
Development of Transient Recombinant Expression and Affinity Chromatography Systems for Human Fibrinogen


 ,
,  , and
, and
Abstract
:1. Introduction
2. Results
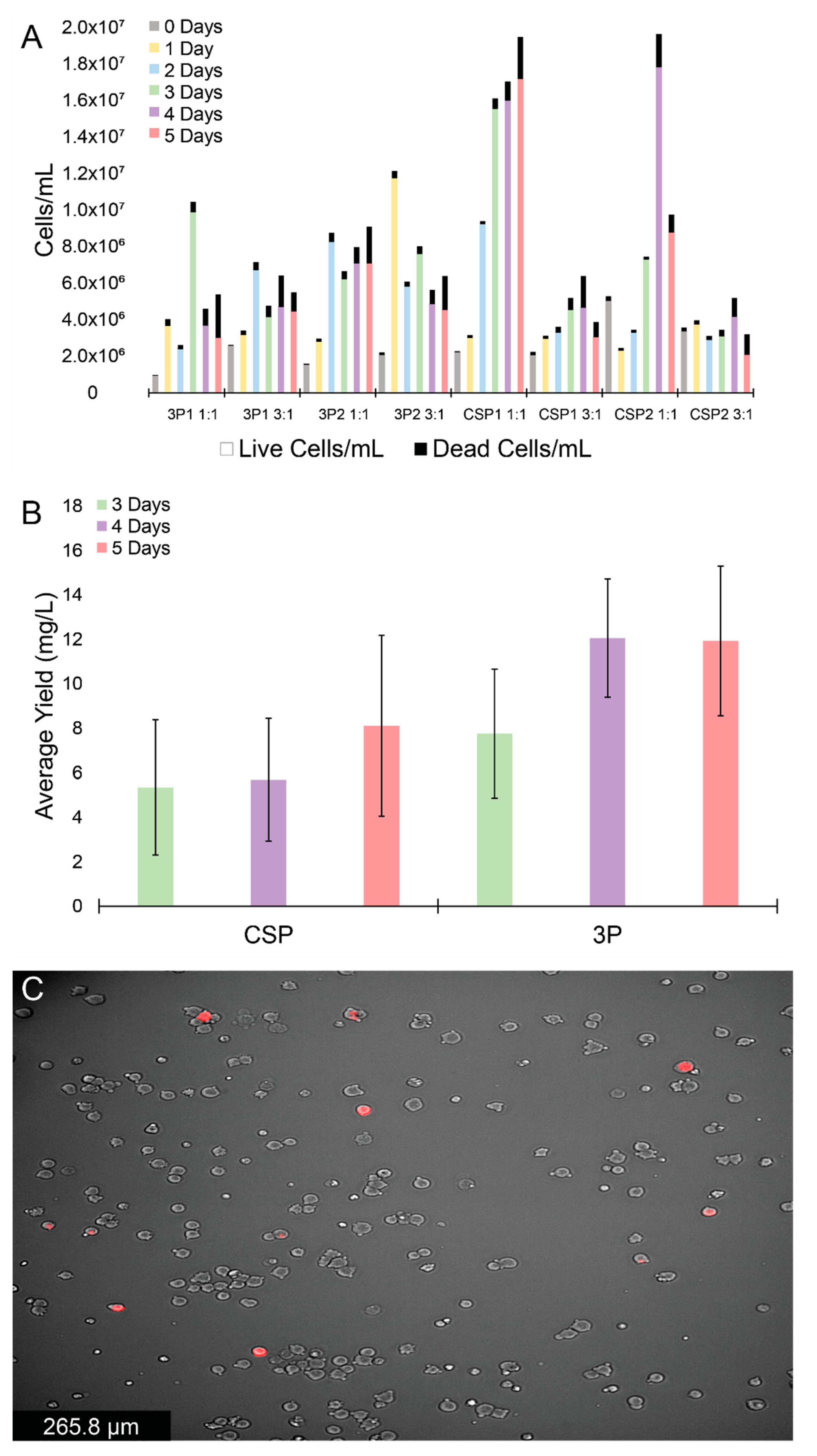
2.1. HEK Expression Tests
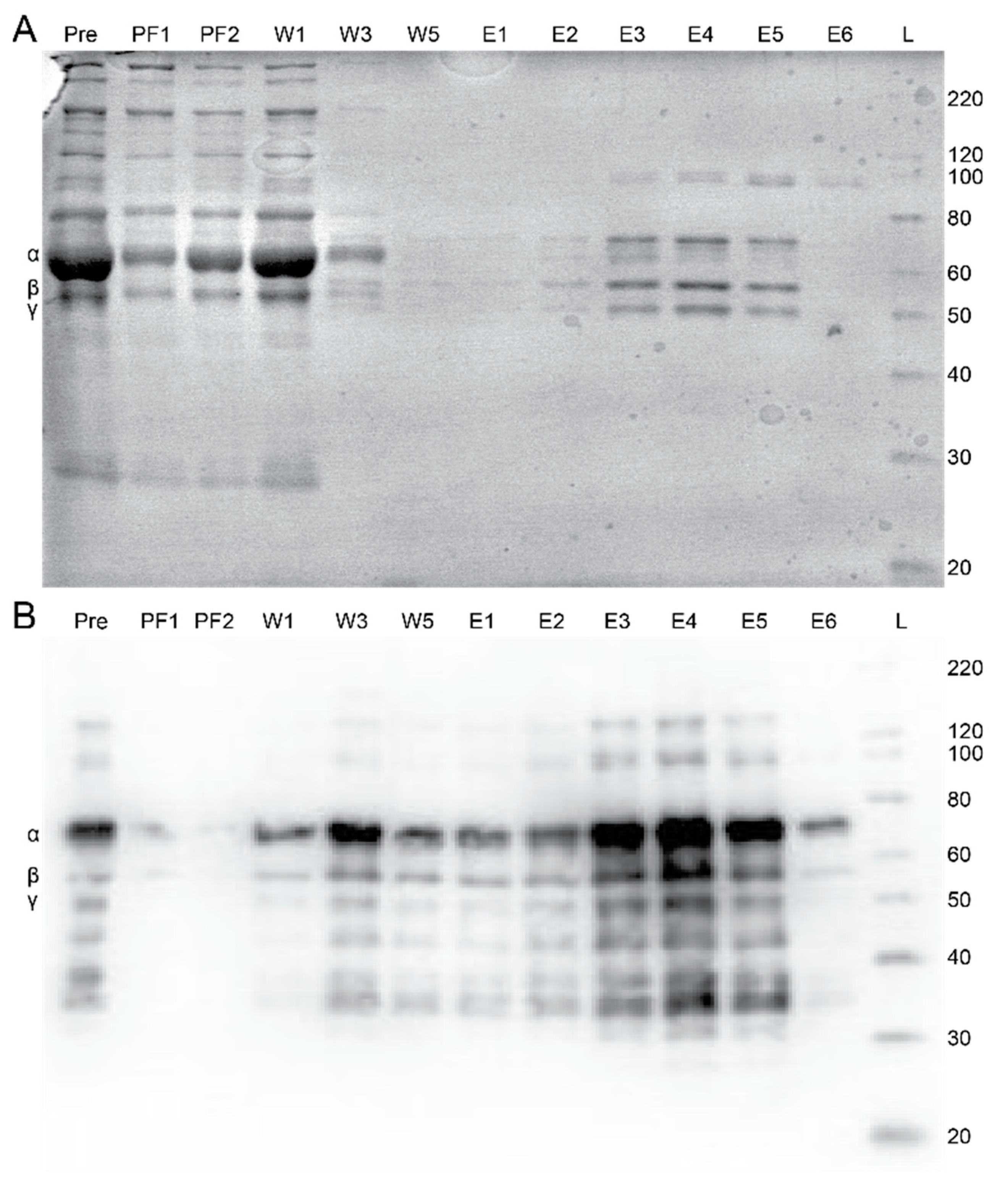
2.2. Peptide Affinity Purification
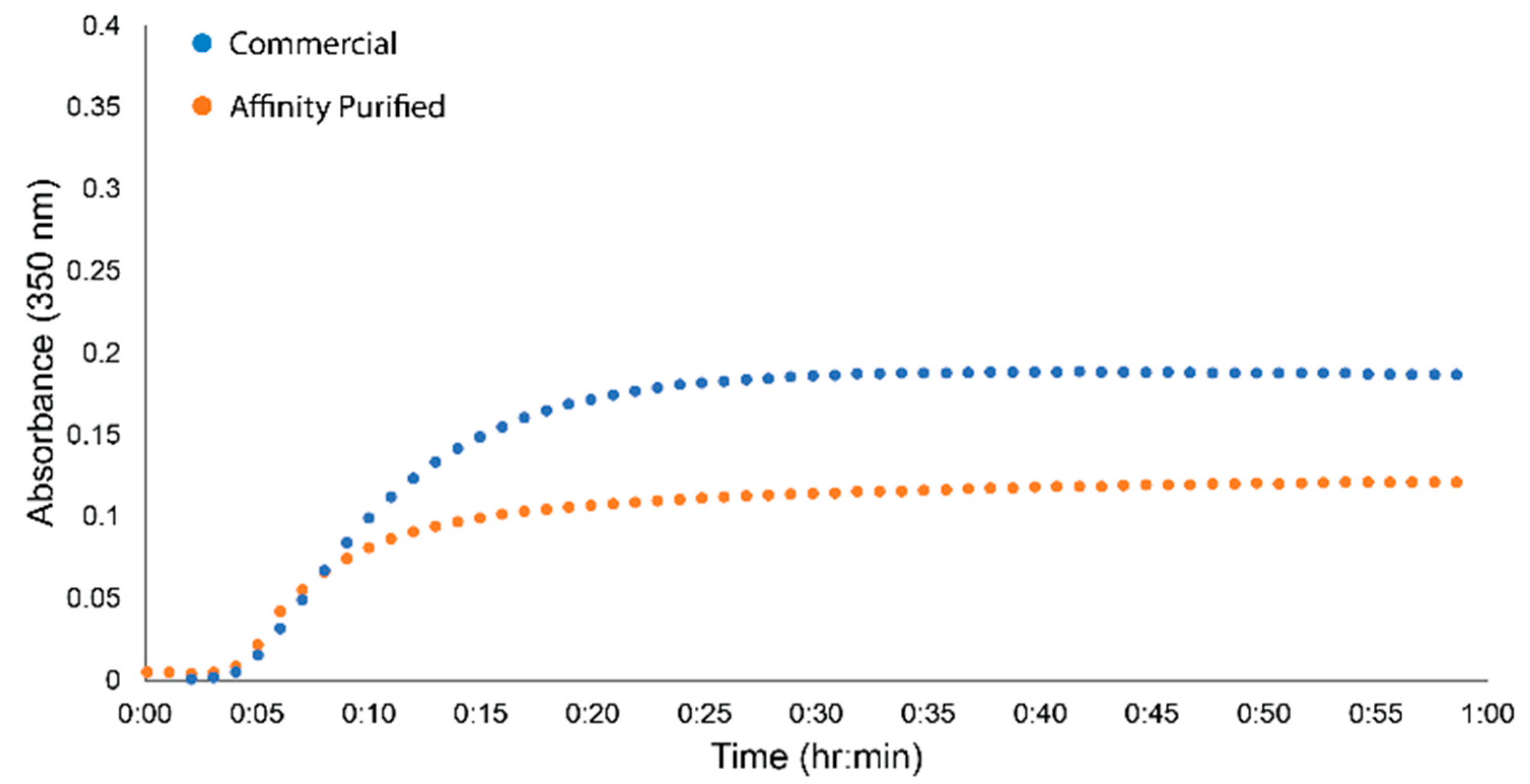
2.3. Plasma Purification
3. Discussion
4. Materials and Methods
4.1. Plasmid Descriptions
4.1.1. Three Plasmid (3P) System
4.1.2. Combined Single Plasmid (CSP) System
4.2. Mammalian Transfections
4.2.1. Polyethylenimine (PEI) Transfections
4.2.2. ExpiFectamine™ Transfections
4.3. Microscopy
Fluorescence Cell Image Processing
4.4. Assessment of Expression Systems
4.4.1. ELISA Assays
4.4.2. SDS-PAGE and Coomassie Gels
4.4.3. Western Blot
4.5. Development of Fibrinogen Peptide-Based Affinity Column
4.6. Purification of Fibrinogen with the Peptide-Based Affinity Column
4.6.1. Affinity Column Purification Tests
4.6.2. Purification of Fibrinogen from Plasma
4.6.3. Purification of Fibrinogen from Plasma with Ethanol Precipitation
4.6.4. Clottability and Turbidity Assays
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Simurda, T.; Asselta, R.; Zolkova, J.; Brunclikova, M.; Dobrotova, M.; Kolkova, Z.; Loderer, D.; Skornova, I.; Hudecek, J.; Lasabova, Z.; et al. Congenital Afibrinogenemia and Hypofibrinogenemia: Laboratory and Genetic Testing in Rare Bleeding Disorders with Life-Threatening Clinical Manifestations and Challenging Management. Diagnostics 2021, 11, 2140. [Google Scholar] [CrossRef]
