
Mitochondrial Processing Peptidases—Structure, Function and the Role in Human Diseases

 , , , , , , and
, , , , , , and
Abstract
1. Introduction
2. Mitochondrial Processing Peptidase


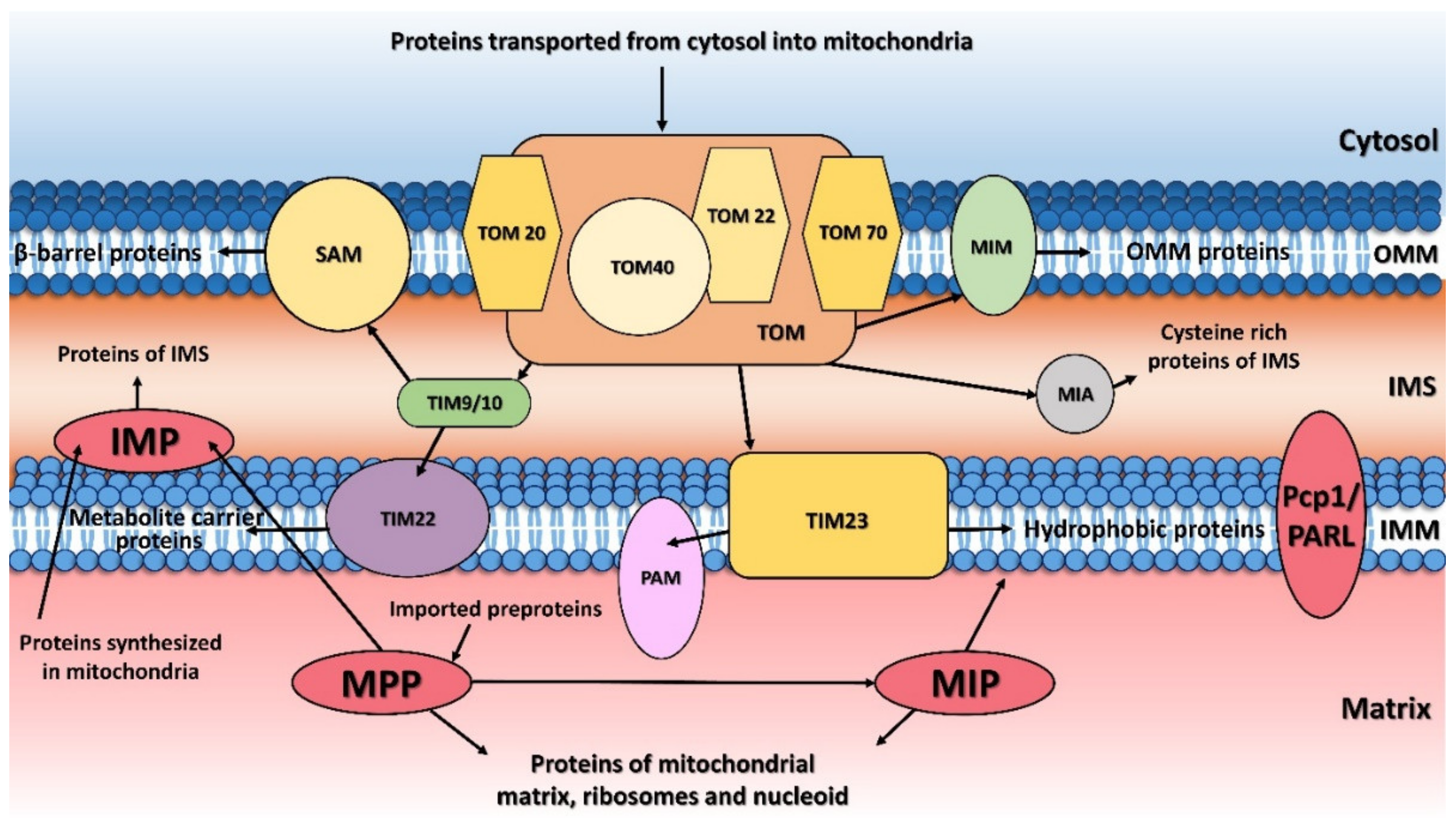{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Processing Peptidase | Protein Variant | Disease, Symptoms | Ref. |
|---|---|---|---|
| PMPCA | Homozygous mutation: c.1129G>A (p.Ala377Thr) Heterozygous mutations: c.287C>T (p.Ser96Leu) with c.1543G>A (p.Gly515Arg) | SCAR2 with non- or slowly progressive cerebellar ataxia and developmental delay | [38] |
| Homozygous mutation: c.766G>A (p.Val256Met) | slowly progressive SCAR2 without intellectual disability | [47] | |
| Heterozygous mutation: c.677C>T (p.Arg223Cys) with c.853del (p.Asp285Ilefs*16) | SCAR2 with progressive cerebellar ataxia and onset in infancy | [18] | |
| Heterozygous mutations: c.1066G>A (p.Gly356Ser) with c.1129G>A (p.Ala377Thr) | SCAR2 with progressive, extensive brain atrophy, muscle weakness, visual impairment, respiratory defects | [48] | |
| Homozygous mutation: c.553C>T (p.Arg185Thr) | SCAR2 with psychomotor delay | [46] | |
| PMPCB | Heterozygous mutations: c.523C>T (p.Arg175Cys) with c.601G>C (p.Ala201Pro); c.524G>A (p.Arg175His) with c.530T>G (p.Val177Gly) Homozygous mutation: c.1265T>C (p.Ile422Thr) | Prominent cerebellar atrophy in early childhood | [50] |
| IMMP2L | Duplication: 46,XY,dup(7)(q22.1-q31.1) | GTS/TS | [15] |
| Deletions ranged from ~49 kb to ~337 kb | Neurological disorders (ADHD, GTS/TS, OCD, ASD, Asperger′s syndrome, schizophrenia and developmental delay) | [53,54,55,56] | |
| Base pair change | Autism | [57] | |
| Copy number variation | Alzheimer′s disease | [58] | |
| Downregulation | Prostate cancer | [59] | |
| MIP | Homozygous SNV: p.K343E Heterozygous SNVs: p.L582R with p.L71Q; p.E602* with p.L306 and p.H512D with 1.4-Mb deletion of 13q12.12 | LVNC and developmental delay, seizures, hypotonia | [60] |
| Heterozygous mutation: c.916C > T (p.Leu306Phe) with c.1970 + 2 T>A (p.Ala658Lysfs*38) | Developmental delay, hypotonia and intellectual disability | [61] | |
| Hypomethylation | Metabolic syndrome | [62] | |
| Downregulation | Prostate cancer | [59] | |
| PARL | Reduced levels | Type 2 diabetes | [19] |
| Leu262Val polymorphism | Increased plasma insulin concentration | [63] | |
| Mutation: c.230G>A (p.Ser77Asn) | Parkinson′s disease | [64] |
3. Mitochondrial Inner Membrane Peptidase
4. Mitochondrial Intermediate Peptidase
5. Mitochondrial Rhomboid Protease

6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ADHD | attention-deficit hyperactivity disorder |
| AIF | apoptosis-inducing factor |
| Arg5 | N-acetyl-gamma-glutamyl-phosphate reductase, yeast |
| Arg6 | acetylglutamate kinase, yeast |
| ASD | autism spectrum disorder |
| AUTS1 | Autism, Susceptibility To, 1 locus |
| Ccp1 | cytochrome c peroxidase, yeast |
| CLPB | caseinolytic peptidase B |
| Cox2 | cytochrome c oxidase subunit 2 |
| Cox4 | cytochrome c oxidase subunit 4 |
| Cyb2 | cytochrome b2 |
| Cyt1 | cytochrome c1 |
| Fe-S | iron-sulfur |
| FRDA | Friedreich’s ataxia |
| FXN | frataxin |
| GPD2 | glycerol-3-phosphate dehydrogenase 2 |
| GRL | glycine-rich loop |
| GTS/TS | Gills de la Tourette syndrome/Tourette’s syndrome |
| HCC | hepatocellular carcinoma |
| HTRA2 | high temperature requirement factor A2 |
| HUVECs | human umbilical vein endothelial cells |
| IMM | inner mitochondrial membrane |
| IMMP1L | inner mitochondrial membrane peptidase 1-like, human |
| IMMP2L | inner mitochondrial membrane peptidase 2-like, human |
| IMP | inner membrane peptidase |
| Imp1 | mitochondrial inner membrane protease subunit 1, yeast |
| Imp2 | mitochondrial inner membrane protease subunit 2, yeast |
| LRR | leucine-rich repeat |
| LVNC | left ventricular non-compaction |
| m-AAA | mitochondrial matrix-exposed ATPase associated with diverse cellular activities |
| MCL-1 | induced myeloid leukemia cell differentiation protein 1 |
| Mcr1 | mitochondrial cytochrome b reductase, yeast |
| Mgm1 | mitochondrial genome maintenance 1, yeast |
| MIP | mitochondrial intermediate peptidase |
| MPP | mitochondrial processing peptidase |
| MPPα | mitochondrial processing peptidase subunit alpha |
| MPPβ | mitochondrial processing peptidase subunit beta |
| MrpS28 | mitochondrial 37S ribosomal protein subunit 28, yeast |
| MTS | mitochondrial targeting sequence |
| NEM | N-ethylmaleimide |
| NO | nitric oxide |
