitchell, K.D.; Navar, L.G. Increased angiotensinogen expression, urinary angiotensinogen excretion, and tissue injury in nonclipped kidneys of two-kidney, one-clip hypertensive rats. Am. J. Physiol. Renal. Physiol. 2016, 311, F278–F290. [Google Scholar] [CrossRef] [Green Version]
- Ryan, M.J.; Didion, S.P.; Mathur, S.; Faraci, F.M.; Sigmund, C.D. Angiotensin II-induced vascular dysfunction is medi-ated by the AT1A receptor in mice. Hypertension 2004, 43, 1074–1079. [Google Scholar] [CrossRef] [Green Version]
- Sparks, M.A.; Dilmen, E.; Ralph, D.L.; Rianto, F.; Hoang, T.A.; Hollis, A.; Diaz, E.J.; Adhikari, R.; Chew, G.; Petretto, E.G.; et al. Vascular control of kidney epithelial transporters. Am. J. Physiol. Renal. Physiol. 2021, 320, F1080–F1092. [Google Scholar] [CrossRef]
- Sparks, M.A.; Stegbauer, J.; Chen, D.; Gomez, J.A.; Griffiths, R.C.; Azad, H.A.; Herrera, M.; Gurley, S.B.; Coffman, T.M. Vascular Type 1A angiotensin II receptors control BP by regulating renal blood flow and urinary sodium excretion. J. Am. Soc. Nephrol. 2015, 26, 2953–2962. [Google Scholar] [CrossRef] [Green Version]
- Lincevicius, G.S.; Shimoura, C.G.; Nishi, E.E.; Perry, J.C.; Casarini, D.E.; Gomes, G.N.; Bergamaschi, C.T.; Campos, R.R. Aldosterone contributes to sympathoexcitation in renovascular hypertension. Am. J. Hypertens. 2015, 28, 1083–1090. [Google Scholar] [CrossRef] [Green Version]
- Jancovski, N.; Bassi, J.K.; Carter, D.A.; Choong, Y.-T.; Connelly, A.; Nguyen, T.-P.; Chen, D.; Lukoshkova, E.V.; Menuet, C.; Head, G.A.; et al. Stimulation of angiotensin type 1A receptors on catecholaminergic cells contributes to angiotensin-dependent hypertension. Hypertension 2013, 62, 866–871. [Google Scholar] [CrossRef] [Green Version]
- Galli, S.M.; Phillips, M.I. Angiotensin II AT1A receptor antisense lowers blood pressure in acute 2-kidney, 1-clip hypertension. Hypertension 2001, 38 Pt 2, 674–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.C.; Soleimani, M.; Zhu, D.; Rubera, I.; Tauc, M.; Zheng, X.; Zhang, J.; Chen, X.; Zhuo, J.L. Proximal tubule-specific deletion of the NHE3 (Na+/H+ exchanger 3) promotes the pressure-natriuresis response and lowers blood pressure in mice. Hypertension 2018, 72, 1328–1336. [Google Scholar] [CrossRef] [PubMed]
- Li, X.C.; Zhu, D.; Chen, X.; Zheng, X.; Zhao, C.; Zhang, J.; Soleimani, M.; Rubera, I.; Tauc, M.; Zhou, X.; et al. Proximal tubule-specific deletion of the NHE3 (Na+/H+ exchanger 3) in the kidney attenuates Ang II (angiotensin II)-induced hypertension in mice. Hypertension 2019, 74, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Li, X.C.; Leite, A.P.O.; Zheng, X.; Zhao, C.; Chen, X.; Zhang, L.; Zhou, X.; Rubera, I.; Tauc, M.; Zhuo, J.L. Proximal tubule-specific deletion of angiotensin II type 1a receptors in the kidney attenuates circulating and intratubular angiotensin II-induced hypertension in PT-Agtr1a−/− mice. Hypertension 2021, 77, 1285–1298. [Google Scholar] [CrossRef] [PubMed]