- Neerman-Arbez, M.; Casini, A. Clinical Consequences and Molecular Bases of Low Fibrinogen Levels. Int. J. Mol. Sci. 2018, 19, 192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pieters, M.; Wolberg, A.S. Fibrinogen and Fibrin: An Illustrated Review. Res. Pract. Thromb. Haemost. 2019, 3, 161–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyu Ryu, J.; Rafalski, V.A.; Meyer-Franke, A.; Adams, R.A.; Poda, S.B.; Rios Coronado, P.E.; Østergaard Pedersen, L.; Menon, V.; Baeten, K.M.; Sikorski, S.L.; et al. Fibrin-Targeting Immunotherapy Protects Against Neuroinflammation and Neurodegeneration. Nat. Immunol. 2018, 19, 1212–1223. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.A.; Ryu, J.K.; Akassoglou, K. Fibrinogen in Neurological Diseases: Mechanisms, Imaging and Therapeutics. Nature reviews. Neuroscience 2018, 19, 283–301. [Google Scholar]
- Weisel, J.W.; Litvinov, R.I. Fibrin Formation, Structure and Properties. Sub-Cell. Biochem. 2017, 82, 405–456. [Google Scholar]
- Hudson, N.E.; Ding, F.; Bucay, I.; O’Brien, E.T.; Gorkun, O.V.; Superfine, R.; Lord, S.T.; Dokholyan, N.V.; Falvo, M.R. Submillisecond Elastic Recoil Reveals Molecular Origins of Fibrin Fiber Mechanics. Biophys. J. 2013, 104, 2671–2680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houser, J.R.; Hudson, N.E.; Ping, L.; O’Brien, E.T.; Superfine, R.; Lord, S.T.; Falvo, M.R. Evidence that αC Region is Origin of Low Modulus, High Extensibility, and Strain Stiffening in Fibrin Fibers. Biophys. J. 2010, 99, 3038–3047. