
Uric Acid and Oxidative Stress—Relationship with Cardiovascular, Metabolic, and Renal Impairment

 , , , and
, , , and
Abstract
1. Introduction
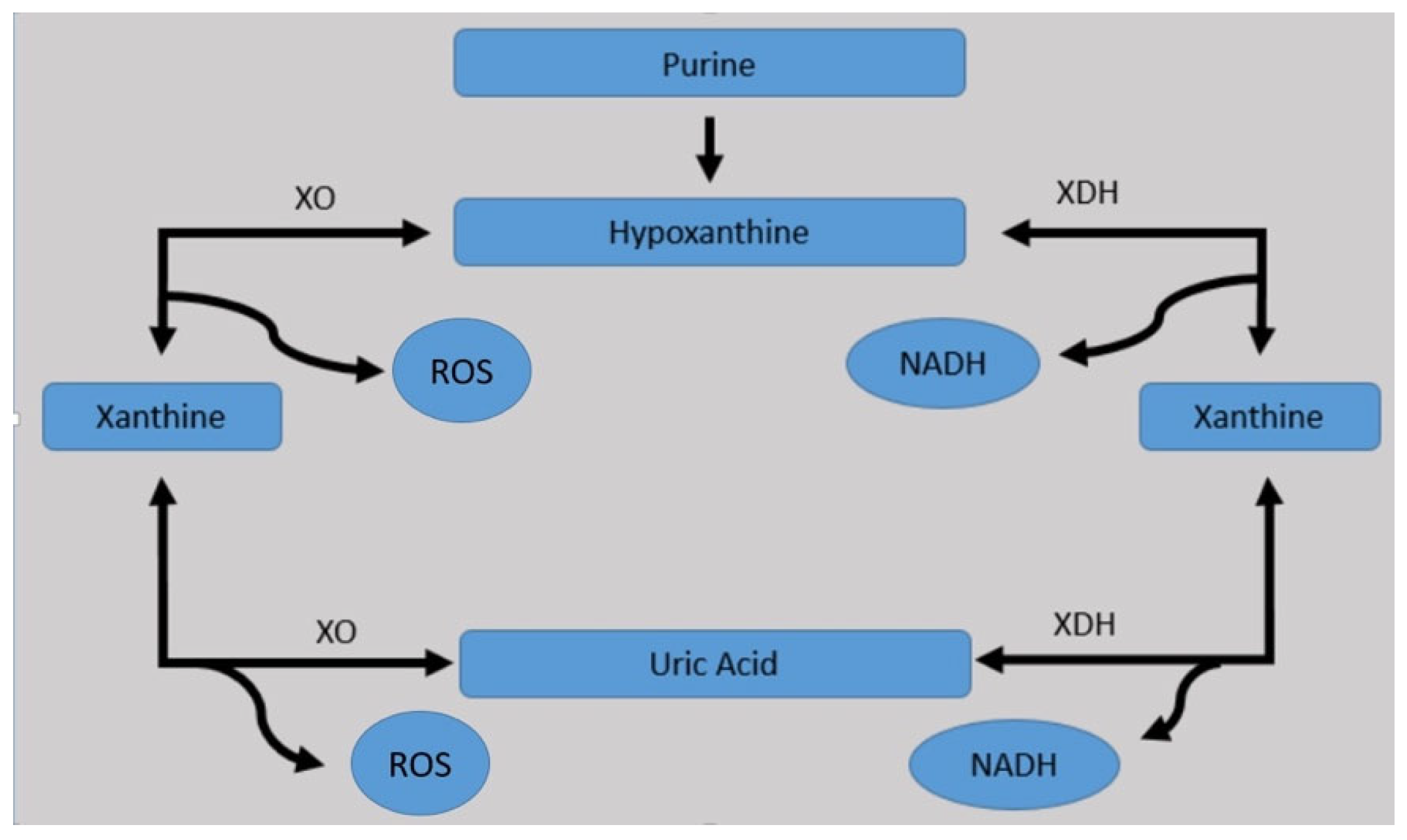
2. Uric Acid Synthesis
- White crystals or powder aspect,
- A molar weight of 168.11 g/mol,
- A heavy atom count of 12,
- A melting point >300 °C,
- A water solubility of 60 mg/L (at a temperature of 20 °C).
- Adenine pathway:
- ○
- Adenosine monophosphate (AMP) is converted by nucleotidase into adenosine, which is further converted by purine nucleoside phosphorylase into adenine, which, through deamination, is degraded to hypoxanthine.
- ○
- AMP can also present a deamination reaction, being converted into inosine monophosphate (IMP), which is converted by nucleotidase into inosine, which is further degraded by purine nucleoside phosphorylase into hypoxanthine.
- ○
- The resulting hypoxanthine, under the action of xanthine oxidase, is converted to xanthine.
- Guanine pathway:
- ○
- Guanosine monophosphate (GMP) is converted by nucleotidase into guanosine, which is further converted by purine nucleoside phosphorylase into guanine, which, through deamination, is degraded to xanthine.
