
Investigating the Molecular Mechanisms of Renal Hepcidin Induction and Protection upon Hemoglobin-Induced Acute Kidney Injury


 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
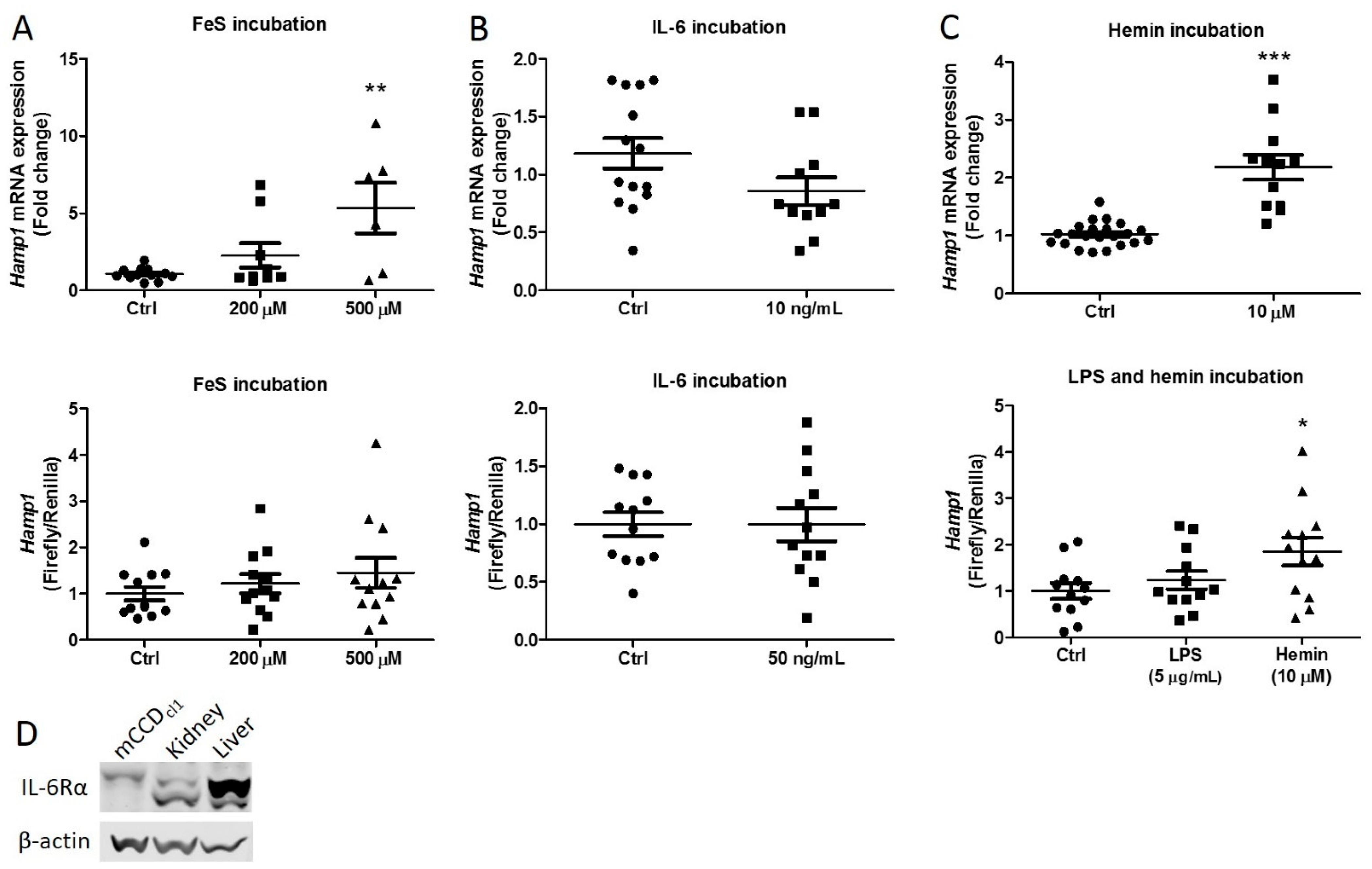
2.1. Iron Rather Than Inflammation Induces Hepcidin Synthesis
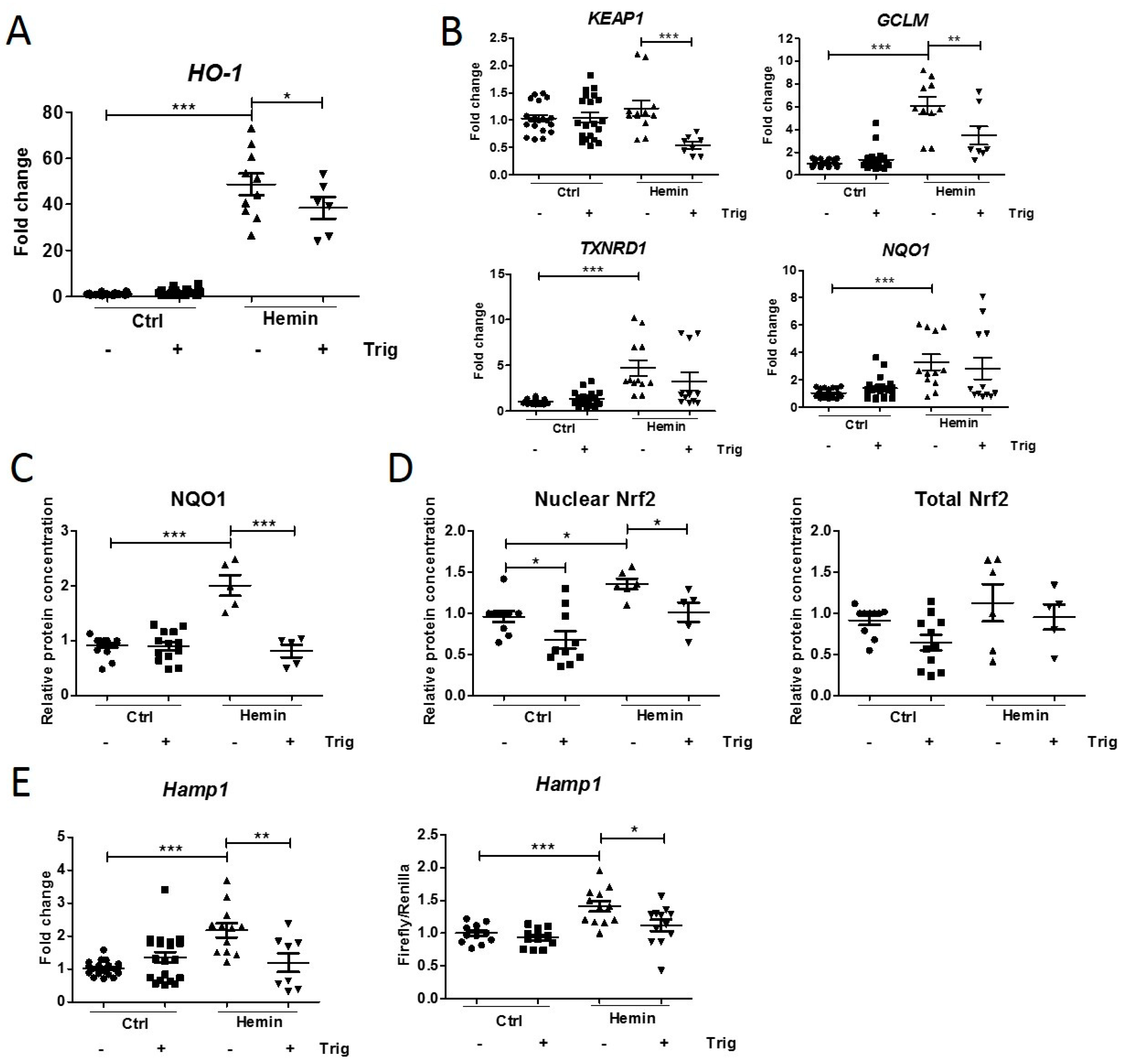
2.2. Downregulation of Hemin-Induced Nrf2 Pathway Activation Decreases Hepcidin Synthesis
2.3. The Effect of a Renal Hepcidin KO
3. Discussion
4. Methods
4.1. Cell Culture
4.2. Protein Isolation and Immunoblotting
4.3. Luciferase Assay
4.4. Animal Studies
4.5. Immunohistochemistry
4.6. ELISA
4.7. Genotyping
4.8. DNA Isolation and PCR
4.9. Plasma and Urine Hepcidin Analysis
4.10. Plasma and Urine Non-Heme Iron Assay
4.11. RNA Isolation and qPCR
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Uchino, S.; Kellum, J.A.; Bellomo, R.; Doig, G.S.; Morimatsu, H.; Morgera, S.; Schetz, M.; Tan, I.; Bouman, C.; Macedo, E.; et al. Acute renal failure in critically ill patients: A multinational, multicenter study. JAMA 2005, 294, 813–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, H.; Katz, N.; Ariyanon, W.; Blanca-Martos, L.; Adybelli, Z.; Giuliani, A.; Danesi, T.H.; Kim, J.C.; Nayak, A.; Neri, M.; et al. Cardiac surgery-associated acute kidney injury. Blood Purif. 2014, 37 (Suppl. S2), 34–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vercaemst, L. Hemolysis in cardiac surgery patients undergoing cardiopulmonary bypass: A review in search of a treatment algorithm. J. Extra-Corpor. Technol. 2008, 40, 257–267. [Google Scholar] [PubMed]
- Moat, N.E.; Evans, T.E.; Quinlan, G.J.; Gutteridge, J.M. Chelatable iron and copper can be released from extracorporeally circulated blood during cardiopulmonary bypass. FEBS Lett. 1993, 328, 103–106. [Google Scholar] [CrossRef] [Green Version]
- Schaer, D.J.; Buehler, P.W.; Alayash, A.I.; Belcher, J.D.; Vercellotti, G.M. Hemolysis and free hemoglobin revisited: Exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins. Blood 2013, 121, 1276–1284. [Google Scholar] [CrossRef] [Green Version]
- Haase, M.; Bellomo, R.; Haase-Fielitz, A. Novel biomarkers, oxidative stress, and the role of labile iron toxicity in cardiopulmonary bypass-associated acute kidney injury. J. Am. Coll. Cardiol. 2010, 55, 2024–2033. [Google Scholar] [CrossRef] [Green Version]
