
The Molecular Mechanisms and Function of miR-15a/16 Dysregulation in Fibrotic Diseases

Abstract
:1. Introduction
2. Biological Characteristics of the miR-15 Family
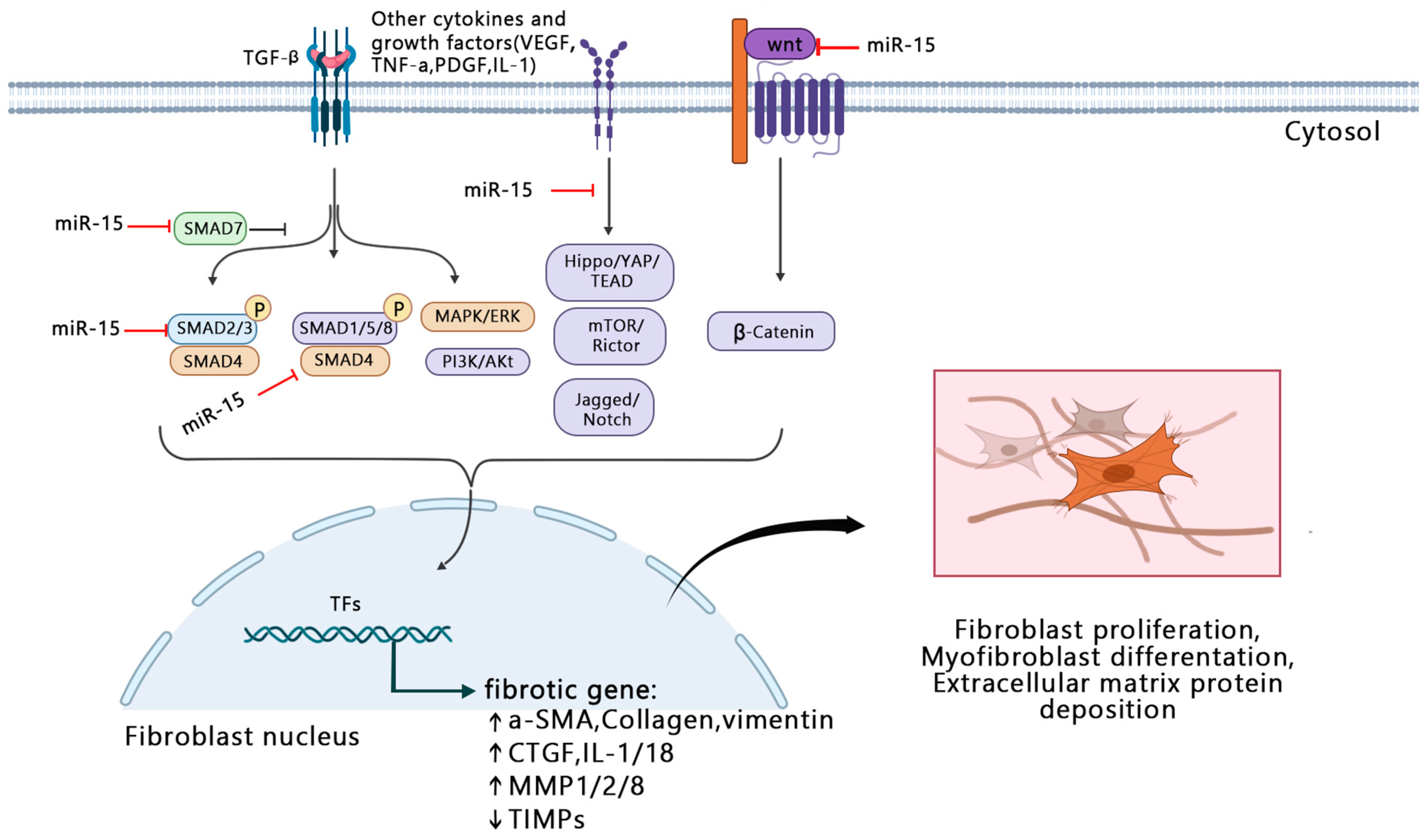
3. The Role and Mechanism of miR-15a/16 in Fibrogenesis
3.1. Regulation of Cell Transformation
3.2. Regulation of the Synthesis and Degradation of ECM
3.3. Regulation of the Release of Fibrotic Mediators
4. miR-15a/16 and Fibrotic Disease
4.1. Liver Fibrosis
4.2. Pulmonary Fibrosis
4.3. Cardiac Fibrosis
4.4. Renal Fibrosis
4.5. Other Fibrotic Diseases
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Lytle, J.R.; Yario, T.A.; Steitz, J.A. Target mRNAs are repressed as efficiently by microRNA-binding sites in the 5’ UTR as in the 3’ UTR. Proc. Natl. Acad. Sci. USA 2007, 104, 9667–9672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caporali, A.; Emanueli, C. MicroRNA-503 and the extended microRNA-16 family in angiogenesis. Trends Cardiovasc. Med. 2011, 21, 162–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uddin, A.; Chakraborty, S. Role of miRNAs in lung cancer. Cell Physiol. 2018; ahead of print. [Google Scholar] [CrossRef]
- Patel, V.; Noureddine, L. MicroRNAs and fibrosis. Curr. Opin. Nephrol. Hypertens. 2012, 21, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Kim, V.N. Processing of intronic microRNAs. EMBO 2007, 26, 775–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franklin, T. Therapeutic approaches to organ fibrosis. Int. Biochem. Cell Biol. 1997, 29, 79–89. [Google Scholar] [CrossRef]
- Finnerty, J.R.; Wang, W.X.; Hébert, S.S.; Wilfred, B.R.; Mao, G.; Nelson, P.T. The miR-15/107 group of microRNA genes: Evolutionary biology, cellular functions, and roles in human diseases. Mol. Biol. 2010, 402, 491–509. [Google Scholar] [CrossRef] [Green Version]
- Hullinger, T.G.; Montgomery, R.L.; Seto, A.G.; Dickinson, B.A.; Semus, H.M.; Lynch, J.M.; Dalby, C.M.; Robinson, K.; Stack, C.; Latimer, P.A.; et al. Inhibition of miR-15 protects against cardiac ischemic injury. Circ. Res. 2012, 110, 71–81. [Google Scholar] [CrossRef]
- Liu, L.F.; Wang, Y. Cellular function of microRNA-15 family. Sheng Li Xue Bao 2012, 64, 101–106. [Google Scholar]
- Lerner, M.; Harada, M.; Lovén, J.; Castro, J.; Davis, Z.; Oscier, D.; Henriksson, M.; Sangfelt, O.; Grandér, D.; Corcoran, M.M. DLEU2, frequently deleted in malignancy, functions as a critical host gene of the cell cycle inhibitory microRNAs miR-15a and miR-16-1. Exp. Cell Res. 2009, 315, 2941–2952. [Google Scholar] [CrossRef]