| OCD | obsessive-compulsive disorder |
| OPA1 | optic atrophy protein 1 |
| OXA1L | oxidase assembly1-like protein, human |
| OXPHOS | oxidative phosphorylation |
| PARL | presenilin-associated rhomboid like |
| Parkin | E3 ubiquitin-protein ligase protein |
| Pcp1 | processing of cytochrome c peroxidase protein 1 |
| PD | Parkinson’s disease |
| PGAM5 | phosphoglycerate mutase family member 5 |
| PINK1 | PTEN-induced putative kinase 1 |
| PMPCA | mitochondrial processing peptidase subunit alpha |
| PMPCB | mitochondrial processing peptidase subunit beta |
| Rim1 | single-stranded DNA-binding protein Rim1 (Replication in Mitochondria), yeast |
| ROS | reactive oxygen species |
| SCAR2 | autosomal recessive spinocerebellar ataxia type 2 |
| shRNA | short hairpin RNA |
| siRNA | small interfering RNA |
| Smac | second mitochondria-derived activator of caspase, human |
| SNVs | single nucleotide variants |
| Som1 | Sorting mitochondrial protein 1, yeast |
| STARD7 | mitochondrial StAR-related lipid transfer protein 7, human |
| T2D | type 2 diabetes |
| TIM | translocase of the inner mitochondrial membrane |
| TOM | translocase of the outer mitochondrial membrane |
| TRX2 | thioredoxin 2 |
| TTC19 | tetratricopeptide repeat protein 19 |
| Tuf1 | mitochondrial translation elongation factor Tu, yeast |
References
- Chacinska, A.; Koehler, C.M.; Milenkovic, D.; Lithgow, T.; Pfanner, N. Importing Mitochondrial Proteins: Machineries and Mechanisms. Cell 2009, 138, 628–644. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, N.; Pfanner, N. Mitochondrial Machineries for Protein Import and Assembly. Annu. Rev. Biochem. 2017, 86, 685–714. [Google Scholar] [CrossRef] [PubMed]
- Kulawiak, B.; Höpker, J.; Gebert, M.; Guiard, B.; Wiedemann, N.; Gebert, N. The mitochondrial protein import machinery has multiple connections to the respiratory chain. Biochim. Biophys. Acta BBA Bioenerg. 2013, 1827, 612–626. [Google Scholar] [CrossRef] [PubMed]
- Shiota, T.; Imai, K.; Qiu, J.; Hewitt, V.L.; Tan, K.; Shen, H.-H.; Sakiyama, N.; Fukasawa, Y.; Hayat, S.; Kamiya, M.; et al. Molecular architecture of the active mitochondrial protein gate. Science 2015, 349, 1544–1548. [Google Scholar] [CrossRef]
- Straub, S.P.; Stiller, S.B.; Wiedemann, N.; Pfanner, N. Dynamic organization of the mitochondrial protein import machinery. Biol. Chem. 2016, 397, 1097–1114. [Google Scholar] [CrossRef]
- Moulin, C.; Caumont-Sarcos, A.; Ieva, R. Mitochondrial presequence import: Multiple regulatory knobs fine-tune mitochondrial biogenesis and homeostasis. Biochim. Biophys. Acta BBA Bioenerg. 2019, 1866, 930–944. [Google Scholar] [CrossRef]
- Nicolas, E.; Tricarico, R.; Savage, M.; Golemis, E.A.; Hall, M.J. Disease-Associated Genetic Variation in Human Mitochondrial Protein Import. Am. J. Hum. Genet. 2019, 104, 784–801. [Google Scholar] [CrossRef]
- Priesnitz, C.; Becker, T. Pathways to balance mitochondrial translation and protein import. Genes Dev. 2018, 32, 1285–1296. [Google Scholar] [CrossRef]
- Poveda-Huertes, D.; Mulica, P.; Vögtle, F.N. The versatility of the mitochondrial presequence processing machinery: Cleavage, quality control and turnover. Cell Tissue Res. 2017, 367, 73–81. [Google Scholar] [CrossRef]
- Becker, T.; Vögtle, F.-N.; Stojanovski, D.; Meisinger, C. Sorting and assembly of mitochondrial outer membrane proteins. Biochim. Biophys. Acta BBA Bioenerg. 2008, 1777, 557–563. [Google Scholar] [CrossRef]
- Gakh, O.; Cavadini, P.; Isaya, G. Mitochondrial processing peptidases. Biochim. Biophys. Acta BBA Bioenerg. 2002, 1592, 63–77. [Google Scholar] [CrossRef]
- Kutejová, E.; Kučera, T.; Matušková, A.; Janata, J. Mitochondrial Processing Peptidase. In Handbook of Proteolytic Enzymes, 3rd ed.; Neil, D., Rawlings, G.S., Eds.; Academic Press: Cambridge, MA, USA, 2013; pp. 1435–1442. [Google Scholar] [CrossRef]
- Spinazzi, M.; De Strooper, B. PARL: The mitochondrial rhomboid protease. Semin. Cell Dev. Biol. 2016, 60, 19–28. [Google Scholar] [CrossRef]
- Petek, E.; Schwarzbraun, T.; Noor, A.; Patel, M.; Nakabayashi, K.; Choufani, S.; Windpassinger, C.; Stamenkovic, M.; Robertson, M.M.; Aschauer, H.N.; et al. Molecular and genomic studies of IMMP2L and mutation screening in autism and Tourette syndrome. Mol. Genet. Genom. 2006, 277, 71–81. [Google Scholar] [CrossRef]
- Petek, E.; Windpassinger, C.; Vincent, J.B.; Cheung, J.; Boright, A.P.; Scherer, S.; Kroisel, P.M.; Wagner, K. Disruption of a Novel Gene (IMMP2L) by a Breakpoint in 7q31 Associated with Tourette Syndrome. Am. J. Hum. Genet. 2001, 68, 848–858. [Google Scholar] [CrossRef]