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Jawerth, L.M.; Sparks, E.A.; Falvo, M.R.; Hantgan, R.R.; Superfine, R.; Lord, S.T.; Guthold, M. Fibrin Fibers have Extraordinary Extensibility and Elasticity. Science 2006, 313, 634. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Carlisle, C.R.; Sparks, E.A.; Guthold, M. The Mechanical Properties of Single Fibrin Fibers. J. Thromb. Haemost. 2010, 8, 1030–1036. [Google Scholar] [CrossRef] [Green Version]
- Krug, C.; Beer, A.; Hartmann, B.; Prein, C.; Clause-Schaumann, H.; Holzbach, T.; Aszodi, A.; Giunta, R.E.; Saller, M.M.; Volkmer, E. Fibrin Glue Displays Promising in Vitro Characteristics as a Potential Carrier of Adipose Progenitor Cells for Tissue Regeneration. J. Tissue Eng. Regen. Med. 2019, 13, 359–368. [Google Scholar] [CrossRef]
- Scull, G.; Fisher, M.B.; Brown, A.C. Fibrin-based Biomaterial Systems to Enhance Anterior Cruciate Ligament Healing. Med. Devices Sens. 2021, 4, e10147. [Google Scholar] [CrossRef]
- Shashank, B.; Bhushan, M. Injectable Platelet-Rich Fibrin (PRF): The Newest Biomaterial and its use in various Dermatological Conditions in our Practice: A Case Series. J. Cosmet. Dermatol. 2021, 20, 1421–1426. [Google Scholar] [CrossRef] [PubMed]
- Reiner, A.P. Fibrin Glue Increasingly Popular for Topical Surgical Hemostasis. Lab. Med. 1999, 30, 189–193. [Google Scholar] [CrossRef] [Green Version]