- Chen, C.Q.; Chen, C.S.; Chen, J.J.; Zhou, L.-P.; Xu, H.-L.; Jin, W.-W.; Wu, J.-B.; Gao, S.-M. Histone deacetylases inhibitor trichostatin A increases the expression of Dleu2/miR-15a/16-1 via HDAC3 in non-small cell lung cancer. Mol. Cell Biochem. 2013, 383, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, X.; Lin, J.; Lwin, T.; Wright, G.; Moscinski, L.C.; Dalton, W.S.; Seto, E.; Wright, K.; Sotomayor, E.; et al. Myc represses miR-15a/miR-16-1 expression through recruitment of HDAC3 in mantle cell and other non-Hodgkin B-cell lymphomas. Oncogene 2012, 31, 3002–3008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasar, S.; Underbayev, C.; Yuan, Y.; Hanlon, M.; Aly, S.; Khan, H.; Chang, V.; Batish, M.; Gavrilova, T.; Badiane, F.; et al. Therapeutic implications of activation of the host gene (Dleu2) promoter for miR-15a/16-1 in chronic lymphocytic leukemia. Oncogene 2014, 33, 3307–3315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Zhou, J.; Wang, S.; Song, Y.; Zhou, J.; Ren, F. Long non-coding RNA SNHG12 promotes proliferation and invasion of colorectal cancer cells by acting as a molecular sponge of microRNA-16. Exp. Ther. Med. 2019, 18, 1212–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Su, W.; Zhao, X.; Shan, T.; Jin, T.; Guo, Y.; Li, C.; Li, R.; Zhou, Y.; Shan, H.; et al. LncRNA PFAR contributes to fibrogenesis in lung fibroblasts through competitively binding to miR-15a. Biosci. Rep. 2019, 39, BSR20190280. [Google Scholar] [CrossRef] [Green Version]
- Pardali, E.; Sanchez-Duffhues, G.; Gomez-Puerto, M.C.; Dijke, P.T. TGF-beta-Induced Endothelial-Mesenchymal Transition in Fibrotic Diseases. Int. Mol. Sci. 2017, 18, 2157. [Google Scholar] [CrossRef] [Green Version]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef]
- Zhu, B.; Wei, X.X.; Wang, T.-B.; Zhou, Y.-C.; Liu, A.-M.; Zhang, G.-W. Increased miR-16 expression induced by hepatitis C virus infection promotes liver fibrosis through downregulation of hepatocyte growth factor and Smad7. Arch. Virol. 2015, 160, 2043–2050. [Google Scholar] [CrossRef]
- Dooley, S.; Hamzavi, J.; Breitkopf, K.; Wiercinska, E.; Said, H.M.; Lorenzen, J.; Dijke, P.T.; Gressner, A.M. Smad7 prevents activation of hepatic stellate cells and liver fibrosis in rats. Gastroenterology 2003, 125, 178–191. [Google Scholar] [CrossRef]
- Pan, Q.; Guo, C.-J.; Xu, Q.-Y.; Wang, J.-Z.; Li, H.; Fang, C.-H. miR-16 integrates signal pathways in myofibroblasts: Determinant of cell fate necessary for fibrosis resolution. Cell Death Dis. 2020, 11, 639. [Google Scholar] [CrossRef]