- Leite, A.P.O.; Li, X.C.; Hassan, R.; Zheng, X.; Alexander, B.; Casarini, D.E.; Zhuo, J.L. Sex differences in angiotensin II-induced hypertension and kidney injury: Role of AT1a receptors in the proximal tubule of the kidney. Clin. Sci. 2021, 135, 1825–1843. [Google Scholar] [CrossRef] [PubMed]
- Li, X.C.; Navar, L.G.; Shao, Y.; Zhuo, J.L. Genetic deletion of AT1a receptors attenuates intracellular accumulation of ANG II in the kidney of AT1a receptor-deficient mice. Am. J. Physiol. Renal. Physiol. 2007, 293, F586–F593. [Google Scholar] [CrossRef] [Green Version]
- Li, X.C.; Shao, Y.; Zhuo, J.L. AT1a receptor knockout in mice impairs urine concentration by reducing basal vasopressin levels and its receptor signaling proteins in the inner medulla. Kidney Int. 2009, 76, 169–177. [Google Scholar] [CrossRef] [Green Version]
- Li, X.C.; Zhuo, J.L. Phosphoproteomic analysis of AT1 receptor-mediated signaling responses in proximal tubules of angiotensin II-induced hypertensive rats. Kidney Int. 2011, 80, 620–632. [Google Scholar] [CrossRef] [Green Version]
- Zhuo, J.L.; Alcorn, D.; McCausland, J.; Casley, D.; Mendelsohn, F.A. In vivo occupancy of angiotensin II subtype 1 re-ceptors in rat renal medullary interstitial cells. Hypertension 1994, 23 Pt 2, 838–843. [Google Scholar] [CrossRef] [Green Version]
- Li, X.C.; Zhuo, J.L. In vivo regulation of AT1a receptor-mediated intracellular uptake of [125I]Val5-Ang II in the kidneys and adrenals of AT1a receptor-deficient mice. Am. J. Physiol. Renal. Physiol. 2008, 294, F293–F302. [Google Scholar] [CrossRef] [Green Version]
- Crowley, S.D.; Gurley, S.B.; Oliverio, M.I.; Pazmino, A.K.; Griffiths, R.; Flannery, P.J.; Spurney, R.F.; Kim, H.S.; Smithies, O.; Le, T.H.; et al. Distinct roles for the kidney and systemic tissues in blood pressure regulation by the ren-in-angiotensin system. J. Clin. Investig. 2005, 115, 1092–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crowley, S.D.; Gurley, S.B.; Herrera, M.J.; Ruiz, P.; Griffiths, R.; Kumar, A.P.; Kim, H.-S.; Smithies, O.; Le, T.H.; Coffman, T.M. Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc. Natl. Acad. Sci. USA 2006, 103, 17985–17990. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, F.A.; Quirion, R.; Saavedra, J.M.; Aguilera, G.; Catt, K.J. Autoradiographic localization of angiotensin II receptors in rat brain. Proc. Natl. Acad. Sci. USA 1984, 81, 1575–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sechi, L.A.; Griffin, C.A.; Grady, E.F.; Kalinyak, J.E.; Schambelan, M. Characterization of angiotensin II receptor sub-types in rat heart. Circ. Res. 1992, 71, 1482–1489. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, S.; Ishida, A.; Moriya, H.; Mori, N.; Fukuda, T.; Takamura, T. Angiotensin II receptor blockade limits kidney injury in two-kidney, one-clip Goldblatt hypertensive rats with special reference to phenotypic changes. J. Lab. Clin. Med. 1999, 133, 134–143. [Google Scholar] [CrossRef]