- Spotnitz, W.D. Fibrin Sealant: Past, Present, and Future: A Brief Review. World J. Surg. 2009, 34, 632–634. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.C.; Barker, T.H. Fibrin-Based Biomaterials: Modulation of Macroscopic Properties through Rational Design at the Molecular Level. Acta Biomater. 2014, 10, 1502–1514. [Google Scholar] [CrossRef] [Green Version]
- Bowley, S.R.; Okumura, N.; Lord, S.T. Impaired Protofibril Formation in Fibrinogen γN308K is due to Altered D:D and “A:A” Interactions. Biochemistry 2009, 48, 8656–8663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simurda, T.; Vilar, R.; Zolkova, J.; Ceznerova, E.; Kolkova, Z.; Loderer, D.; Neerman-Arbez, M.; Casini, A.; Brunclikova, M.; Skornova, I.; et al. A Novel Nonsense Mutation in FGB (C.1421G>A.; P.Trp474Ter) in the Beta Chain of Fibrinogen Causing Hypofibrinogenemia with Bleeding Phenotype. Biomedicines 2020, 8, 605. [Google Scholar] [CrossRef] [PubMed]
- Duga, S.; Asselta, R.; Santagostino, E.; Zeinali, S.; Simonic, T.; Malcovati, M.; Mannucci, P.M.; Tenchini, M.L. Missense Mutations in the Human Β Fibrinogen Gene Cause Congenital Afibrinogenemia by Impairing Fibrinogen Secretion. Blood 2000, 95, 1336–1341. [Google Scholar] [CrossRef]
- Simurda, T.; Snahnicanova, Z.; Loderer, D.; Sokol, J.; Stasko, J.; Lasabova, Z.; Kubisz, P. Fibrinogen Martin: A Novel Mutation in FGB (Gln180Stop) Causing Congenital Afibrinogenemia. Semin. Thromb. Hemost. 2016, 42, 455–458. [Google Scholar]
- Medved, L.; Weisel, J.W. Recommendations for Nomenclature on Fibrinogen and Fibrin. J. Thromb. Haemost. 2009, 7, 355–359. [Google Scholar] [CrossRef] [Green Version]