- ○
- GMP can also present a deamination reaction, being converted into xanthosine monophosphate (XMP), which is converted by nucleotidase into xanthosine, which is further degraded by purine nucleoside phosphorylase into xanthine.
- The resulting xanthine, through adenine and guanine pathway, under the action of xanthine oxidase, is oxidized to uric acid, which in the normal human body’s physiological conditions, exists as urate, with the following normal range of levels, which are different for men and women: 2.5–7.0 mg/dL in male gender and 1.5–6.0 mg/dL in female gender, respectively. Furthermore, the urate is easily transformed to allantoic acid and ammonia, allowing its renal excretion (almost 200–300 mg/day).

3. Uric Acid Rich Diets
4. Possible Genetic Disorders Involved in Uric Acid Over-Synthesis
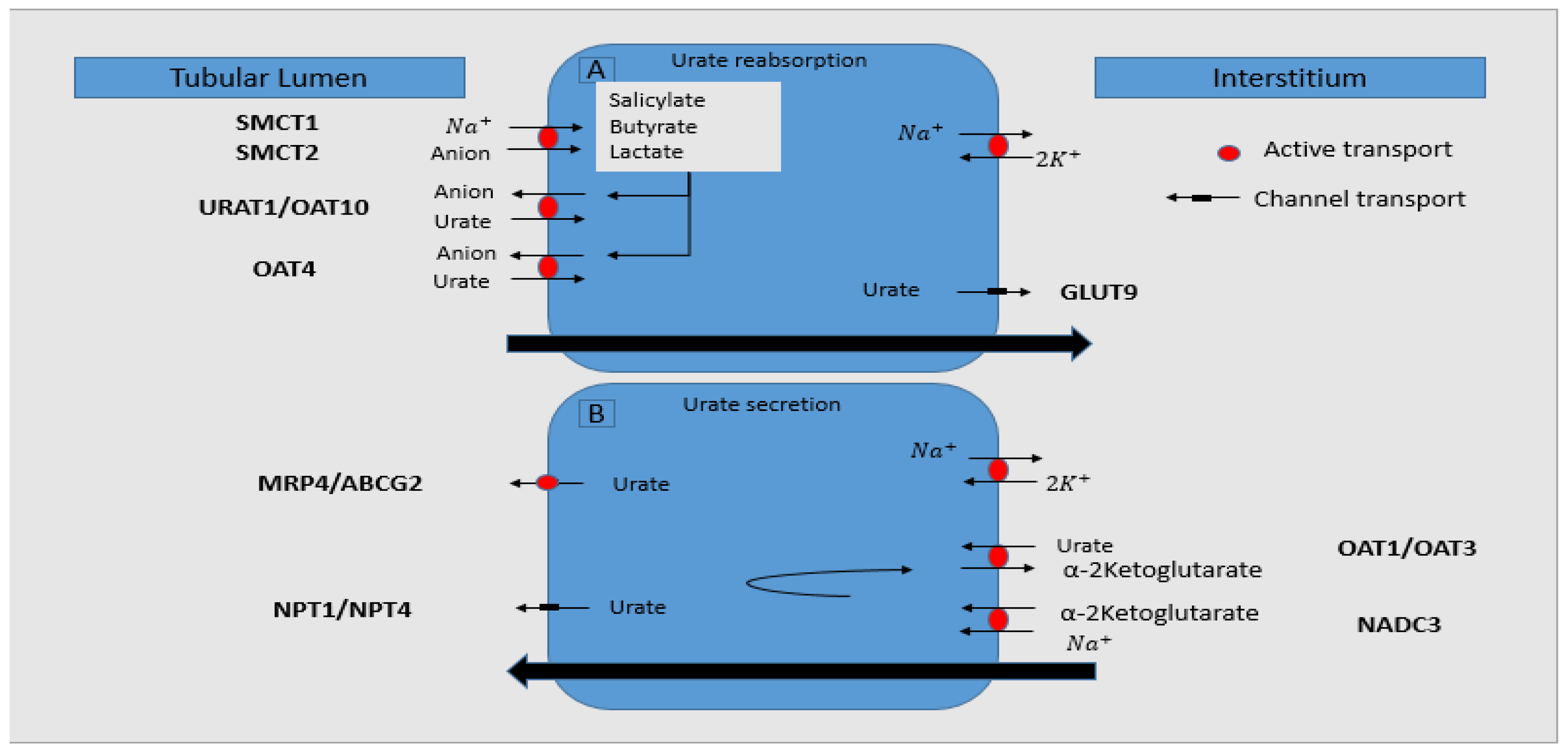
5. Uric Acid Excretion
- Most of the urate is excreted by the kidneys (only one-third is gastro-intestinally eliminated), which involves three phases: filtration, reabsorption, and secretion; knowing that it is not bound to the proteins, it is easily ultra-filtrated by the glomeruli. In the proximal renal tubules, most of it is reabsorbed (~95–98%), and then, secretion occurs, with 10% of filtered uric acid being further excreted by the kidneys [33].