- Zhuo, J.L.; Allen, A.M.; Alcorn, D.; MacGregor, D.; Aldred, G.P.; Mendelsohn, F.A.O. The Distribution of Angiotensin II Receptors. In Hypertension: Pathology, Diagnosis, & Management, 2nd ed.; Laragh, J.H., Brenner, B.M., Eds.; Raven Press: New York, NY, USA, 1995; pp. 1739–1762. [Google Scholar]
- Zhuo, J.; Alcorn, D.; Harris, P.J.; Mendelsohn, F.A. Localization and properties of angiotensin II receptors in rat kidney. Kidney Int. Suppl. 1993, 42, S40–S46. [Google Scholar]
- Kakinuma, Y.; Fogo, A.; Inagami, T.; Ichikawa, I. Intrarenal localization of angiotensin II type 1 receptor mRNA in the rat. Kidney Int. 1993, 43, 1229–1235. [Google Scholar] [CrossRef] [Green Version]
- Healy, D.P.; Ye, M.Q.; Troyanovskaya, M. Localization of angiotensin II type 1 receptor subtype mRNA in rat kid-ney. Am. J. Physiol. 1995, 268 Pt 2, F220–F226. [Google Scholar]
- Skorecki, K.L.; Ballermann, B.J.; Rennke, H.G.; Brenner, B.M. Angiotensin II receptor regulation in isolated renal glomeruli. Fed. Proc. 1983, 42, 3064–3070. [Google Scholar]
- Zhuo, J.; Alcorn, D.; McCausland, J.; Mendelsohn, F.A. Localization and regulation of angiotensin II receptors in renomedullary interstitial cells. Kidney Int. 1994, 46, 1483–1485. [Google Scholar] [CrossRef] [Green Version]
- Zhuo, J.; Maric, C.; Harris, P.J.; Alcorn, D.; Mendelsohn, F.A. Localization and functional properties of angiotensin II AT1 receptors in the Kidney. Focus on renomedullary interstitial cells. Hypertens. Res. 1997, 20, 233–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobori, H.; Nangaku, M.; Navar, L.G.; Nishiyama, A. The Intrarenal Renin-Angiotensin System: From Physiology to the Pathobiology of Hypertension and Kidney Disease. Pharmacol. Rev. 2007, 59, 251–287. [Google Scholar] [CrossRef] [PubMed]
- Matsusaka, T.; Niimura, F.; Shimizu, A.; Pastan, I.; Saito, A.; Kobori, H.; Nishiyama, A.; Ichikawa, I. Liver angiotensinogen is the primary source of renal angiotensin II. J. Am. Soc. Nephrol. 2012, 23, 1181–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navar, L.G.; Satou, R.; Gonzalez-Villalobos, R.A. The Increasing Complexity of the Intratubular Renin-Angiotensin System. J. Am. Soc. Nephrol. 2012, 23, 1130–1132. [Google Scholar] [CrossRef] [Green Version]
- Zhuo, J.L.; Imig, J.D.; Hammond, T.G.; Orengo, S.; Benes, E.; Navar, L.G. Ang II accumulation in rat renal endosomes during Ang II-induced hypertension: Role of AT1 receptor. Hypertension 2002, 39, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Li, X.C.; Hopfer, U.; Zhuo, J.L. Novel signaling mechanisms of intracellular angiotensin II-induced NHE3 expression and activation in mouse proximal tubule cells. Am. J. Physiol. Renal Physiol. 2012, 303, F1617–F1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.C.; Shull, G.E.; Miguel-Qin, E.; Zhuo, J.L. Role of the Na+/H+ exchanger 3 in angiotensin II-induced hypertension. Physiol. Genom. 2015, 47, 479–487. [Google Scholar] [CrossRef] [Green Version]