- Bo, Y.; Liu, B.; Yang, L.; Zhang, L.; Yan, Y. Exosomes derived from miR-16-5p-overexpressing keratinocytes attenuates bleomycin-induced skin fibrosis. Biochem. Biophys. Res. Commun. 2021, 561, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Ji, T.; Feng, W.; Zhang, X.; Zang, K.; Zhu, X.; Shang, F. HDAC inhibitors promote pancreatic stellate cell apoptosis and relieve pancreatic fibrosis by upregulating miR-15/16 in chronic pancreatitis. Hum. Cell 2020, 33, 1006–1016. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; Fan, X.; Yu, H.; Ai, G.; Zhang, H.; Kong, H.; Song, Q.; Huang, Y.; Huang, J.; Ning, Q. MicroRNA-29b-3p prevents Schistosoma japonicum-induced liver fibrosis by targeting COL1A1 and COL3A1. Cell Biochem. 2018, 119, 3199–3209. [Google Scholar] [CrossRef] [PubMed]
- Piccoli, M.-T.; Gupta, S.K.; Viereck, J.; Foinquinos, A.; Samolovac, S.; Kramer, F.L.; Garg, A.; Remke, J.; Zimmer, K.; Batkai, S.; et al. Inhibition of the Cardiac Fibroblast-Enriched lncRNA Meg3 Prevents Cardiac Fibrosis and Diastolic Dysfunction. Circ. Res. 2017, 121, 575–583. [Google Scholar] [CrossRef]
- Yao, Q.; Xing, Y.; Wang, Z.; Liang, J.; Lin, Q.; Huang, M.; Chen, Y.; Lin, B.; Xu, X.; Chen, W. MiR-16-5p suppresses myofibroblast activation in systemic sclerosis by inhibiting NOTCH signaling. Aging 2020, 13, 2640–2654. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, X.; Sun, J.; Su, W.; Zhang, L.; Li, Y.; Liu, Y.; Zhang, L.; Lu, Y.; Shan, H.; et al. YAP1/Twist promotes fibroblast activation and lung fibrosis that conferred by miR-15a loss in IPF. Cell Death Differ. 2019, 26, 1832–1844. [Google Scholar] [CrossRef]
- Inomata, M.; Kamio, K.; Azuma, A.; Matsuda, K.; Usuki, J.; Morinaga, A.; Tanaka, T.; Kashiwada, T.; Atsumi, K.; Hayashi, H.; et al. Rictor-targeting exosomal microRNA-16 ameliorates lung fibrosis by inhibiting the mTORC2-SPARC axis. Exp. Cell Res. 2021, 398, 112416. [Google Scholar] [CrossRef]
- Haque, M.F.; Harris, M.; Meghji, S.; Barrett, A.W. Immunolocalization of cytokines and growth factors in oral submucous fibrosis. Cytokine 1998, 10, 713–719. [Google Scholar] [CrossRef]
- Arakeri, G.; Rai, K.K.; Hunasgi, S.; Merkx, M.; Gao, S.; Brennan, P.A. Oral submucous fibrosis: An update on current theories of pathogenesis. Oral Pathol. Med. 2017, 46, 406–412. [Google Scholar] [CrossRef]
- Wang, M.; Li, J.; Cai, J.; Cheng, L.; Wang, X.; Xu, P.; Li, G.; Liang, X. Overexpression of MicroRNA-16 Alleviates Atherosclerosis by Inhibition of Inflammatory Pathways. Biomed. Res. Int. 2020, 2020, 8504238. [Google Scholar] [CrossRef]
- Liang, X.; Xu, Z.; Yuan, M.; Zhang, Y.; Zhao, B.; Wang, J.; Zhang, A.; Li, G. MicroRNA-16 suppresses the activation of inflammatory macrophages in atherosclerosis by targeting PDCD4. Int. Mol. Med. 2016, 37, 967–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, X.; Li, X.; Shen, Y.; Miao, J.; Liu, H.; Li, G.; Wang, Z. MiR-16 regulates mouse peritoneal macrophage polarization and affects T-cell activation. Cell Mol. Med. 2016, 20, 1898–1907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Yang, F.; Yu, X.; Wang, B.; Yang, Y.; Zhou, X.; Cheng, R.; Xia, S.; Zhou, X. miR-16 inhibits NLRP3 inflammasome activation by directly targeting TLR4 in acute lung injury. Biomed. Pharmacother. 