- These processes of urate elimination and regulation are influenced by different proteins/transporters:
- ○
- URAT1 (urate transporter 1)—it belongs to the organic anion transporter (OAT) family and is a urate exchanger in the apical region of the proximal renal tubules, being encoded by SLC22A12 (solute carrier family 22, organic anion/urate transporter, member 12) gene. It is responsible for the urate reabsorption, therefore having an important key role in maintaining serum UA normal values.
- ○
- BCRP (breast-cancer-resistance protein)—a transporter encoded by ABCG2 (adenosine triphosphate binding cassette subfamily G member 2) gene that recently was observed to have an essential contribution in urate excretion. In CKD, high urate levels induce a more active intestinal transporter of UA through ABCG2 increased expression and/or functionality [34].
- ○
- GLUT9 (glucose transporter 9)—a transporter for urate, with similar roles as URAT1, located in the basolateral region of the proximal renal tubules.
- ○
- SGLT2 (sodium-glucose transporter 2)—recent data suggested its contribution in transporting urate and consequent involvement in its excretion.
6. Pathogenic Potential of Serum Uric Acid in Renal Impairment
7. Bimodal Role of Uric Acid in Oxidative Balance
8. Oxidative Stress and Uric Acid
9. Non-Interventional Studies Focused on Uric Acid
10. Mendelian Randomized Studies Focused on Uric Acid
11. Available Treatment Option for Lowering Uric Acid
- Estradiol and Losartan are also URAT1 and GLUT9 inhibitors, whereas Fenofibrate acts only through URAT1 inhibition [99]. Despite higher prevalence of CKD in women, a recent study from nationwide Swedish Renal Registry—CKD evidenced that males are prone to a higher eGFR decline. Apparently, more and more studies sustain that sex hormones, especially estrogen, exerts renal and cardiovascular protective proprieties. This may also be a reason for lower levels of uric acid in women [100].
- Vitamin E is a nutrient lipid soluble with pleiotropic benefits in protecting the integrity of cell membranes through ROS scavengers and by blocking the chain of oxidative reactions [101].
- Vitamin C or ascorbic acid and NO are involved in a complex relationship. It has been suggested that ascorbic acid is a potent antioxidant and reduces serum UA levels by increasing urinary excretion, inhibiting AU synthesis, and by directly decreasing ROS-derived cell damage. Flavonoids and other polyphenols due to their anti-oxidative capacity act like superoxide and XO inhibitors, resulting in suppression of ROS and UA synthesis [102].
- ʟ-Arginine and N-Acetylcysteine also known as strong antioxidants and have shown repeatedly good results in efficient anti-oxidative properties in CKD. ʟ-Arginine is a substrate for NO synthesis [17].
- An NHANES study in 14,758 subjects sustained that only tea and coffee consumption was associated with low levels of serum UA [103].
12. Interventional Studies Focused on Uric Acid
Current Guidelines Recommendations
- In 2020, the Guideline for Management of Gout by the American College of Rheumatology (ACR) recommended initiating urate-lowering therapy for gout patients with more than one subcutaneous tophus, evidence of radiographical damage attributable to gout, or frequent gout flares (>2 per year) [121].
- Japanese guidelines for gout recommended to treat asymptomatic hyperuricemia in patients with UA serum level over 8 mg/dL and complications, such as CKD, urolithiasis, hypertension, cardiovascular disease, diabetes mellitus, and metabolic syndrome, or in patients with UA serum level over 9 mg/dL [122].
- In 2017, EULAR recommendations for gout management suggested Allopurinol as first-line treatment. If target UA level could not be managed (serum UA < 6 mg/dL), the use of Febuxostat or other uricosuric agent was indicated. As third line, a combination of uricosuric and XO inhibitor was suggested. Pegloticase was recommended only for refractory gout [123].
13. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Adomako, E.; Moe, O.W. Uric Acid and Urate in Urolithiasis: The Innocent Bystander, Instigator, and Perpetrator. Semin. Nephrol. 2020, 40, 564–573. [Google Scholar] [CrossRef] [PubMed]
- Jakše, B.; Jakše, B.; Pajek, M.; Pajek, J. Uric Acid and Plant-Based Nutrition. Nutrients 2019, 11, 1736. [Google Scholar] [CrossRef]
- Caliceti, C.; Calabria, D.; Roda, A.; Cicero, A. Fructose Intake, Serum Uric Acid, and Cardiometabolic Disorders: A Critical Review. Nutrients 2017, 9, 395. [Google Scholar] [CrossRef]
- Choi, H.K.; Mount, D.B.; Reginato, A.M. Pathogenesis of gout. Ann. Intern. Med. 2005, 143, 499–516. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Al Salhi, Y.; Tasca, A.; Palleschi, G.; Fuschi, A.; De Nunzio, C.; Bozzini, G.; Mazzaferro, S.; Pastore, A.L. Obesity and kidney stone disease: A systematic review. Minerva Urol. Nefrol. 2018, 70, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Gaubert, M.; Bardin, T.; Cohen-Solal, A.; Diévart, F.; Fauvel, J.P.; Guieu, R.; Sadrin, S.; Maixent, J.M.; Galinier, M.; Paganelli, F. Hyperuricemia and Hypertension, Coronary Artery Disease, Kidney Disease: From Concept to Practice. Int. J. Mol. Sci. 2020, 21, 4066. [Google Scholar] [CrossRef] [PubMed]
- Aktas, G.; Khalid, A.; Kurtkulagi, O.; Duman, T.T.; Bilgin, S.; Kahveci, G.; Atak Tel, B.M.; Sincer, I.; Gunes, Y. Poorly controlled hypertension is associated with elevated serum uric acid to HDL-cholesterol ratio: A cross-sectional cohort study. Postgrad. Med. 2022, 1–6, Epub ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Pandya, B.J.; Choi, H.K. Comorbidities of gout and hyperuricemia in the US general population: NHANES 2007–2008. Am. J. Med. 2012, 125, 679–687.e1. [Google Scholar] [CrossRef] [PubMed]
- Novak, S.; Melkonian, A.K.; Patel, P.A.; Kleinman, N.L.; Joseph-Ridge, N.; Brook, R.A. Metabolic syndrome-related conditions among people with and without gout: Prevalence and resource use. Curr. Med. Res. Opin. 2007, 23, 623–630. [Google Scholar] [CrossRef]
- Heda, R.; Yazawa, M.; Shi, M.; Bhaskaran, M.; Aloor, F.Z.; Thuluvath, P.J.; Satapathy, S.K. Non-alcoholic fatty liver and chronic kidney disease: Retrospect, introspect, and prospect. World J. Gastroenterol. 2021, 27, 1864–1882. [Google Scholar] [CrossRef]
- Franco, Á.O.; Starosta, R.T.; Roriz-Cruz, M. The specific impact of uremic toxins upon cognitive domains: A review. Braz. J. Nephrol. 2019, 41, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Jo, Y.I.; Lee, J.H. Renal effects of uric acid: Hyperuricemia and hypouricemia. Korean J. Intern. Med. 2020, 35, 1291–1304. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Guo, Y.; Luo, W.; Lin, C.; Ding, M. Serum urate and the risk of Parkinson’s disease: Results from a meta-analysis. Can. J. Neurol. Sci. 2013, 40, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Moccia, M.; Lanzillo, R.; Costabile, T.; Russo, C.; Carotenuto, A.; Sasso, G.; Postiglione, E.; De Luca Picione, C.; Vastola, M.; Maniscalco, G.T.; et al. Uric acid in relapsing-remitting multiple sclerosis: A 2-year longitudinal study. J. Neurol. 2015, 262, 961–967. [Google Scholar] [CrossRef]
- Bolayir, A.; Cigdem, B.; Gokce, S.F.; Yilmaz, D. The relationship between neutrophil/lymphocyte ratio and uric acid levels in multiple sclerosis patients. Bratisl. Lek. Listy. 2021, 122, 357–361. [Google Scholar] [CrossRef]
- Lu, N.; Dubreuil, M.; Zhang, Y.; Neogi, T.; Rai, S.K.; Ascherio, A.; Hernán, M.A.; Choi, H.K. Gout and the risk of Alzheimer’s disease: A population-based, BMI-matched cohort study. Ann. Rheum. Dis. 2016, 75, 547–551. [Google Scholar] [CrossRef]
- Roumeliotis, S.; Roumeliotis, A.; Dounousi, E.; Eleftheriadis, T.; Liakopoulos, V. Dietary Antioxidant Supplements and Uric Acid in Chronic Kidney Disease: A Review. Nutrients 2019, 11, 1911. [Google Scholar] [CrossRef]
- Keenan, R.T. The biology of urate. Semin. Arthritis Rheum. 2020, 50, S2–S10. [Google Scholar] [CrossRef] [PubMed]
- Available online. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Uric-acid#section=Dissociation-Constants (accessed on 16 January 2022).