- Gburek, J.; Verroust, P.J.; Willnow, T.E.; Fyfe, J.C.; Nowacki, W.; Jacobsen, C.; Moestrup, S.K.; Christensen, E.I. Megalin and cubilin are endocytic receptors involved in renal clearance of hemoglobin. J. Am. Soc. Nephrol. 2002, 13, 423–430. [Google Scholar] [CrossRef]
- van Swelm, R.P.L.; Vos, M.; Verhoeven, F.; Thevenod, F.; Swinkels, D.W. Endogenous hepcidin synthesis protects the distal nephron against hemin and hemoglobin mediated necroptosis. Cell Death Dis. 2018, 9, 550. [Google Scholar] [CrossRef]
- Nath, K.A. Heme oxygenase-1 and acute kidney injury. Curr. Opin. Nephrol. Hypertens. 2014, 23, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Kartikasari, A.E.R.; Wagener, F.A.D.T.G.; Yachie, A.; Wiegerinck, E.T.G.; Kemna, E.H.J.M.; Swinkels, D.W. Hepcidin suppression and defective iron recycling account for dysregulation of iron homeostasis in heme oxygenase-1 deficiency. J. Cell. Mol. Med. 2009, 13, 3091–3102. [Google Scholar] [CrossRef] [Green Version]
- Billings, F.T.; Ball, S.K.; Roberts, L.J., 2nd; Pretorius, M. Postoperative acute kidney injury is associated with hemoglobinemia and an enhanced oxidative stress response. Free Radic. Biol. Med. 2011, 50, 1480–1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, J.; Reslerova, M.; Gali, B.; Gao, A.; Bestland, J.; Rush, D.N.; Nickerson, P.W.; Rigatto, C. Urinary hepcidin-25 and risk of acute kidney injury following cardiopulmonary bypass. Clin. J. Am. Soc. Nephrol. 2011, 6, 2340–2346. [Google Scholar] [CrossRef] [PubMed]
- Prowle, J.R.; Ostland, V.; Calzavacca, P.; Licari, E.; Ligabo, E.V.; Echeverri, J.E.; Bagshaw, S.M.; Haase-Fielitz, A.; Haase, M.; Westerman, M.; et al. Greater increase in urinary hepcidin predicts protection from acute kidney injury after cardiopulmonary bypass. Nephrol. Dial. Transplant. 2012, 27, 595–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haase-Fielitz, A.; Mertens, P.R.; Plass, M.; Kuppe, H.; Hetzer, R.; Westerman, M.; Ostland, V.; Prowle, J.R.; Bellomo, R.; Haase, M. Urine hepcidin has additive value in ruling out cardiopulmonary bypass-associated acute kidney injury: An observational cohort study. Crit. Care 2011, 15, R186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Swelm, R.P.; Wetzels, J.F.; Verweij, V.G.; Laarakkers, C.M.; Pertijs, J.C.; van der Wijst, J.; Thévenod, F.; Masereeuw, R.; Swinkels, D.W. Renal Handling of Circulating and Renal-Synthesized Hepcidin and Its Protective Effects against Hemoglobin-Mediated Kidney Injury. J. Am. Soc. Nephrol. 2016, 27, 2720–2732. [Google Scholar] [CrossRef] [Green Version]