2019, 112, 108664. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.F.; Liang, Z.; Lv, Z.R.; Liu, X.H.; Bai, J.; Chen, J.; Chen, C.; Wang, Y. MicroRNA-15a/b are up-regulated in response to myocardial ischemia/reperfusion injury. Geriatr. Cardiol. 2012, 9, 28–32. [Google Scholar] [CrossRef]
- Xu, J.; Chen, X.; Nie, W. miR-15b-5p regulates the NLRP3 inflammasome signal through targeting SIRT3 to regulate hypoxia/reoxygenation-induced cardiomyocyte pyroptosis process. Shock 2022, 58, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.-J.; Pan, Q.; Li, D.-G.; Sun, H.; Liu, B.-W. miR-15b and miR-16 are implicated in activation of the rat hepatic stellate cell: An essential role for apoptosis. J. Hepatol. 2009, 50, 766–778. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-L.; Pan, Q.; Zhang, R.-N.; Shen, F.; Yan, S.-Y.; Sun, C.; Xu, Z.-J.; Chen, Y.-W.; Fan, J.-G. Disease-specific miR-34a as diagnostic marker of non-alcoholic steatohepatitis in a Chinese population. World Gastroenterol. 2016, 22, 9844–9852. [Google Scholar] [CrossRef]
- Pan, Q.; Guo, C.; Sun, C.; Fan, J.; Fang, C. Integrative analysis of the transcriptome and targetome identifies the regulatory network of miR-16: An inhibitory role against the activation of hepatic stellate cells. Biomed. Mater. Eng. 2014, 24, 3863–3871. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Liu, J.; Xiao, E.; Ning, H.; Li, K.; Shang, J.; Kang, Y. MiR-15b and miR-16 suppress TGF-beta1-induced proliferation and fibrogenesis by regulating LOXL1 in hepatic stellate cells. Life Sci. 2021, 270, 119144. [Google Scholar] [CrossRef]
- Kim, K.M.; Han, C.Y.; Kim, J.Y.; Cho, S.S.; Kim, Y.S.; Koo, J.H.; Lee, J.M.; Lim, S.C.; Kang, K.W.; Kim, J.-S.; et al. Galpha12 overexpression induced by miR-16 dysregulation contributes to liver fibrosis by promoting autophagy in hepatic stellate cells. J. Hepatol. 2018, 68, 493–504. [Google Scholar] [CrossRef]
- Lacedonia, D.; Scioscia, G.; Soccio, P.; Conese, M.; Catucci, L.; Palladino, G.P.; Simone, F.; Quarato, C.M.I.; Di Gioia, S.; Rana, R.; et al. Downregulation of exosomal let-7d and miR-16 in idiopathic pulmonary fibrosis. BMC Pulm. Med. 2021, 21, 188. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.; Liang, J.; Guo, R.; Liu, N.; Noble, P.W.; Jiang, D. Comprehensive microRNA analysis in bleomycin-induced pulmonary fibrosis identifies multiple sites of molecular regulation. Physiol. Genom. 2011, 43, 479–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadota, T.; Fujita, Y.; Araya, J.; Watanabe, N.; Fujimoto, S.; Kawamoto, H.; Minagawa, S.; Hara, H.; Ohtsuka, T.; Yamamoto, Y.; et al. Human bronchial epithelial cell-derived extracellular vesicle therapy for pulmonary fibrosis via inhibition of TGF-beta-WNT crosstalk. Extracell Vesicles 2021, 10, e12124. [Google Scholar] [CrossRef]