- Gurley, S.B.; Riquier-Brison, A.D.; Schnermann, J.; Sparks, M.A.; Allen, A.M.; Haase, V.H.; Snouwaert, J.N.; Le, T.H.; McDonough, A.A.; Koller, B.H.; et al. AT1A Angiotensin Receptors in the Renal Proximal Tubule Regulate Blood Pressure. Cell Metab. 2011, 13, 469–475. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Weatherford, E.T.; Davis, D.R.; Keen, H.L.; Grobe, J.L.; Daugherty, A.; Cassis, L.A.; Allen, A.M.; Sigmund, C.D. Renal proximal tubule angiotensin AT1A receptors regulate blood pressure. Am. J. Physiol. Renal. Integr. Comp. Physiol. 2011, 301, R1067–R1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, R.J.; Alpers, C.E.; Yoshimura, A.; Lombardi, D.; Pritzl, P.; Floege, J.; Schwartz, S.M. Renal injury from angiotensin II-mediated hypertension. Hypertension 1992, 19, 464–474. [Google Scholar] [CrossRef] [Green Version]
- Crowley, S.D.; Frey, C.W.; Gould, S.K.; Griffiths, R.; Ruiz, P.; Burchette, J.L.; Howell, D.N.; Makhanova, N.; Yan, M.; Kim, H.-S.; et al. Stimulation of lymphocyte responses by angiotensin II promotes kidney injury in hypertension. Am. J. Physiol. Renal. Physiol. 2008, 295, F515–F524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreiber, A.; Shulhevich, Y.; Geraci, S.; Hesser, J.; Stsepankou, D.; Neudecker, S.; Koenig, S.; Heinrich, R.; Hoecklin, F.; Pill, J.; et al. Transcutaneous measurement of renal function in conscious mice. Am. J. Physiol. Renal. Physiol. 2012, 303, F783–F788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schock-Kusch, D.; Xie, Q.; Shulhevich, Y.; Hesser, J.; Stsepankou, D.; Sadick, M.; Koenig, S.; Hoecklin, F.; Pill, J.; Gretz, N. Transcutaneous assessment of renal function in conscious rats with a device for measuring FITC-sinistrin disappearance curves. Kidney Int. 2011, 79, 1254–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satou, R.; Cypress, M.W.; Woods, T.C.; Katsurada, A.; Dugas, C.M.; Fonseca, V.A.; Navar, L.G. Blockade of sodi-um-glucose cotransporter 2 suppresses high glucose-induced angiotensinogen augmentation in renal proximal tubular cells. Am. J. Physiol. Renal Physiol. 2020, 318, F67–F75. [Google Scholar] [CrossRef] [PubMed]
- Woods, T.C.; Satou, R.; Miyata, K.; Katsurada, A.; Dugas, C.M.; Klingenberg, N.C.; Fonseca, V.A.; Navar, L.G. Canagliflozin Prevents Intrarenal Angiotensinogen Augmentation and Mitigates Kidney Injury and Hypertension in Mouse Model of Type 2 Diabetes Mellitus. Am. J. Nephrol. 2019, 49, 331–342. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | WT Control N = 13 | Agtr1a−/− Control N = 9 | PT-Agtr1a−/− Control N = 13 | PT-Nhe3−/− Control N = 13 |
|---|---|---|---|---|
| Body wt., g | 28.6 ± 0.4 | 29.2 ± 0.7 | 29.3 ± 0.4 | 31.8 ± 1.2 |
| Heart wt., g | 0.14 ± 0.01 | 0.14 ± 0.01 | 0.15 ± 0.01 | 0.16 ± 0.01 |
| Heart wt./Body wt. ratio | 0.50 ± 0.02 | 0.47 ± 0.01 * | 0.52 ± 0.01 † | 0.52 ± 0.02 † |