- Kulaksiz, H.; Theilig, F.; Bachmann, S.; Gehrke, S.G.; Rost, D.; Janetzko, A.; Cetin, Y.; Stremmel, W. The iron-regulatory peptide hormone hepcidin: Expression and cellular localization in the mammalian kidney. J. Endocrinol. 2005, 184, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Scindia, Y.; Dey, P.; Thirunagari, A.; Liping, H.; Rosin, D.L.; Floris, M.; Okusa, M.D.; Swaminathan, S. Hepcidin Mitigates Renal Ischemia-Reperfusion Injury by Modulating Systemic Iron Homeostasis. J. Am. Soc. Nephrol. 2015, 26, 2800–2814. [Google Scholar] [CrossRef] [Green Version]
- Kemna, E.; Pickkers, P.; Nemeth, E.; van der Hoeven, H.; Swinkels, D. Time-course analysis of hepcidin, serum iron, and plasma cytokine levels in humans injected with LPS. Blood 2005, 106, 1864–1866. [Google Scholar] [CrossRef]
- Lim, P.J.; Duarte, T.L.; Arezes, J.; Garcia-Santos, D.; Hamdi, A.; Pasricha, S.-R.; Armitage, A.E.; Mehta, H.; Wideman, S.; Santos, A.G.; et al. Nrf2 controls iron homeostasis in haemochromatosis and thalassaemia via Bmp6 and hepcidin. Nat. Metab. 2019, 1, 519–531. [Google Scholar] [CrossRef]
- Suzuki, T.; Yamamoto, M. Molecular basis of the Keap1-Nrf2 system. Free Radic. Biol. Med. 2015, 88 Pt B, 93–100. [Google Scholar] [CrossRef] [Green Version]
- Bayele, H.K.; Balesaria, S.; Srai, S.K.S. Phytoestrogens modulate hepcidin expression by Nrf2: Implications for dietary control of iron absorption. Free Radic. Biol. Med. 2015, 89, 1192–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Tan, Y.; Yang, B.; Wu, Y.; Fan, B.; Zhu, S.; Song, E.; Song, Y. Polychlorinated biphenyl quinone induces hepatocytes iron overload through up-regulating hepcidin expression. Environ. Int. 2020, 139, 105701. [Google Scholar] [CrossRef] [PubMed]
- van Raaij, S.E.G.; Masereeuw, R.; Swinkels, D.W.; van Swelm, R.P.L. Inhibition of Nrf2 alters cell stress induced by chronic iron exposure in human proximal tubular epithelial cells. Toxicol. Lett. 2018, 295, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Gozzelino, R.; Jeney, V.; Soares, M.P. Mechanisms of cell protection by heme oxygenase-1. Annu. Rev. Pharm. Toxicol. 2010, 50, 323–354. [Google Scholar] [CrossRef] [Green Version]