- Stojanović, S.D.; Fuchs, M.; Fiedler, J.; Xiao, K.; Meinecke, A.; Just, A.; Pich, A.; Thum, T.; Kunz, M. Comprehensive Bioinformatics Identifies Key microRNA Players in ATG7-Deficient Lung Fibroblasts. Int. Mol. Sci. 2020, 21, 4126. [Google Scholar] [CrossRef] [PubMed]
- Dehmel, S.; Weiss, K.J.; El-Merhie, N.; Callegari, J.; Konrad, B.; Mutze, K.; Eickelberg, O.; Königshoff, M.; Krauss-Etschmann, S. microRNA Expression Profile of Purified Alveolar Epithelial Type II Cells. Genes 2022, 13, 1420. [Google Scholar] [CrossRef]
- Fang, L.; Ellims, A.H.; Moore, X.-L.; White, D.A.; Taylor, A.J.; Chin-Dusting, J.; Dart, A.M. Circulating microRNAs as biomarkers for diffuse myocardial fibrosis in patients with hypertrophic cardiomyopathy. Transl. Med. 2015, 13, 314. [Google Scholar] [CrossRef]
- Porrello, E.; Johnson, B.A.; Aurora, A.B.; Simpson, E.; Nam, Y.-J.; Matkovich, S.; Dorn, G.W., 2nd; Van Rooij, E.; Olson, E.N. MiR-15 family regulates postnatal mitotic arrest of cardiomyocytes. Circ. Res. 2011, 109, 670–679. [Google Scholar] [CrossRef] [Green Version]
- He, D.; Ruan, Z.-B.; Song, G.-X.; Chen, G.-C.; Wang, F.; Wang, M.-X.; Yuan, M.-K.; Zhu, L. miR-15a-5p regulates myocardial fibrosis in atrial fibrillation by targeting Smad7. PeerJ 2021, 9, e12686. [Google Scholar] [CrossRef]
- He, L.; Huang, C. MiR-19b and miR-16 cooperatively signaling target the regulator ADRA1A in Hypertensive heart disease. Biomed. Pharmacother. 2017, 91, 1178–1183. [Google Scholar] [CrossRef]
- Rawal, S.; Munasinghe, P.E.; Nagesh, P.T.; Lew, J.K.S.; Jones, G.T.; Williams, M.; Davis, P.; Bunton, D.; Galvin, I.F.; Manning, P.; et al. Down-regulation of miR-15a/b accelerates fibrotic remodelling in the Type 2 diabetic human and mouse heart. Clin. Sci. 2017, 131, 847–863. [Google Scholar] [CrossRef]
- Mohan, A.; Singh, R.S.; Kumari, M.; Garg, D.; Upadhyay, A.; Ecelbarger, C.M.; Tripathy, S.; Tiwari, S. Urinary Exosomal microRNA-451-5p Is a Potential Early Biomarker of Diabetic Nephropathy in Rats. PLoS ONE 2016, 11, e0154055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, H.; Cao, Q.; Liu, Q. MicroRNA-16 directly binds to DEC2 and inactivates the TLR4 signaling pathway to inhibit lupus nephritis-induced kidney tissue hyperplasia and mesangial cell proliferation. Int. Immunopharmacol. 2020, 88, 106859. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhang, D.; Liu, F.; Xiang, X.; Ling, G.; Xiao, L.; Liu, Y.; Zhu, X.; Zhan, M.; Yang, Y.; et al. Low-dose paclitaxel ameliorates fibrosis in the remnant kidney model by down-regulating miR-192. J. Pathol. 2011, 225, 364–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolcino, M.; Pelosi, A.; Fiore, P.F.; Patuzzo, G.; Tinazzi, E.; Lunardi, C.; Puccetti, A. Gene Profiling in Patients with Systemic Sclerosis Reveals the Presence of Oncogenic Gene Signatures. Front. Immunol. 2018, 9, 449. [Google Scholar] [CrossRef] [Green Version]