- Kollman, J.M.; Pandi, L.; Sawaya, M.R.; Riley, M.; Doolittle, R.F. Crystal Structure of Human Fibrinogen. Biochemistry 2009, 48, 3877–3886. [Google Scholar] [CrossRef] [PubMed]
- Laudano, A.P.; Doolittle, R.F. Influence of Calcium Ion on the Binding of Fibrin Amino Terminal Peptides to Fibrinogen. Science 1981, 212, 457–459. [Google Scholar] [CrossRef]
- Laudano, A.P.; Doolittle, R.F. Synthetic Peptide Derivatives that Bind to Fibrinogen and Prevent the Polymerization of Fibrin Monomers. Proc. Natl. Acad. Sci. USA 1978, 75, 3085–3089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spraggon, G.; Everse, S.J.; Doolittle, R.F. Crystal Structures of Fragment D from Human Fibrinogen and its Crosslinked Counterpart from Fibrin. Nature 1997, 389, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Mochalkin, I.; Veerapandian, L.; Riley, M.; Doolittle, R.F. Crystal Structure of Native Chicken Fibrinogen at 5.5-Å Resolution. Proc. Natl. Acad. Sci. USA 2000, 97, 3907–3912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burton, R.A.; Tsurupa, G.; Medved, L.; Tjandra, N. Identification of an Ordered Compact Structure within the Recombinant Bovine Fibrinogen αC-Domain Fragment by NMR. Biochemistry 2006, 45, 2257–2266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binnie, C.G.; Hettasch, J.M.; Strickland, E.; Lord, S.T. Characterization of Purified Recombinant Fibrinogen: Partial Phosphorylation of Fibrinopeptide A. Biochemistry 1993, 32, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, R.; Danishefsky, K.J. Studies on the Assembly and Secretion of Fibrinogen. J. Biol. Chem. 1991, 266, 6578–6585. [Google Scholar] [CrossRef]