| Left kidney wt., g | 0.18 ± 0.01 | 0.14 ± 0.01 ** | 0.17 ± 0.01 †† | 0.19 ± 0.01 †† |
| Let kidney wt./body wt. ratio | 0.62 ± 0.03 | 0.49 ± 0.02 ** | 0.59 ± 0.03 †† | 0.62 ± 0.02 †† |
| Blood volume, mL | 0.72 ± 0.04 | 0.73 ± 0.02 | 0.76 ± 0.03 | 0.82 ± 0.05 †† |
| Hematocrit, % | 44 ± 0.7 | 43 ± 0.6 | 45 ± 0.4 | 46 ± 0.5 |
| Urine excretion, mL/24 h | 1.47 ± 0.10 | 1.90 ± 0.09 ** | 1.40 ± 0.03 † | 1.33 ± 0.09 †† |
| Na+ excretion, µmol/24 h | 156.2 ± 4.8 | 237.3 ± 10.7 ** | 222.6 ± 6.9 ** | 186.1 ± 7.4 *† |
| K+ excretion, µmol/24 h | 221.0 ± 7.4 | 384.8 ± 14.7 ** | 317.2 ± 14.1 † | 264.9 ± 17.4 †† |
| Urine Cl− excretion, µmol/24 h | 242.1 ± 11.6 | 404.2 ± 45.9 ** | 344.7 ± 25.2 † | 260 ± 11.1 †† |
| Parameter | WT + 2K1C N = 15 | Agtr1a−/− + 2K1C N = 9 | PT-Agtr1a−/− + 2K1C N = 15 | PT-Nhe3−/− + 2K1C N = 12 |
|---|---|---|---|---|
| Body wt., g | 27.0 ± 0.5 | 28.4 ± 0.6 | 29.1 ± 0.4 | 28.3 ± 0.6 |
| Heart wt., g | 0.14 ± 0.01 | 0.13 ± 0.01 | 0.15 ± 0.01 | 0.13 ± 0.01 |
| Heart wt./Body wt. ratio | 0.53 ± 0.02 | 0.47 ± 0.01 ** | 0.52 ± 0.01 † | 0.47 ± 0.01 |
| Left kidney wt., g | 0.14 ± 0.01 | 0.15 ± 0.02 | 0.16 ± 0.01 | 0.15 ± 0.01 |
| Let kidney wt./body wt. ratio | 0.54 ± 0.03 | 0.53 ± 0.01 | 0.56 ± 0.02 | 0.57 ± 0.02 |
| Blood volume, mL | 0.66 ± 0.04 | 0.80 ± 0.02 ** | 0.72 ± 0.03 *† | 0.71 ± 0.03 *† |
| Hematocrit, % | 47 ± 0.5 | 43 ± 0.4 ** | 45 ± 0.4 | 45 ± 0.5 |
| Urine excretion, mL/24 h | 0.93 ± 0.04 | 1.80 ± 0.06 ** | 1.65 ± 0.04 ** | 1.54 ± 0.03 ** |
| Na+ excretion, µmol/24 h | 134.1 ± 2.6 | 156.8 ± 4.8 ** | 219.2 ± 5.7 **†† | 200.3 ± 5.0 **†† |
| K+ excretion, µmol/24 h | 170.6 ± 3.8 | 281.9 ± 10.5 ** | 372.3 ± 11.6 **†† | 312.2 ± 14.1 ** |
| Urine Cl− excretion, µmol/24 h | 163.9 ± 3.8 | 506.0 ± 39.6 ** | 336.3 ± 9.8 **†† | 303.6 ± 7.6 **†† |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.C.; Hassan, R.; Leite, A.P.O.; Katsurada, A.; Dugas, C.; Sato, R.; Zhuo, J.L. Genetic Deletion of AT1a Receptor or Na+/H+ Exchanger 3 Selectively in the Proximal Tubules of the Kidney Attenuates Two-Kidney, One-Clip Goldblatt Hypertension in Mice. Int. J. Mol. Sci. 2022, 23, 15798. https://doi.org/10.3390/ijms232415798
Li XC, Hassan R, Leite APO, Katsurada A, Dugas C, Sato R, Zhuo JL. Genetic Deletion of AT1a Receptor or Na+/H+ Exchanger 3 Selectively in the Proximal Tubules of the Kidney Attenuates Two-Kidney, One-Clip Goldblatt Hypertension in Mice. International Journal of Molecular Sciences. 2022; 23(24):15798. https://doi.org/10.3390/ijms232415798
Chicago/Turabian StyleLi, Xiao Chun, Rumana Hassan, Ana Paula O. Leite, Akemi Katsurada, Courtney Dugas, Ryosuke Sato, and Jia Long Zhuo. 2022. "Genetic Deletion of AT1a Receptor or Na+/H+ Exchanger 3 Selectively in the Proximal Tubules of the Kidney Attenuates Two-Kidney, One-Clip Goldblatt Hypertension in Mice" International Journal of Molecular Sciences 23, no. 24: 15798. https://doi.org/10.3390/ijms232415798