- Manesh, P.V.; Farazmand, A.; Gharibdoost, F.; Vanaki, N.; Mostafaei, S.; Kavosi, H.; Mahmoudi, M.B.; Mahmoudi, M. Downregulation of miR-542-3p Contributes to Apoptosis Resistance in Dermal Fibroblasts from Systemic Sclerosis Patients via Survivin Overexpression. Iran Allergy Asthma Immunol. 2019, 18, 173–181. [Google Scholar]
- Landgraf, P.; Rusu, M.; Sheridan, R.; Sewer, A.; Iovino, N.; Aravin, A.; Pfeffer, S.; Rice, A.; Kamphorst, A.O.; Landthaler, M.; et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 2007, 129, 1401–1414. [Google Scholar] [CrossRef] [Green Version]
- Gallant-Behm, C.L.; Piper, J.; Lynch, J.M.; Seto, A.G.; Hong, S.J.; Mustoe, T.A.; Maari, C.; Pestano, L.A.; Dalby, C.M.; Jackson, A.L.; et al. A MicroRNA-29 Mimic (Remlarsen) Represses Extracellular Matrix Expression and Fibroplasia in the Skin. Invest. Dermatol. 2019, 139, 1073–1081. [Google Scholar] [CrossRef]
- Qin, W.; Chung, A.C.; Huang, X.R.; Meng, X.-M.; Hui, D.; Yu, C.-M.; Sung, J.J.Y.; Lan, H.Y. TGF-beta/Smad3 signaling promotes renal fibrosis by inhibiting miR-29. Am. Soc. Nephrol. 2011, 22, 1462–1474. [Google Scholar] [CrossRef] [Green Version]
- Takeshita, F.; Patrawala, L.; Osaki, M.; Takahashi, R.-U.; Yamamoto, Y.; Kosaka, N.; Kawamata, M.; Kelnar, K.; Bader, A.G.; Brown, D.; et al. Systemic delivery of synthetic microRNA-16 inhibits the growth of metastatic prostate tumors via downregulation of multiple cell-cycle genes. Mol. Ther. 2010, 18, 181–187. [Google Scholar] [CrossRef]
- Hao, Z.; Fan, W.; Hao, J.; Wu, X.; Zeng, G.Q.; Zhang, L.J.; Nie, S.F.; Wang, X.D. Efficient delivery of micro RNA to bone-metastatic prostate tumors by using aptamer-conjugated atelocollagen in vitro and in vivo. Drug Deliv. 2016, 23, 874–881. [Google Scholar] [CrossRef]
- Thakral, S.; Ghoshal, K. miR-122 is a unique molecule with great potential in diagnosis, prognosis of liver disease, and therapy both as miRNA mimic and antimir. Curr. Gene Ther. 2015, 15, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Ivkovic, T.C.; Voss, G.; Cornella, H.; Ceder, Y. microRNAs as cancer therapeutics: A step closer to clinical application. Cancer Lett. 2017, 407, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Romano, G.; Acunzo, M.; Nana-Sinkam, P. microRNAs as Novel Therapeutics in Cancer. Cancers 2021, 13, 1526. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| miRNA | The First Nucleotide of a Mature miRNA Sequence | Seed Regions | The Last Nucleotide of the Seed Sequence | The Remaining Sequence of the Mature miRNA |