- Whatley, S.A.; Curti, D.; Marchbanks, R.M. Mitochondrial involvement in schizophrenia and other functional psychoses. Neurochem. Res. 1996, 21, 995–1004. [Google Scholar] [CrossRef]
- Santos, R.; Lefevre, S.D.; Sliwa, D.; Seguin, A.; Camadro, J.-M.; Lesuisse, E. Friedreich Ataxia: Molecular Mechanisms, Redox Considerations, and Therapeutic Opportunities. Antioxid. Redox Signal. 2010, 13, 651–690. [Google Scholar] [CrossRef]
- Takahashi, Y.; Kubota, M.; Kosaki, R.; Kosaki, K.; Ishiguro, A. A severe form of autosomal recessive spinocerebellar ataxia associated with novel PMPCA variants. Brain Dev. 2021, 43, 464–469. [Google Scholar] [CrossRef]
- Civitarese, A.E.; MacLean, P.; Carling, S.; Kerr-Bayles, L.; McMillan, R.P.; Pierce, A.; Becker, T.C.; Moro, C.; Finlayson, J.; Lefort, N.; et al. Regulation of Skeletal Muscle Oxidative Capacity and Insulin Signaling by the Mitochondrial Rhomboid Protease PARL. Cell Metab. 2010, 11, 412–426. [Google Scholar] [CrossRef]
- Taylor, A.B.; Smith, B.S.; Kitada, S.; Kojima, K.; Miyaura, H.; Otwinowski, Z.; Ito, A.; Deisenhofer, J. Crystal Structures of Mitochondrial Processing Peptidase Reveal the Mode for Specific Cleavage of Import Signal Sequences. Structure 2001, 9, 615–625. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [PubMed]
- Dvořáková-Holá, K.; Matušková, A.; Kubala, M.; Otyepka, M.; Kucera, T.; Večeř, J.; Herman, P.; Parkhomenko, N.; Kutejova, E.; Janata, J. Glycine-Rich Loop of Mitochondrial Processing Peptidase α-Subunit Is Responsible for Substrate Recognition by a Mechanism Analogous to Mitochondrial Receptor Tom20. J. Mol. Biol. 2010, 396, 1197–1210. [Google Scholar] [CrossRef] [PubMed]
- Nagao, Y.; Kitada, S.; Kojima, K.; Toh, H.; Kuhara, S.; Ogishima, T.; Ito, A. Glycine-rich Region of Mitochondrial Processing Peptidase α-Subunit Is Essential for Binding and Cleavage of the Precursor Proteins. J. Biol. Chem. 2000, 275, 34552–34556. [Google Scholar] [CrossRef] [PubMed]
- Kučera, T.; Otyepka, M.; Matušková, A.; Samad, A.; Kutejová, E.; Janata, J. A Computational Study of the Glycine-Rich Loop of Mitochondrial Processing Peptidase. PLoS ONE 2013, 8, e74518. [Google Scholar] [CrossRef]
- Roise, D.; Schatz, G. Mitochondrial presequences. J. Biol. Chem. 1988, 263, 4509–4511. [Google Scholar] [CrossRef]
- Roise, D.; Theiler, F.; Horvath, S.J.; Tomich, J.M.; Richards, J.H.; Allison, D.S.; Schatz, G. Amphiphilicity is essential for mitochondrial presequence function. EMBO J. 1988, 7, 649–653. [Google Scholar] [CrossRef] [PubMed]
- Gavel, Y.; Von Heijne, G. Cleavage-site motifs in mitochondrial targeting peptides. Protein Eng. Des. Sel. 1990, 4, 33–37. [Google Scholar] [CrossRef]
- Friedl, J.; Knopp, M.R.; Groh, C.; Paz, E.; Gould, S.B.; Herrmann, J.M.; Boos, F. More than just a ticket canceller: The mitochondrial processing peptidase tailors complex precursor proteins at internal cleavage sites. Mol. Biol. Cell 2020, 31, 2657–2668. [Google Scholar] [CrossRef]
- Hendriks, I.A.; Lyon, D.; Su, D.; Skotte, N.H.; Daniel, J.A.; Jensen, L.J.; Nielsen, M.L. Site-specific characterization of endogenous SUMOylation across species and organs. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef]
- Hendriks, I.A.; Lyon, D.; Young, C.; Jensen, L.J.; Vertegaal, A.C.O.; Nielsen, M.L. Site-specific mapping of the human SUMO proteome reveals co-modification with phosphorylation. Nat. Struct. Mol. Biol. 2017, 24, 325–336. [Google Scholar] [CrossRef]
- Chen, C.; Wang, K.; Zhang, H.; Zhou, H.J.; Chen, Y.; Min, W. A Unique SUMO-Interacting Motif of Trx2 Is Critical for Its Mitochondrial Presequence Processing and Anti-oxidant Activity. Front. Physiol. 2019, 10, 1089. [Google Scholar] [CrossRef] [PubMed]
- Pollock, R.A.; Hartl, F.U.; Cheng, M.Y.; Ostermann, J.; Horwich, A.; Neupert, W. The processing peptidase of yeast mitochondria: The two co-operating components MPP and PEP are structurally related. EMBO J. 1988, 7, 3493–3500. [Google Scholar] [CrossRef] [PubMed]
- Witte, C.; Jensen, R.E.; Yaffe, M.P.; Schatz, G. MAS1, a gene essential for yeast mitochondrial assembly, encodes a subunit of the mitochondrial processing protease. EMBO J. 1988, 7, 1439–1447. [Google Scholar] [CrossRef] [PubMed]
- Lightowlers, R.N.; Taylor, R.W.; Turnbull, D.M. Mutations causing mitochondrial disease: What is new and what challenges remain? Science 2015, 349, 1494–1499. [Google Scholar] [CrossRef] [PubMed]