- Muckenthaler, M.U.; Galy, B.; Hentze, M.W. Systemic Iron Homeostasis and the Iron-Responsive Element/Iron-Regulatory Protein (IRE/IRP) Regulatory Network. Annu. Rev. Nutr. 2008, 28, 197–213. [Google Scholar] [CrossRef]
- Nezu, M.; Suzuki, N.; Yamamoto, M. Targeting the KEAP1-NRF@ system to prevent kidney disease progression. Am. J. Nephrol. 2017, 45, 473–483. [Google Scholar] [CrossRef]
- Zheng, H.; Whitman, S.A.; Wu, W.; Wondrak, G.T.; Wong, P.K.; Fang, D.; Zhang, D.D. Therapeutic potential of Nrf2 activators in streptozotocin-induced diabetic nephropathy. Diabetes 2011, 60, 3055–3066. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q.; Battelli, L.; Hubbs, A.F. Multiorgan autoimmune inflammation, enhanced lymphoproliferation, and impaired homeostasis of reactive oxygen species in mice lacking the antioxidant-activated transcription factor Nrf2. Am. J. Pathol. 2006, 168, 1960–1974. [Google Scholar] [CrossRef] [Green Version]
- Nezu, M.; Souma, T.; Yu, L.; Suzuki, T.; Saigusa, D.; Ito, S.; Yamamoto, M. Transcription factor Nrf2 hyperactivation in early-phase renal ischemia-reperfusion injury prevents tubular damage progression. Kidney Int. 2017, 91, 387–401. [Google Scholar] [CrossRef]
- Pergola, P.E.; Raskin, P.; Toto, R.D.; Meyer, C.J.; Huff, J.W.; Grossman, E.B.; Ruiz, S.; Audhya, P.; Wittes, J.; Warnock, D.G.; et al. Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N. Engl. J. Med. 2011, 365, 327–336. [Google Scholar] [CrossRef] [Green Version]
- Kerins, M.J.; Ooi, A. The Roles of NRF2 in Modulating Cellular Iron Homeostasis. Antioxid. Redox Signal. 2018, 29, 1756–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duarte, T.L.; Talbot, N.P.; Drakesmith, H. NRF2 and Hypoxia-Inducible Factors: Key Players in the Redox Control of Systemic Iron Homeostasis. Antioxid. Redox Signal. 2020, 35, 433–452. [Google Scholar] [CrossRef] [PubMed]