|---|---|---|---|---|
| hsa-miR-15a-5p | U | AGCAGCA | C | AUAAUGGUUUGUG |
| hsa-miR-15b-5p | U | AGCAGCA | C | AUCAUGGUUUACA |
| hsa-miR-15a-3p | C | CAGGCCA | U | AUUGUGCUGCCUCA |
| hsa-miR-15b-3p | C | GAAUCAU | U | AUUUGCUGCUCUA |
| hsa-miR-16 | U | AGCAGCA | C | GUAAAUAUUGGCG |
| hsa-miR-195 | U | AGCAGCA | C | AGAAAUAUUGGC |
| hsa-miR-424 | C | AGCAGCAAU | U | CAUGUUUUGAA |
| hsa-miR-497 | C | AGCAGCA | C | ACUGUGGUUUGU |
| hsa-miR-503 | U | AGCAGCG | G | GAACAGUUCUGCAG |
| miRNA | Disease Type | Expression | Target Gene | Biological Processes Affected | Study Model/Cell | Reference |
|---|---|---|---|---|---|---|
| miR-16 | Liver fibrosis | Upregulation | HGF and Smad7 | Affects the TGF-β/Smad signaling pathway and promotes liver fibrosis | QSG-7701 cells and Huh-7.5.1 cells | [19] |
| miR-16 | Liver fibrosis | Downregulation | Smad2/3 | Inhibits the TGF-β/Smad pathway | HSCs | [21] |
| miR-16 | Skin fibrosis | Downregulation | Smad3 | Inhibits the TGF-β/Smad pathway | Exosomes from HaCaT cells and HDFs | [22] |
| miR-16 | Systemic sclerosis | Downregulation | Notch2 | Inhibits Notch signaling and reduces the synthesis of collagen and other ECM components | HDFs and HSFs | [26] |
| miR-15a | Pulmonary fibrosis | Downregulation | YAP1/Twist | Modulates the lncRNA PFAR/YAP1/Twist axis to inhibit cell activation and ECM deposition | Primary lung fibroblasts | [16,27] |
| miR-16 | Pulmonary fibrosis | Downregulation | Rictor | Inhibits the mTORC2 signaling pathway, downregulates SPARC expression, and reduces ECM synthesis | Human fetal lung fibroblast (HFL-1) cells | [28] |
| miR-16 | Pulmonary fibrosis | Downregulation | TLR4 | Inhibits the TLR4/NF-kB pathway and the NLRP3 inflammasome signaling pathway | A549 cells and 293 T cells | [34] |
| miR-16 | Liver fibrosis | Downregulation | Bcl-2 | Affects the mitochondrial apoptosis pathway and induces apoptosis | HSCs | [37,38,39] |
| miR-16 | Liver fibrosis | Downregulation | LOXL1 | Inhibits Smad2/3 phosphorylation and inhibits the TGF-β/Smad pathway | LX2 cells | [40] |
| miR-16 | Liver fibrosis | Downregulation | Gα12 | Affects its binding to ATG12-5 and induces autophagy | HSCs | [41] |
| miR-16 | Pulmonary fibrosis | Downregulation | ND | ND | Serum exosomes of IPF patients | [42] |
| miR-16 | Pulmonary fibrosis | Downregulation | Bcl2, NF-kB, IL-1, JNK, Smad3/7, COL-I, and TGFβ-R | Affects apoptosis, Wnt signaling, TLR signaling, and the TGF-β/Smad pathway | Bleomycin mouse model of IPF | [43] |