- Park, C.B.; Larsson, N.-G. Mitochondrial DNA mutations in disease and aging. J. Cell Biol. 2011, 193, 809–818. [Google Scholar] [CrossRef]
- Shoubridge, E.A. Nuclear genetic defects of oxidative phosphorylation. Hum. Mol. Genet. 2001, 10, 2277–2284. [Google Scholar] [CrossRef]
- Jobling, R.K.; Assoum, M.; Gakh, O.; Blaser, S.; Raiman, J.A.; Mignot, C.; Roze, E.; Durr, A.; Brice, A.; Lévy, N.; et al. PMPCAmutations cause abnormal mitochondrial protein processing in patients with non-progressive cerebellar ataxia. Brain 2015, 138, 1505–1517. [Google Scholar] [CrossRef]
- Das, D.; Patra, S.; Bridwell-Rabb, J.; Barondeau, D.P. Mechanism of frataxin “bypass” in human iron–sulfur cluster biosynthesis with implications for Friedreich’s ataxia. J. Biol. Chem. 2019, 294, 9276–9284. [Google Scholar] [CrossRef]
- Kumar, V.; Kitaeff, N.; Hampton, M.B.; Cannell, M.B.; Winterbourn, C.C. Reversible oxidation of mitochondrial peroxiredoxin 3 in mouse heart subjected to ischemia and reperfusion. FEBS Lett. 2009, 583, 997–1000. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Koutnikova, H.; Campuzano, V.; Koenig, M. Maturation of wild-type and mutated frataxin by the mitochondrial processing peptidase. Hum. Mol. Genet. 1998, 7, 1485–1489. [Google Scholar] [CrossRef]
- Isaya, G.; Miklos, D.; A Rollins, R. MIP1, a new yeast gene homologous to the rat mitochondrial intermediate peptidase gene, is required for oxidative metabolism in Saccharomyces cerevisiae. Mol. Cell. Biol. 1994, 14, 5603–5616. [Google Scholar] [CrossRef]
- Muthuswamy, S.; Agarwal, S. Friedreich Ataxia. Neurologist 2015, 20, 51–55. [Google Scholar] [CrossRef]
- Lynch, D.R.; Farmer, J.M.; Balcer, L.J.; Wilson, R.B. Friedreich Ataxia. Arch. Neurol. 2002, 59, 743–747. [Google Scholar] [CrossRef]
- Rubegni, A.; Pasquariello, R.; Dosi, C.; Astrea, G.; Canapicchi, R.; Santorelli, F.M.; Nesti, C. Teaching NeuroImages: Leigh-like features expand the picture of PMPCA-related disorders. Neurologist 2019, 92, e168–e169. [Google Scholar] [CrossRef]
- Choquet, K.; Zurita-Rendón, O.; La Piana, R.; Yang, S.; Dicaire, M.-J.; Boycott, K.M.; Majewski, J.; Shoubridge, E.A.; Brais, B.; Tétreault, M.; et al. Autosomal recessive cerebellar ataxia caused by a homozygous mutation in PMPCA. Brain 2015, 139, e19. [Google Scholar] [CrossRef]
- Joshi, M.; Anselm, I.; Shi, J.; Bale, T.A.; Towne, M.; Schmitz-Abe, K.; Crowley, L.; Giani, F.C.; Kazerounian, S.; Markianos, K.; et al. Mutations in the substrate binding glycine-rich loop of the mitochondrial processing peptidase-α protein (PMPCA) cause a severe mitochondrial disease. Mol. Case Stud. 2016, 2, a000786. [Google Scholar] [CrossRef]
- Ondrovičová, G.; Liu, T.; Singh, K.; Tian, B.; Li, H.; Gakh, O.; Perečko, D.; Janata, J.; Granot, Z.; Orly, J.; et al. Cleavage Site Selection within a Folded Substrate by the ATP-dependent Lon Protease. J. Biol. Chem. 2005, 280, 25103–25110. [Google Scholar] [CrossRef]
- Vögtle, F.-N.; Brändl, B.; Larson, A.; Pendziwiat, M.; Friederich, M.W.; White, S.M.; Basinger, A.; Kücükköse, C.; Muhle, H.; Jähn, J.A.; et al. Mutations in PMPCB Encoding the Catalytic Subunit of the Mitochondrial Presequence Protease Cause Neurodegeneration in Early Childhood. Am. J. Hum. Genet. 2018, 102, 557–573. [Google Scholar] [CrossRef]
- Rudalska, R.; Dauch, D.; Longerich, T.; McJunkin, K.; Wuestefeld, T.; Kang, T.-W.; Hohmeyer, A.; Pesic, M.; Leibold, J.; Von Thun, A.; et al. In vivo RNAi screening identifies a mechanism of sorafenib resistance in liver cancer. Nat. Med. 2014, 20, 1138–1146. [Google Scholar] [CrossRef]
- Zheng, J.-F.; He, S.; Zeng, Z.; Gu, X.; Cai, L.; Qi, G. PMPCB Silencing Sensitizes HCC Tumor Cells to Sorafenib Therapy. Mol. Ther. 2019, 27, 1784–1795. [Google Scholar] [CrossRef] [PubMed]
- Bertelsen, B.; Melchior, L.; Jensen, L.R.; Groth, C.; Glenthøj, B.; Rizzo, R.; Debes, N.M.; Skov, L.; Brøndum-Nielsen, K.; Paschou, P.; et al. Intragenic deletions affecting two alternative transcripts of the IMMP2L gene in patients with Tourette syndrome. Eur. J. Hum. Genet. 2014, 22, 1283–1289. [Google Scholar] [CrossRef] [PubMed]
- Gimelli, S.; Capra, V.; Di Rocco, M.; Leoni, M.; Mirabelli-Badenier, M.; Schiaffino, M.C.; Fiorio, P.; Cuoco, C.; Gimelli, G.; Tassano, E. Interstitial 7q31.1 copy number variations disrupting IMMP2L gene are associated with a wide spectrum of neurodevelopmental disorders. Mol. Cytogenet. 2014, 7, 54. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Y.; Zarrei, M.; Tong, W.; Dong, R.; Wang, Y.; Zhang, H.; Yang, X.; Macdonald, J.R.; Uddin, M.; et al. Association of IMMP2L deletions with autism spectrum disorder: A trio family study and meta-analysis. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2018, 177, 93–100. [Google Scholar] [CrossRef]