- Puntarulo, S. Iron, oxidative stress and human health. Mol. Asp. Med. 2005, 26, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Dendooven, A.; van Oostrom, O.; van der Giezen, D.M.; Leeuwis, J.W.; Snijckers, C.; Joles, J.A.; Robertson, E.J.; Verhaar, M.C.; Nguyen, T.Q.; Goldschmeding, R. Loss of endogenous bone morphogenetic protein-6 aggravates renal fibrosis. Am. J. Pathol. 2011, 178, 1069–1079. [Google Scholar] [CrossRef] [PubMed]
- El-Achkar, T.M.; Dagher, P.C. Tubular cross talk in acute kidney injury: A story of sense and sensibility. Am. J. Physiol. Ren. Physiol. 2015, 308, F1317–F1323. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Rivera, S.; Gabayan, V.; Keller, C.; Taudorf, S.; Pedersen, B.K.; Ganz, T. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J. Clin. Investig. 2004, 113, 1271–1276. [Google Scholar] [CrossRef] [Green Version]
- Traykova-Brauch, M.; Schönig, K.; Greiner, O.; Miloud, T.; Jauch, A.; Bode, M.; Felsher, D.W.; Glick, A.B.; Kwiatkowski, D.J.; Bujard, H.; et al. An efficient and versatile system for acute and chronic modulation of renal tubular function in transgenic mice. Nat. Med. 2008, 14, 979–984. [Google Scholar] [CrossRef] [Green Version]
- Coffey, R.; Ganz, T. Erythroferrone: An Erythroid Regulator of Hepcidin and Iron Metabolism. HemaSphere 2018, 2, e35. [Google Scholar] [CrossRef]
- Kapitsinou, P.P.; Liu, Q.; Unger, T.L.; Rha, J.; Davidoff, O.; Keith, B.; Epstein, J.A.; Moores, S.L.; Erickson-Miller, C.L.; Haase, V.H. Hepatic HIF-2 regulates erythropoietic responses to hypoxia in renal anemia. Blood 2010, 116, 3039–3048. [Google Scholar] [CrossRef] [Green Version]
- Stoffel, N.U.; Lazrak, M.; Bellitir, S.; Mir, N.E.; Hamdouchi, A.E.; Barkat, A.; Moretti, D.; Aguenaou, H.; Zimmermann, M.B. The opposing effects of acute inflammation and iron deficiency anemia on serum hepcidin and iron absorption in young women. Haematologica 2019, 104, 1143–1149. [Google Scholar] [CrossRef] [Green Version]
- Yousef Yengej, F.A.; Jansen, J.; Rookmaaker, M.B.; Verhaar, M.C.; Clevers, H. Kidney Organoids and Tubuloids. Cells 2020, 9, 1326. [Google Scholar] [CrossRef] [PubMed]
- Gaeggeler, H.P.; Gonzalez-Rodriguez, E.; Jaeger, N.F.; Loffing-Cueni, D.; Norregaard, R.; Loffing, J.; Horisberger, J.-D.; Rossier, B.C. Mineralocorticoid versus glucocorticoid receptor occupancy mediating aldosterone-stimulated sodium transport in a novel renal cell line. J. Am. Soc. Nephrol. 