| miR-16 | Pulmonary fibrosis | Downregulation | Wnt ligands (including Wnt10b and Wnt3a) | Attenuates the Wnt signaling pathway, inhibits fibroblast transformation, and induces epithelial senescence | HBEC EVs and mouse models of BLM-induced lung fibrosis | [44] |
| miR-16 | Pulmonary fibrosis | Downregulation | ATG5/7 | Affects mitochondrial dysfunction and cellar senescence | MRC-5 cells and endothelial cells | [45] |
| miR-16 | Pulmonary fibrosis | Downregulation | MAP2K1, MAP2K4, JUN, and BCL2 | Inhibits the TGF-β/Smad pathway | A549 cells and primary murine ATII cells | [46] |
| miR-15a | Cardiac fibrosis | Upregulation | ND | Promotes the MAPK and TGF-β/Smad pathways | Serum of diffuse myocardial fibrosis patients | [47] |
| miR-15a, miR-16 | Cardiac fibrosis | Upregulation | ND | Represses cardiomyocyte proliferation | C57BL/6 mice | [48] |
| miR-15a | Cardiac fibrosis | Upregulation | SMAD7 | Promotes the TGF-β/Smad pathway | Lipopolysaccharide (LPS)-stimulated H9C2 cells | [49] |
| miR-16 | Hypertensive heart disease | Upregulation | ADRA1A | Promotes cardiomyocyte apoptosis | Primary myocardial cells and H9C2 cells | [50] |
| miR-15a | Cardiac fibrosis | Downregulation | ND | Affects the TGF-β/Smad and p53 pathways | Human primary cardiac fibroblasts and Type 2 diabetic mice | [51] |
| miR-16 | Renal fibrosis | Downregulation | MMP-9 and IL-6 | Affects the fibrotic/inflammatory genes in the kidney tissue | Urinary exosomes (UEs) of diabetic rats | [52] |
| miR-16 | Renal fibrosis | Downregulation | DEC2 | Inhibits the TLR4 signaling pathway | SV40 MES-13 cells and Fcgamma receptor II-b-deficient (Fcgr2b−/−) mouse | [53] |
| miR-15a | Renal fibrosis | Downregulation | ND | Affects the TGF-β/Smad signaling pathway | Remnant kidney model and NRK-52E cells | [54] |
| miR-16 | Systemic sclerosis | Upregulation | ND | Induces apoptosis | Serum of systemic sclerosis patients | [55] |
| miR-16 | Systemic sclerosis | Downregulation | Survivin and p53 | Inhibits cell proliferation and promotes cell apoptosis | HSFs | [56] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wen, D.; Zhang, H.; Zhou, Y.; Wang, J. The Molecular Mechanisms and Function of miR-15a/16 Dysregulation in Fibrotic Diseases. Int. J. Mol. Sci. 2022, 23, 16041. https://doi.org/10.3390/ijms232416041
Wen D, Zhang H, Zhou Y, Wang J. The Molecular Mechanisms and Function of miR-15a/16 Dysregulation in Fibrotic Diseases. International Journal of Molecular Sciences. 2022; 23(24):16041. https://doi.org/10.3390/ijms232416041
Chicago/Turabian StyleWen, Dada, Huamin Zhang, Yutong Zhou, and Jie Wang. 2022. "The Molecular Mechanisms and Function of miR-15a/16 Dysregulation in Fibrotic Diseases" International Journal of Molecular Sciences 23, no. 24: 16041. https://doi.org/10.3390/ijms232416041