- Elia, J.; Gai, X.; Xie, H.M.; Perin, J.; Geiger, E.; Glessner, J.; D’Arcy, M.; DeBerardinis, R.; Frackelton, E.; Kim, C.; et al. Rare structural variants found in attention-deficit hyperactivity disorder are preferentially associated with neurodevelopmental genes. Mol. Psychiatry 2009, 15, 637–646, Corrigendum in Mol. Psychiatry 2010, 15, 637–646. [Google Scholar] [CrossRef]
- Liang, S.; Wang, X.-L.; Zou, M.-Y.; Wang, H.; Zhou, X.; Sun, C.-H.; Xia, W.; Wu, L.-J.; Fujisawa, T.X.; Tomoda, A. Family-based association study of ZNF533, DOCK4 and IMMP2L gene polymorphisms linked to autism in a northeastern Chinese Han population. J. Zhejiang Univ. Sci. B 2014, 15, 264–271. [Google Scholar] [CrossRef]
- Swaminathan, S.; Shen, L.; Kim, S.; Inlow, M.; West, J.D.; Faber, K.M.; Foroud, T.; Mayeux, R.; Saykin, A.J. Analysis of Copy Number Variation in Alzheimer’s Disease: The NIALOAD/NCRAD Family Study. Curr. Alzheimer Res. 2012, 9, 801–814. [Google Scholar] [CrossRef]
- Hsiao, C.-P.; Wang, D.; Kaushal, A.; Saligan, L. Mitochondria-Related Gene Expression Changes Are Associated With Fatigue in Patients With Nonmetastatic Prostate Cancer Receiving External Beam Radiation Therapy. Cancer Nurs. 2013, 36, 189–197. [Google Scholar] [CrossRef]
- Eldomery, M.K.; Akdemir, Z.C.; Vögtle, F.-N.; Charng, W.-L.; Mulica, P.; Rosenfeld, J.A.; Gambin, T.; Gu, S.; Burrage, L.C.; Al Shamsi, A.; et al. MIPEP recessive variants cause a syndrome of left ventricular non-compaction, hypotonia, and infantile death. Genome Med. 2016, 8, 106. [Google Scholar] [CrossRef]
- Pulman, J.; Ruzzenente, B.; Horak, M.; Barcia, G.; Boddaert, N.; Munnich, A.; Rötig, A.; Metodiev, M.D. Variants in the MIPEP gene presenting with complex neurological phenotype without cardiomyopathy, impair OXPHOS protein maturation and lead to a reduced OXPHOS abundance in patient cells. Mol. Genet. Metab. 2021, 134, 267–273. [Google Scholar] [CrossRef]
- Chitrala, K.N.; Hernandez, D.G.; Nalls, M.A.; Mode, N.A.; Zonderman, A.B.; Ezike, N.; Evans, M.K. Race-specific alterations in DNA methylation among middle-aged African Americans and Whites with metabolic syndrome. Epigenetics 2020, 15, 462–482. [Google Scholar] [CrossRef]
- Hatunic, M.; Stapleton, M.; Hand, E.; DeLong, C.; Crowley, V.; Nolan, J. The Leu262Val polymorphism of presenilin associated rhomboid like protein (PARL) is associated with earlier onset of type 2 diabetes and increased urinary microalbumin creatinine ratio in an Irish case–control population. Diabetes Res. Clin. Pr. 2009, 83, 316–319. [Google Scholar] [CrossRef]
- Shi, G.; Lee, J.R.; Grimes, D.A.; Racacho, L.; Ye, D.; Yang, H.; Ross, O.A.; Farrer, M.; McQuibban, G.A.; Bulman, D.E. Functional alteration of PARL contributes to mitochondrial dysregulation in Parkinson’s disease. Hum. Mol. Genet. 2011, 20, 1966–1974. [Google Scholar] [CrossRef]
- Branda, S.S.; Isaya, G. Prediction and Identification of New Natural Substrates of the Yeast Mitochondrial Intermediate Peptidase. J. Biol. Chem. 1995, 270, 27366–27373. [Google Scholar] [CrossRef]
- Burri, L.; Strahm, Y.; Hawkins, C.J.; Gentle, I.E.; Puryer, M.A.; Verhagen, A.; Callus, B.; Vaux, D.; Lithgow, T. Mature DIABLO/Smac Is Produced by the IMP Protease Complex on the Mitochondrial Inner Membrane. Mol. Biol. Cell 2005, 16, 2926–2933. [Google Scholar] [CrossRef]
- Esser, K.; Jan, P.-S.; Pratje, E.; Michaelis, G. The mitochondrial IMP peptidase of yeast: Functional analysis of domains and identification of Gut2 as a new natural substrate. Mol. Genet. Genom. 2004, 271, 616–626. [Google Scholar] [CrossRef]
- Jan, P.-S.; Esser, K.; Pratje, E.; Michaelis, G. Som1, a third component of the yeast mitochondrial inner membrane peptidase complex that contains Imp1 and Imp2. Mol. Genet. Genom. 2000, 263, 483–491. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2021, 50, D439–D444. [Google Scholar] [CrossRef]
- Nunnari, J.; Fox, T.D.; Walter, P. A Mitochondrial Protease with Two Catalytic Subunits of Nonoverlapping Specificities. Science 1993, 262, 1997–2004. [Google Scholar] [CrossRef]
- Schneider, A.; Behrens, M.; Scherer, P.; Pratje, E.; Michaelis, G.; Schatz, G. Inner membrane protease I, an enzyme mediating intramitochondrial protein sorting in yeast. EMBO J. 1991, 10, 247–254. [Google Scholar] [CrossRef]