- El Ridi, R.; Tallima, H. Physiological functions and pathogenic potential of uric acid: A review. J. Adv. Res. 2017, 8, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Yang, F.; Yang, I.; Yin, Y.; Luo, J.J.; Wang, H.; Yang, X.F. Uric acid, hyperuricemia and vascular diseases. Front. Biosci. Landmark Ed. 2012, 17, 656–669. [Google Scholar] [CrossRef]
- Kimura, Y.; Tsukui, D.; Kono, H. Uric Acid in Inflammation and the Pathogenesis of Atherosclerosis. Int. J. Mol. Sci. 2021, 22, 12394. [Google Scholar] [CrossRef] [PubMed]
- Waheed, Y.; Yang, F.; Sun, D. Role of asymptomatic hyperuricemia in the progression of chronic kidney disease and cardiovascular disease. Korean J. Intern. Med. 2021, 36, 1281–1293. [Google Scholar] [CrossRef]
- Kim, K.M.; Henderson, G.N.; Ouyang, X.; Frye, R.F.; Sautin, Y.Y.; Feig, D.I.; Johnson, R.J. A sensitive and specific liquid chromatography-tandem mass spectrometry method for the determination of intracellular and extracellular uric acid. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2009, 877, 2032–2038. [Google Scholar] [CrossRef] [PubMed]
- Su, H.Y.; Yang, C.; Liang, D.; Liu, H.F. Research Advances in the Mechanisms of Hyperuricemia-Induced Renal Injury. Biomed. Res. Int. 2020, 2020, 5817348. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.G.; Hafen, P.S.; Brault, J.J. Increased Adenine Nucleotide Degradation in Skeletal Muscle Atrophy. Int. J. Mol. Sci. 2019, 21, 88. [Google Scholar] [CrossRef] [PubMed]
- Petreski, T.; Ekart, R.; Hojs, R.; Bevc, S. Hyperuricemia, the heart, and the kidneys—To treat or not to treat? Ren. Fail. 2020, 42, 978–986. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Sanchez-Lozada, L.G.; Andres-Hernando, A.; Kojima, H.; Kasahara, M.; Rodriguez-Iturbe, B.; Bjornstad, P.; Lanaspa, M.A.; Johnson, R.J. Endogenous Fructose Metabolism Could Explain the Warburg Effect and the Protection of SGLT2 Inhibitors in Chronic Kidney Disease. Front. Immunol. 2021, 12, 694457. [Google Scholar] [CrossRef]
- Major, T.J.; Topless, R.K.; Dalbeth, N.; Merriman, T.R. Evaluation of the diet wide contribution to serum urate levels: Meta-analysis of population based cohorts. BMJ 2018, 363, k3951. [Google Scholar] [CrossRef]
- Mandal, A.K.; Mount, D.B. The molecular physiology of uric acid homeostasis. Annu. Rev. Physiol. 2015, 77, 323–345. [Google Scholar] [CrossRef]
- Wong, K.; Briddon, S.J.; Holliday, N.D.; Kerr, I.D. Plasma membrane dynamics and tetrameric organisation of ABCG2 transporters in mammalian cells revealed by single particle imaging techniques. Biochim. Biophys. Acta 2016, 1863, 19–29. [Google Scholar] [CrossRef]
- Mahaffey, K.W.; Li, J.; Badve, S.V.; Zhou, Z.; Oh, R.; Lee, M.; Perkovic, V.; de Zeeuw, D.; Fulcher, G.; Matthews, D.R.; et al. The effects of canagliflozin on uric acid and gout in patients with type 2 diabetes in the CANVAS programme. Diabetologia 2019, 62, S340–S341. [Google Scholar]
- Roch-Ramel, F.; Guisan, B. Renal transport of urate in humans. News Physiol. Sci. 1999, 14, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Nigam, S.K.; Bhatnagar, V. The systems biology of uric acid transporters. Curr. Opin. Nephrol. Hypertens. 2018, 27, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.J.; Bakris, G.L.; Borghi, C.; Chonchol, M.B.; Feldman, D.; Lanaspa, M.A.; Merriman, T.R.; Moe, O.W.; Mount, D.B.; Sanchez Lozada, L.G.; et al. Hyperuricemia, Acute and Chronic Kidney Disease, Hypertension, and Cardiovascular Disease: Report of a Scientific Workshop Organized by the National Kidney Foundation. Am. J. Kidney Dis. 2018, 71, 851–865. [Google Scholar] [CrossRef]
- Fanelli, G.M.; Bohn, D.; Stafford, S. Functional characteristics of renal urate transport in the Cebus monkey. Am. J. Physiol. 1970, 218, 627–636. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gershon, S.L.; Fox, I.H. Pharmacologic effects of nicotinic acid on human purine metabolism. J. Lab. Clin. Med. 1974, 84, 179–186. [Google Scholar]
- Köttgen, A.; Albrecht, E.; Teumer, A.; Vitart, V.; Krumsiek, J.; Hundertmark, C.; Pistis, G.; Ruggiero, D.; O’Seaghdha, C.M.; Haller, T.; et al. Genome-wide association analyses identify 18 new loci associated with serum urate concentrations. Nat. Genet. 2013, 45, 145–154. [Google Scholar] [CrossRef]