- Piechocka, I.K.; Kurniawan, N.A.; Grimbergen, J.; Koopman, J.; Koenderink, G.H. Recombinant Fibrinogen Reveals the Differential Roles of Α- and Γ-Chain Cross-Linking and Molecular Heterogeneity in Fibrin Clot Strain-Stiffening. J. Thromb. Haemost. 2017, 15, 938–949. [Google Scholar] [CrossRef] [Green Version]
- Gorkun, O.V.; Veklich, Y.I.; Weisel, J.W.; Lord, S.T. The Conversion of Fibrinogen to Fibrin: Recombinant Fibrinogen Typifies Plasma Fibrinogen. Blood 1997, 89, 4407. [Google Scholar] [CrossRef] [PubMed]
- Hirashima, M.; Imamura, T.; Yano, K.; Kawamura, R.; Meta, A.; Tokieda, Y.; Nakashima, T. High-Level Expression and Preparation of Recombinant Human Fibrinogen as Biopharmaceuticals. J. Biochem. 2016, 159, 261–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stabenfeldt, S.E.; Gossett, J.J.; Barker, T.H. Building Better Fibrin Knob Mimics: An Investigation of Synthetic Fibrin Knob Peptide Structures in Solution and their Dynamic Binding with Fibrinogen/Fibrin Holes. Blood 2010, 116, 1352–1359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorkun, O.V.; Henschen-Edman, A.H.; Ping, L.F.; Lord, S.T. Analysis of A Alpha 251 Fibrinogen: The Alpha C Domain has a Role in Polymerization, Albeit More Subtle than Anticipated from the Analogous Proteolytic Fragment X. Biochemistry 1998, 37, 15434. [Google Scholar] [CrossRef]
- Va, S.; Liu, C.Y.; Cisneros, I.; Jones, M.; Van Le, S.; Zmuda, J.; Rogers, J. Expi293 Expression System 2016.