- Schneider, A.; Oppliger, W.; Jenö, P. Purified inner membrane protease I of yeast mitochondria is a heterodimer. J. Biol. Chem. 1994, 269, 8635–8638. [Google Scholar] [CrossRef]
- Daum, G.; Gasser, S.; Schatz, G. Import of proteins into mitochondria. Energy-dependent, two-step processing of the intermembrane space enzyme cytochrome b2 by isolated yeast mitochondria. J. Biol. Chem. 1982, 257, 13075–13080. [Google Scholar] [CrossRef]
- Esser, K.; Pratje, E.; Michaelis, G. SOM 1, a small new gene required for mitochondrial inner membrane peptidase function inSaccharomyces cerevisiae. Mol. Genet. Genom. 1996, 252, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Girirajan, S.; Brkanac, Z.; Coe, B.P.; Baker, C.; Vives, L.; Vu, T.H.; Shafer, N.; Bernier, R.; Ferrero, G.B.; Silengo, M.; et al. Relative Burden of Large CNVs on a Range of Neurodevelopmental Phenotypes. PLoS Genet. 2011, 7, e1002334. [Google Scholar] [CrossRef] [PubMed]
- Goes, F.S.; McGrath, J.A.; Avramopoulos, D.; Wolyniec, P.; Pirooznia, M.; Ruczinski, I.; Nestadt, G.; Kenny, E.E.; Vacic, V.; Peters, I.; et al. Genome-wide association study of schizophrenia in Ashkenazi Jews. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2015, 168, 649–659. [Google Scholar] [CrossRef]
- Maestrini, E.; Pagnamenta, A.T.; Lamb, J.A.; Bacchelli, E.; Sykes, N.H.; Sousa, I.; Toma, C.; Barnby, G.; Butler, H.; et al.; Imgsac High-density SNP association study and copy number variation analysis of the AUTS1 and AUTS5 loci implicate the IMMP2L–DOCK4 gene region in autism susceptibility. Mol. Psychiatry 2010, 15, 954–968. [Google Scholar] [CrossRef]
- Gokool, A.; Fang, Z.; Braidy, N.; Lin, P.; Pardy, C.J.; Eapen, V.; Clarke, R.; Voineagu, I. Transcriptional response to mitochondrial protease IMMP2L knockdown in human primary astrocytes. Biochem. Biophys. Res. Commun. 2017, 482, 1252–1258. [Google Scholar] [CrossRef]
- Luo, W.; Fang, H.; Green, N. Substrate specificity of inner membrane peptidase in yeast mitochondria. Mol. Genet. Genom. 2006, 275, 431–436. [Google Scholar] [CrossRef]
- Lu, B.; Poirier, C.; Gaspar, T.; Gratzke, C.; Harrison, W.; Busija, D.; Matzuk, M.M.; Andersson, K.-E.; Overbeek, P.; Bishop, C.E. A Mutation in the Inner Mitochondrial Membrane Peptidase 2-Like Gene (Immp2l) Affects Mitochondrial Function and Impairs Fertility in Mice1. Biol. Reprod. 2008, 78, 601–610. [Google Scholar] [CrossRef]
- Bharadwaj, M.S.; Zhou, Y.; Molina, A.J.; Criswell, T.; Lu, B. Examination of bioenergetic function in the inner mitochondrial membrane peptidase 2-like (Immp2l) mutant mice. Redox Biol. 2014, 2, 1008–1015. [Google Scholar] [CrossRef]
- Soler, R.; Füllhase, C.; Lu, B.; Bishop, C.E.; Andersson, K.-E. Bladder Dysfunction in a New Mutant Mouse Model With Increased Superoxide—Lack of Nitric Oxide? J. Urol. 2010, 183, 780–785. [Google Scholar] [CrossRef][Green Version]
- Han, C.; Zhao, Q.; Lu, B. The role of nitric oxide signaling in food intake; insights from the inner mitochondrial membrane peptidase 2 mutant mice. Redox Biol. 2013, 1, 498–507. [Google Scholar] [CrossRef][Green Version]
- George, S.K.; Jiao, Y.; Bishop, C.E.; Lu, B. Mitochondrial peptidase IMMP2L mutation causes early onset of age-associated disorders and impairs adult stem cell self-renewal. Aging Cell 2011, 10, 584–594. [Google Scholar] [CrossRef]
- Liu, C.; Li, X.; Lu, B. The Immp2l mutation causes age-dependent degeneration of cerebellar granule neurons prevented by antioxidant treatment. Aging Cell 2016, 15, 167–176. [Google Scholar] [CrossRef][Green Version]
- Ma, Y.; Mehta, S.L.; Lu, B.; Li, P.A. Deficiency in the inner mitochondrial membrane peptidase 2-like (Immp21) gene increases ischemic brain damage and impairs mitochondrial function. Neurobiol. Dis. 2011, 44, 270–276. [Google Scholar] [CrossRef]
- Yuan, L.; Zhai, L.; Qian, L.; Huang, D.; Ding, Y.; Xiang, H.; Liu, X.; Thompson, J.W.; Liu, J.; He, Y.; et al. Switching off IMMP2L signaling drives senescence via simultaneous metabolic alteration and blockage of cell death. Cell Res. 2018, 28, 625–643. [Google Scholar] [CrossRef] [PubMed]
- Kalousek, F.; Isaya, G.; Rosenberg, L. Rat liver mitochondrial intermediate peptidase (MIP): Purification and initial characterization. EMBO J. 1992, 11, 2803–2809. [Google Scholar] [CrossRef]