- Goldfinger, S.; Klinenberg, E.; Seegmiller, J.E. Renal retention of uric acid induced by infusion of beta-hydroxybutyrate and acetoacetate. N. Engl. J. Med. 1965, 272, 351–355. [Google Scholar] [CrossRef]
- Uetake, D.; Ohno, I.; Ichida, K.; Yamaguchi, Y.; Saikawa, H.; Endou, H.; Hosoya, T. Effect of fenofibrate on uric acid metabolism and urate transporter 1. Intern. Med. 2010, 49, 89–94. [Google Scholar] [CrossRef]
- Nakashima, M.; Uematsu, T.; Kosuge, K.; Kanamaru, M. Pilot study of the uricosuric effect of DuP-753, a new angiotensin II receptor antagonist, in healthy subjects. Eur. J. Clin. Pharmacol. 1992, 42, 333–335. [Google Scholar] [CrossRef]
- Dinour, D.; Gray, N.K.; Campbell, S.; Shu, X.; Sawyer, L.; Richardson, W.; Rechavi, G.; Amariglio, N.; Ganon, L.; Sela, B.A.; et al. Homozygous SLC2A9 mutations cause severe renal hypouricemia. J. Am. Soc. Nephrol. 2010, 21, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Woodward, O.M.; Köttgen, A.; Coresh, J.; Boerwinkle, E.; Guggino, W.B.; Köttgen, M. Identification of a urate transporter, ABCG2, with a common functional polymorphism causing gout. Proc. Natl. Acad. Sci. USA 2009, 106, 10338–10342. [Google Scholar] [CrossRef] [PubMed]
- Woodward, O.M.; Tukaye, D.N.; Cui, J.; Greenwell, P.; Constantoulakis, L.M.; Parker, B.S.; Rao, A.; Köttgen, M.; Maloney, P.C.; Guggino, W.B. Gout-causing Q141K mutation in ABCG2 leads to instability of the nucleotide-binding domain and can be corrected with small molecules. Proc. Natl. Acad. Sci. USA 2013, 110, 5223–5228. [Google Scholar] [CrossRef] [PubMed]
- Jutabha, P.; Anzai, N.; Kitamura, K.; Taniguchi, A.; Kaneko, S.; Yan, K.; Yamada, H.; Shimada, H.; Kimura, T.; Katada, T.; et al. Human sodium phosphate transporter 4 (hNPT4/SLC17A3) as a common renal secretory pathway for drugs and urate. J. Biol. Chem. 2010, 285, 35123–35132. [Google Scholar] [CrossRef] [PubMed]
- Maesaka, J.K.; Fishbane, S. Regulation of renal urate excretion: A critical review. Am. J. Kidney Dis. 1998, 32, 917–933. [Google Scholar] [CrossRef]
- Maiuolo, J.; Oppedisano, F.; Gratteri, S.; Muscoli, C.; Mollace, V. Regulation of uric acid metabolism and excretion. Int. J. Cardiol. 2016, 213, 8–14. [Google Scholar] [CrossRef]
- Egan, B.M.; Lackland, D.T. Biochemical and Metabolic Effects of Very-Low-Salt Diets. Am. J. Med. Sci. 2000, 320, 233–239. [Google Scholar] [CrossRef]
- Xin, P.; Jiang, G.H.; Zheng, W.L.; Fan, L.L.; Li, C.K.; Wang, D.Z. Study on the diet balance index and its relationship with blood uric acid of smoking adults in Tianjin. Zhonghua Liu Xing Bing Xue Za Zhi 2021, 42, 1076–1079. [Google Scholar]
- Quiñones Galvan, A.; Natali, A.; Baldi, S.; Frascerra, S.; Sanna, G.; Ciociaro, D.; Ferrannini, E. Effect of insulin on uric acid excretion in humans. Am. J. Physiol. 1995, 268, E1–E5. [Google Scholar] [CrossRef]
- Flisiński, M.; Brymora, A.; Skoczylas-Makowska, N.; Stefańska, A.; Manitius, J. Fructose-Rich Diet Is a Risk Factor for Metabolic Syndrome, Proximal Tubule Injury and Urolithiasis in Rats. Int. J. Mol. Sci. 2021, 23, 203. [Google Scholar] [CrossRef]
- Chino, Y.; Kuwabara, M.; Hisatome, I. Factors Influencing Change in Serum Uric Acid After Administration of the Sodium-Glucose Cotransporter 2 Inhibitor Luseogliflozin in Patients with Type 2 Diabetes Mellitus. J. Clin. Pharmacol. 2021, 62, 366–375, Epub ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Moriwaki, Y.; Yamamoto, T.; Tsutsumi, Z.; Takahashi, S.; Hada, T. Effects of angiotensin II infusion on renal excretion of purine bases and oxypurinol. Metabolism 2002, 51, 893–895. [Google Scholar] [CrossRef] [PubMed]
- Hui, J.Y.; Choi, J.W.J.; Mount, D.B.; Zhu, Y.; Zhang, Y.; Choi, H.K. The independent association between parathyroid hormone levels and hyperuricemia: A national population study. Arthritis Res. Ther. 2012, 14, R56. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Dnyanmote, A.V.; Nigam, S.K. Remote communication through solute carriers and ATP binding cassette drug transporter pathways: An update on the Remote Sensing and Signaling Hypothesis. Mol. Pharmacol. 2011, 79, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Zanoli, L.; Lentini, P.; Briet, M.; Castellino, P.; House, A.A.; London, G.M.; Malatino, L.; McCullough, P.A.; Mikhailidis, D.P.; Boutouyrie, P. Arterial Stiffness in the Heart Disease of CKD. J. Am. Soc. Nephrol. 2019, 30, 918–928. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.H.; Park, S.K.; Lee, I.K.; Johnson, R.J. Uric acid-induced C-reactive protein expression: Implication on cell proliferation and nitric oxide production of human vascular cells. J. Am. Soc. Nephrol. 2005, 16, 3553–3562. [Google Scholar] [CrossRef]