2005, 16, 878–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tenten, V.; Menzel, S.; Kunter, U.; Sicking, E.M.; van Roeyen, C.R.; Sanden, S.K.; Kaldenbach, M.; Boor, P.; Fuss, A.; Uhlig, S.; et al. Albumin is recycled from the primary urine by tubular transcytosis. J. Am. Soc. Nephrol. 2013, 24, 1966–1980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zumerle, S.; Mathieu, J.R.; Delga, S.; Heinis, M.; Viatte, L.; Vaulont, S.; Peyssonnaux, C. Targeted disruption of hepcidin in the liver recapitulates the hemochromatotic phenotype. Blood 2014, 123, 3646–3650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torrance, J.D.; Bothwell, T.H. A simple technique for measuring storage iron concentrations in formalinized liver samples. S. Afr. J. Med. Sci. 1968, 33, 9–11. [Google Scholar]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diepeveen, L.E.; Stegemann, G.; Wiegerinck, E.T.; Roelofs, R.; Naber, M.; Lóreal, O.; Smeets, B.; Thévenod, F.; Swinkels, D.W.; van Swelm, R.P.L. Investigating the Molecular Mechanisms of Renal Hepcidin Induction and Protection upon Hemoglobin-Induced Acute Kidney Injury. Int. J. Mol. Sci. 2022, 23, 1352. https://doi.org/10.3390/ijms23031352
Diepeveen LE, Stegemann G, Wiegerinck ET, Roelofs R, Naber M, Lóreal O, Smeets B, Thévenod F, Swinkels DW, van Swelm RPL. Investigating the Molecular Mechanisms of Renal Hepcidin Induction and Protection upon Hemoglobin-Induced Acute Kidney Injury. International Journal of Molecular Sciences. 2022; 23(3):1352. https://doi.org/10.3390/ijms23031352
Chicago/Turabian StyleDiepeveen, Laura E., Gaby Stegemann, Erwin T. Wiegerinck, Rian Roelofs, Myrthe Naber, Olivier Lóreal, Bart Smeets, Frank Thévenod, Dorine W. Swinkels, and Rachel P. L. van Swelm. 2022. "Investigating the Molecular Mechanisms of Renal Hepcidin Induction and Protection upon Hemoglobin-Induced Acute Kidney Injury" International Journal of Molecular Sciences 23, no. 3: 1352. https://doi.org/10.3390/ijms23031352
APA StyleDiepeveen, L. E., Stegemann, G., Wiegerinck, E. T., Roelofs, R., Naber, M., Lóreal, O., Smeets, B., Thévenod, F., Swinkels, D. W., & van Swelm, R. P. L. (2022). Investigating the Molecular Mechanisms of Renal Hepcidin Induction and Protection upon Hemoglobin-Induced Acute Kidney Injury. International Journal of Molecular Sciences, 23(3), 1352. https://doi.org/10.3390/ijms23031352