- Chew, A.; Buck, E.A.; Peretz, S.; Sirugo, G.; Rinaldo, P.; Isaya, G. Cloning, Expression, and Chromosomal Assignment of the Human Mitochondrial Intermediate Peptidase Gene (MIPEP). Genomics 1997, 40, 493–496. [Google Scholar] [CrossRef]
- Chew, A.; Sirugob, G.; AlsobrookIIc, J.P.; Isaya, G. Functional and Genomic Analysis of the Human Mitochondrial Intermediate Peptidase, a Putative Protein Partner of Frataxin. Genomics 2000, 65, 104–112. [Google Scholar] [CrossRef]
- Meyers, D.E.; Basha, H.I.; Koenig, M.K. Mitochondrial cardiomyopathy: Pathophysiology, diagnosis, and management. Tex. Hear. Inst. J. 2013, 40, 385–394. [Google Scholar]
- Stiburek, L.; Fornuskova, D.; Wenchich, L.; Pejznochova, M.; Hansikova, H.; Zeman, J. Knockdown of Human Oxa1l Impairs the Biogenesis of F1Fo-ATP Synthase and NADH:Ubiquinone Oxidoreductase. J. Mol. Biol. 2007, 374, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Thompson, K.; Mai, N.; Oláhová, M.; Scialó, F.; Formosa, L.; Stroud, D.; Garrett, M.; Lax, N.Z.; Robertson, F.M.; Jou, C.; et al. OXA 1L mutations cause mitochondrial encephalopathy and a combined oxidative phosphorylation defect. EMBO Mol. Med. 2018, 10, e9060. [Google Scholar] [CrossRef] [PubMed]
- Pawłowski, K.M.; Maciejewski, H.; Majchrzak, K.; Dolka, I.; Mol, J.A.; Motyl, T.; Król, M. Five markers useful for the distinction of canine mammary malignancy. BMC Vet. Res. 2013, 9, 138. [Google Scholar] [CrossRef] [PubMed]
- Freeman, M. Rhomboid Proteases and their Biological Functions. Annu. Rev. Genet. 2008, 42, 191–210. [Google Scholar] [CrossRef]
- Voos, W. Chaperone–protease networks in mitochondrial protein homeostasis. Biochim. Biophys. Acta 2013, 1833, 388–399. [Google Scholar] [CrossRef]
- Jeyaraju, D.V.; McBride, H.M.; Hill, R.B.; Pellegrini, L. Structural and mechanistic basis of Parl activity and regulation. Cell Death Differ. 2011, 18, 1531–1539. [Google Scholar] [CrossRef]
- Koonin, E.V. Horizontal gene transfer: The path to maturity. Mol. Microbiol. 2003, 50, 725–727. [Google Scholar] [CrossRef]
- Sík, A.; Passer, B.J.; Koonin, E.V.; Pellegrini, L. Self-regulated Cleavage of the Mitochondrial Intramembrane-cleaving Protease PARL Yields Pβ, a Nuclear-targeted Peptide. J. Biol. Chem. 2004, 279, 15323–15329. [Google Scholar] [CrossRef]
- Jeyaraju, D.V.; Xu, L.; Letellier, M.-C.; Bandaru, S.; Zunino, R.; Berg, E.A.; McBride, H.M.; Pellegrini, L. Phosphorylation and cleavage of presenilin-associated rhomboid-like protein (PARL) promotes changes in mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2006, 103, 18562–18567. [Google Scholar] [CrossRef]
- Lysyk, L.; Brassard, R.; Touret, N.; Lemieux, M.J. PARL Protease: A Glimpse at Intramembrane Proteolysis in the Inner Mitochondrial Membrane. J. Mol. Biol. 2020, 432, 5052–5062. [Google Scholar] [CrossRef]
- Esser, K.; Tursun, B.; Ingenhoven, M.; Michaelis, G.; Pratje, E. A Novel Two-step Mechanism for Removal of a Mitochondrial Signal Sequence Involves the mAAA Complex and the Putative Rhomboid Protease Pcp1. J. Mol. Biol. 2002, 323, 835–843. [Google Scholar] [CrossRef]
- Herlan, M.; Vogel, F.; Bornhövd, C.; Neupert, W.; Reichert, A.S. Processing of Mgm1 by the Rhomboid-type Protease Pcp1 Is Required for Maintenance of Mitochondrial Morphology and of Mitochondrial DNA. J. Biol. Chem. 2003, 278, 27781–27788. [Google Scholar] [CrossRef]
- Herlan, M.; Bornhoövd, C.; Hell, K.; Neupert, W.; Reichert, A.S. Alternative topogenesis of Mgm1 and mitochondrial morphology depend on ATP and a functional import motor. J. Cell Biol. 2004, 165, 167–173. [Google Scholar] [CrossRef]
- McQuibban, G.A.; Saurya, S.; Freeman, M. Mitochondrial membrane remodelling regulated by a conserved rhomboid protease. Nature 2003, 423, 537–541. [Google Scholar] [CrossRef]
- Chao, J.-R.; Parganas, E.; Boyd, K.; Hong, C.Y.; Opferman, J.T.; Ihle, J.N. Hax1-mediated processing of HtrA2 by Parl allows survival of lymphocytes and neurons. Nature 2008, 452, 98–102. [Google Scholar] [CrossRef]
- Yoshioka, H.; Katsu, M.; Sakata, H.; Okami, N.; Wakai, T.; Kinouchi, H.; Chan, P.H. The Role of Parl and HtrA2 in Striatal Neuronal Injury After Transient Global Cerebral Ischemia. Br. J. Pharmacol. 2013, 33, 1658–1665. [Google Scholar] [CrossRef]
- Needleman, S.B.; Wunsch, C.D. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 1970, 48, 443–453. [Google Scholar] [CrossRef]
- Greene, A.W.; Grenier, K.; Aguileta, M.A.; Muise, S.; Farazifard, R.; Haque, M.E.; McBride, H.M.; Park, D.S.; Fon, E.A. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012, 13, 378–385. [Google Scholar] [CrossRef]