- Fang, X.T.; Sehlin, D.; Lannfelt, L.; Syvänen, S.; Hultqvist, G. Efficient and Inexpensive Transient Expression of Multispecific Multivalent Antibodies in Expi293 Cells. Biol. Proced. Online 2017, 19, 11. [Google Scholar] [CrossRef] [PubMed]
- Brennan, M. Fibrin Glue. Blood Rev. 1991, 5, 240–244. [Google Scholar] [CrossRef]
- Liu, C.; Cheng, H.; Chang, L. A New Feasible Method for Fibrinogen Purification Based on the Affinity of Staphylococcus Aureus Clumping Factor A to Fibrinogen. Protein Expr. Purif. 2008, 61, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Kuyas, C.; Haeberli, A.; Walder, P.; Straub, P.W. Isolation of Human Fibrinogen and its Derivatives by Affinity Chromatography on Gly-Pro-Arg-Pro-Lys-Fractogel. Thromb. Haemost. 1990, 63, 439–444. [Google Scholar] [CrossRef]
- Blombäck, B.; Blombäck, M.; Paul, K.; Theorell, H.; Thorell, B. Purification of Bovine and Human Fibrinogen. Acta Chem. Scand. 1956, 10, 147. [Google Scholar] [CrossRef] [Green Version]
- Okude, M.; Yamanaka, A.; Morimoto, Y.; Akihama, S. Sialic Acid in Fibrinogen: Effects of Sialic Acid on Fibrinogen-Fibrin Conversion by Thrombin and Properties of Asialofibrin Clot. Biol. Pharm. Bull. 1993, 16, 448–452. [Google Scholar] [CrossRef] [Green Version]
- Riedelová-Reicheltová, Z.; Kotlín, R.; Suttnar, J.; Geierová, V.; Riedel, T.; Májek, P.; Dyr, J.E. A Novel Natural Mutation AαPhe98Ile in the Fibrinogen Coiled-Coil Affects Fibrinogen Function. Thromb. Haemost. 2014, 111, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Castaman, G.; Giacomelli, S.H.; Biasoli, C.; Contino, L.; Radossi, P. Risk of Bleeding and Thrombosis in Inherited Qualitative Fibrinogen Disorders. Eur. J. Haematol. 2019, 103, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Simurda, T.; Zolkova, J.; Kolkova, Z.; Loderer, D.; Dobrotova, M.; Skornova, I.; Brunclíkova, M.; Grendar, M.; Lasabova, Z.; Stasko, J.; et al. Comparison of Clinical Phenotype with Genetic and Laboratory Results in 31 Patients with Congenital Dysfibrinogenemia in Northern Slovakia. Int. J. Hematol. 2020, 111, 795–802. [Google Scholar] [CrossRef] [PubMed]
- Gibson, D.G.; Young, L.; Chuang, R.; Venter, J.C.; Hutchison, C.A.; Smith, H.O. Enzymatic Assembly of DNA Molecules Up to several Hundred Kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef] [PubMed]
- Bzymek, M.; Lovett, S.T. Instability of Repetitive DNA Sequences: The Role of Replication in Multiple Mechanisms. Proc. Natl. Acad. Sci. USA 2001, 98, 8319–8325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Doolittle, R.F.; Schubert, D.; Schwartz, S.A. Amino Acid Sequence Studies on Artiodactyl Fibrinopeptides: I. Dromedary Camel, Mule Deer, and Cape Buffalo. Arch. Biochem. Biophys. 1967, 118, 456–467. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fibrinogen Source | Clottability (%) |
|---|---|
| Peak 1 fgn (commercial) 1 | 84 ± 3 |
| Plasma, Purified by GPRPFPAWK column | 86 ± 5 |
| Plasma, Purified by ethanol precipitation | 80 ± 6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Popovic, G.; Kirby, N.C.; Dement, T.C.; Peterson, K.M.; Daub, C.E.; Belcher, H.A.; Guthold, M.; Offenbacher, A.R.; Hudson, N.E. Development of Transient Recombinant Expression and Affinity Chromatography Systems for Human Fibrinogen. Int. J. Mol. Sci. 2022, 23, 1054. https://doi.org/10.3390/ijms23031054
Popovic G, Kirby NC, Dement TC, Peterson KM, Daub CE, Belcher HA, Guthold M, Offenbacher AR, Hudson NE. Development of Transient Recombinant Expression and Affinity Chromatography Systems for Human Fibrinogen. International Journal of Molecular Sciences. 2022; 23(3):1054. https://doi.org/10.3390/ijms23031054
Chicago/Turabian StylePopovic, Grega, Nicholas C. Kirby, Taylor C. Dement, Kristine M. Peterson, Caroline E. Daub, Heather A. Belcher, Martin Guthold, Adam R. Offenbacher, and Nathan E. Hudson. 2022. "Development of Transient Recombinant Expression and Affinity Chromatography Systems for Human Fibrinogen" International Journal of Molecular Sciences 23, no. 3: 1054. https://doi.org/10.3390/ijms23031054