- Zhou, Y.; You, H.; Zhang, A.; Jiang, X.; Pu, Z.; Xu, G.; Zhao, M. Lipoxin A4 attenuates uric acid-activated, NADPH oxidase-dependent oxidative stress by interfering with translocation of p47phox in human umbilical vein endothelial cells. Exp. Ther. Med. 2020, 20, 1682–1692. [Google Scholar] [CrossRef]
- Corry, D.B.; Eslami, P.; Yamamoto, K.; Nyby, M.D.; Makino, H.; Tuck, M.L. Uric acid stimulates vascular smooth muscle cell proliferation and oxidative stress via the vascular renin-angiotensin system. J. Hyperten. 2008, 26, 269–275. [Google Scholar] [CrossRef]
- Doğru, S.; Yaşar, E.; Yeşilkaya, A. Uric acid can enhance MAPK pathway-mediated proliferation in rat primary vascular smooth muscle cells via controlling of mitochondria and caspase-dependent cell death. J. Recept. Signal Transduct. Res. 2021, 30, 1–9. [Google Scholar] [CrossRef]
- Ruggiero, C.; Cherubini, A.; Miller, E.; Maggio, M.; Najjar, S.S.; Lauretani, F.; Bandinelli, S.; Senin, U.; Ferrucci, L. Usefulness of Uric Acid to Predict Changes in C-Reactive Protein and Interleukin-6 in 3-Year Period in Italians Aged 21 to 98 Years. Am. J. Cardiol. 2007, 100, 115–121. [Google Scholar] [CrossRef]
- Gruszka, K.; Drożdż, T.; Wojciechowska, W.; Jankowski, P.; Terlecki, M.; Bijak, M.; Hering, D.; Bilo, G.; Drożdż, D.; Rajzer, M. Effects of uric acid-lowering therapy in patients with essential arterial hypertension. Blood Press. Monit. 2022. Epub ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Ejaz, A.A.; Johnson, R.J.; Shimada, M.; Mohandas, R.; Alquadan, K.F.; Beaver, T.M.; Lapsia, V.; Dass, B. The Role of Uric Acid in Acute Kidney Injury. Nephron 2019, 142, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Isaka, Y.; Takabatake, Y.; Takahashi, A.; Saitoh, T.; Yoshimori, T. Hyperuricemia-induced inflammasome and kidney diseases. Nephrol. Dial. Transplant. 2016, 31, 890–896. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Chen, W.; Jalal, D.; Li, Z.; Chen, W.; Mao, H.; Yang, Q.; Johnson, R.J.; Yu, X. Clinical outcome of hyperuricemia in IgA nephropathy: A retrospective cohort study and randomized controlled trial. Kidney Blood Press. Res. 2012, 35, 153–160. [Google Scholar] [CrossRef]
- Joosten, L.A.B.; Crişan, T.O.; Bjornstad, P.; Johnson, R.J. Asymptomatic hyperuricaemia: A silent activator of the innate immune system. Nat. Rev. Rheumatol. 2020, 16, 75–86. [Google Scholar] [CrossRef]
- Miao, Y.; Ottenbros, S.A.; Laverman, G.D.; Brenner, B.M.; Cooper, M.E.; Parving, H.H.; Grobbee, D.E.; Shahinfar, S.; De Zeeuw, D.; Heerspink, H.J.L. Effect of a reduction in uric acid on renal outcomes during losartan treatment: A post hoc analysis of the reduction of endpoints in non-insulin-dependent diabetes mellitus with the angiotensin ii antagonist losartan trial. Hypertension 2011, 58, 2–7. [Google Scholar] [CrossRef]
- Borghi, C.; Rosei, E.A.; Bardin, T.; Dawson, J.; Dominiczak, A.; Kielstein, J.T.; Manolis, A.J.; Perez-Ruiz, F.; Mancia, G. Serum uric acid and the risk of cardiovascular and renal disease. J. Hypertens. 2015, 33, 1729–1741. [Google Scholar] [CrossRef]
- Rapa, S.F.; Di Iorio, B.R.; Campiglia, P.; Heidland, A.; Marzocco, S. Inflammation and oxidative stress in chronic kidney disease—potential therapeutic role of minerals, vitamins and plant-derived metabolites. Int. J. Mol. Sci. 2020, 21, 263. [Google Scholar] [CrossRef]
- Hanna, R.M.; Ghobry, L.; Wassef, O.; Rhee, C.M.; Kalantar-Zadeh, K. A Practical Approach to Nutrition, Protein-Energy Wasting, Sarcopenia, and Cachexia in Patients with Chronic Kidney Disease. Blood Purif. 2020, 49, 202–211. [Google Scholar] [CrossRef]
- Grayson, P.C.; Young Kim, S.; Lavalley, M.; Choi, H.K. Hyperuricemia and incident hypertension: A systematic review and meta-analysis. Arthritis Care Res. 2011, 63, 102–110. [Google Scholar] [CrossRef]