- Deas, E.; Plun-Favreau, H.; Gandhi, S.; Desmond, H.; Kjaer, S.; Loh, S.H.; Renton, A.E.; Harvey, R.J.; Whitworth, A.J.; Martins, L.M.; et al. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum. Mol. Genet. 2010, 20, 867–879. [Google Scholar] [CrossRef]
- Yamano, K.; Youle, R.J. PINK1 is degraded through the N-end rule pathway. Autophagy 2013, 9, 1758–1769. [Google Scholar] [CrossRef]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef] [PubMed]
- Meissner, C.; Lorenz, H.; Weihofen, A.; Selkoe, D.J.; Lemberg, M.K. The mitochondrial intramembrane protease PARL cleaves human Pink1 to regulate Pink1 trafficking. J. Neurochem. 2011, 117, 856–867. [Google Scholar] [CrossRef] [PubMed]
- Sekine, S.; Kanamaru, Y.; Koike, M.; Nishihara, A.; Okada, M.; Kinoshita, H.; Kamiyama, M.; Maruyama, J.; Uchiyama, Y.; Takeda, K.; et al. Rhomboid Protease PARL Mediates the Mitochondrial Membrane Potential Loss-induced Cleavage of PGAM5. J. Biol. Chem. 2012, 287, 34635–34645. [Google Scholar] [CrossRef] [PubMed]
- Saita, S.; Nolte, H.; Fiedler, K.U.; Kashkar, H.; Venne, A.S.; Zahedi, R.; Krüger, M.; Langer, T. PARL mediates Smac proteolytic maturation in mitochondria to promote apoptosis. Nat. Cell Biol. 2017, 19, 318–328. [Google Scholar] [CrossRef]
- Ishihara, N.; Mihara, K. PARL paves the way to apoptosis. Nat. Cell Biol. 2017, 19, 263–265. [Google Scholar] [CrossRef]
- Saita, S.; Tatsuta, T.; Lampe, P.A.; König, T.; Ohba, Y.; Langer, T. PARL partitions the lipid transfer protein STARD7 between the cytosol and mitochondria. EMBO J. 2018, 37, e97909. [Google Scholar] [CrossRef]
- Lysyk, L.; Brassard, R.; Arutyunova, E.; Siebert, V.; Jiang, Z.; Takyi, E.; Morrison, M.; Young, H.S.; Lemberg, M.K.; O’Donoghue, A.J.; et al. Insights into the catalytic properties of the mitochondrial rhomboid protease PARL. J. Biol. Chem. 2021, 296, 100383. [Google Scholar] [CrossRef]
- Cipolat, S.; Rudka, T.; Hartmann, D.; Costa, V.; Serneels, L.; Craessaerts, K.; Metzger, K.; Frezza, C.; Annaert, W.; D’Adamio, L.; et al. Mitochondrial Rhomboid PARL Regulates Cytochrome c Release during Apoptosis via OPA1-Dependent Cristae Remodeling. Cell 2006, 126, 163–175. [Google Scholar] [CrossRef]
- Fawcett, K.A.; Wareham, N.J.; Luan, J.; Syddall, H.; Cooper, C.; O’Rahilly, S.; Day, I.N.M.; Sandhu, M.S.; Barroso, I. PARL Leu262Val is not associated with fasting insulin levels in UK populations. Diabetologia 2006, 49, 2649–2652. [Google Scholar] [CrossRef]
- Heinitz, S.; Klein, C.; Djarmati, A. The p.S77N presenilin-associated rhomboid-like protein mutation is not a frequent cause of early-onset Parkinson’s disease. Mov. Disord. 2011, 26, 2441–2442. [Google Scholar] [CrossRef]
- Wüst, R.; Maurer, B.; Hauser, K.; Woitalla, D.; Sharma, M.; Krüger, R. Mutation analyses and association studies to assess the role of the presenilin-associated rhomboid-like gene in Parkinson’s disease. Neurobiol. Aging 2016, 39, 217.e13–217.e15. [Google Scholar] [CrossRef]
- Kücükköse, C.; Taskin, A.A.; Marada, A.; Brummer, T.; Dennerlein, S.; Vögtle, F. Functional coupling of presequence processing and degradation in human mitochondria. FEBS J. 2021, 288, 600–613. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kunová, N.; Havalová, H.; Ondrovičová, G.; Stojkovičová, B.; Bauer, J.A.; Bauerová-Hlinková, V.; Pevala, V.; Kutejová, E. Mitochondrial Processing Peptidases—Structure, Function and the Role in Human Diseases. Int. J. Mol. Sci. 2022, 23, 1297. https://doi.org/10.3390/ijms23031297
Kunová N, Havalová H, Ondrovičová G, Stojkovičová B, Bauer JA, Bauerová-Hlinková V, Pevala V, Kutejová E. Mitochondrial Processing Peptidases—Structure, Function and the Role in Human Diseases. International Journal of Molecular Sciences. 2022; 23(3):1297. https://doi.org/10.3390/ijms23031297
Chicago/Turabian StyleKunová, Nina, Henrieta Havalová, Gabriela Ondrovičová, Barbora Stojkovičová, Jacob A. Bauer, Vladena Bauerová-Hlinková, Vladimir Pevala, and Eva Kutejová. 2022. "Mitochondrial Processing Peptidases—Structure, Function and the Role in Human Diseases" International Journal of Molecular Sciences 23, no. 3: 1297. https://doi.org/10.3390/ijms23031297
APA StyleKunová, N., Havalová, H., Ondrovičová, G., Stojkovičová, B., Bauer, J. A., Bauerová-Hlinková, V., Pevala, V., & Kutejová, E. (2022). Mitochondrial Processing Peptidases—Structure, Function and the Role in Human Diseases. International Journal of Molecular Sciences, 23(3), 1297. https://doi.org/10.3390/ijms23031297