- Wang, J.; Qin, T.; Chen, J.; Li, Y.; Wang, L.; Huang, H.; Li, J. Hyperuricemia and risk of incident hypertension: A systematic review and meta-analysis of observational studies. PLoS ONE 2014, 9, e114259. [Google Scholar] [CrossRef] [PubMed]
- Lv, Q.; Meng, X.F.; He, F.F.; Chen, S.; Su, H.; Xiong, J.; Gao, P.; Tian, X.J.; Liu, J.S.; Zhu, Z.H.; et al. High Serum Uric Acid and Increased Risk of Type 2 Diabetes: A Systemic Review and Meta-Analysis of Prospective Cohort Studies. PLoS ONE 2013, 8, e56864. [Google Scholar] [CrossRef]
- Kielstein, J.T.; Pontremoli, R.; Burnier, M. Management of Hyperuricemia in Patients with Chronic Kidney Disease: A Focus on Renal Protection. Curr. Hypertens. Rep. 2020, 22, 102. [Google Scholar] [CrossRef]
- Chonchol, M.; Shlipak, M.G.; Katz, R.; Sarnak, M.J.; Newman, A.B.; Siscovick, D.S.; Kestenbaum, B.; Carney, J.K.; Fried, L.F. Relationship of uric acid with progression of kidney disease. Am. J. Kidney Dis. 2007, 50, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Xia, X.; Li, B.; Lin, Z.; Yu, X.; Huang, F. Serum uric acid and cardiovascular mortality in chronic kidney disease: A meta-analysis. BMC Nephrol. 2019, 20, 18. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.W.; Lin, S.Y.; Kuo, C.C.; Huang, C.C. Serum Uric Acid and Progression of Kidney Disease: A Longitudinal Analysis and Mini-Review. PLoS ONE 2017, 12, e0170393. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhao, M.; Pu, Z.; Xu, G.; Li, X. Relationship between oxidative stress and inflammation in hyperuricemia Analysis based on asymptomatic young patients with primary hyperuricemia. Medicine 2018, 97, e13108. [Google Scholar] [CrossRef]
- Yang, T.; Ding, X.; Wang, Y.; Zeng, C.; Wie, J.; Li, H.; Xiong, Y.L.; Gao, S.G.; Li, Y.S.; Lei, G.H. Association between high-sensitivity C-reactive protein and hyperuricemia. Rheumatol. Int. 2016, 36, 561–566. [Google Scholar] [CrossRef]
- Viazzi, F.; Piscitelli, P.; Giorda, C.; Ceriello, A.; Genovese, S.; Russo, G.; Guida, P.; Fioretto, P.; De Cosmo, S.; Pontremoli, R. Metabolic syndrome, serum uric acid and renal risk in patients with T2D. PLoS ONE 2017, 12, e0176058. [Google Scholar] [CrossRef]
- Oikonen, M.; Wendelin-Saarenhovi, M.; Lyytikäinen, L.P.; Siitonen, N.; Loo, B.M.; Jula, A.; Seppälä, I.; Saarikoski, L.; Lehtimäki, T.; Hutri-Kähönen, N.; et al. Associations between serum uric acid and markers of subclinical atherosclerosis in young adults. The cardiovascular risk in Young Finns study. Atherosclerosis 2012, 223, 497–503. [Google Scholar] [CrossRef]
- Palmer, T.M.; Nordestgaard, B.G.; Benn, M.; Tybjarg-Hansen, A.; Smith, G.D.; Lawlor, D.A.; Timpson, N.J. Association of plasma uric acid with ischaemic heart disease and blood pressure: Mendelian randomization analysis of two large cohorts. BMJ 2013, 347, f4262. [Google Scholar] [CrossRef] [PubMed]
- Sedaghat, S.; Pazoki, R.; Uitterlinden, A.G.; Hofman, A.; Stricker, B.H.C.; Ikram, M.A.; Franco, O.H.; Dehghan, A. Association of uric acid genetic risk score with blood pressure: The rotterdam study. Hypertension 2014, 64, 1061–1066. [Google Scholar] [CrossRef] [PubMed]
- Mallamaci, F.; Testa, A.; Leonardis, D.; Tripepi, R.; Pisano, A.; Spoto, B.; Sanguedolce, M.C.; Parlongo, R.M.; Tripepi, G.; Zoccali, C. A polymorphism in the major gene regulating serum uric acid associates with clinic SBP and the white-coat effect in a family-based study. J. Hypertens. 2014, 32, 1621–1628. [Google Scholar] [CrossRef]
- Kleber, M.E.; Delgado, G.; Grammer, T.B.; Silbernagel, G.; Huang, J.; Krämer, B.K.; Ritz, E.; März, W. Uric acid and cardiovascular events: A Mendelian randomization study. J. Am. Soc. Nephrol. 2015, 26, 2831–2838. [Google Scholar] [CrossRef] [PubMed]
- Hughes, K.; Flynn, T.; De Zoysa, J.; Dalbeth, N.; Merriman, T.R. Mendelian randomization analysis associates increased serum urate, due to genetic variation in uric acid transporters, with improved renal function. Kidney Int. 2014, 85, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Niu, S.W.; Hung, C.C.; Lin, H.Y.; Kuo, I.C.; Huang, J.C.; He, J.S.; Wen, Z.H.; Liang, P.I.; Chiu, Y.W.; Chang, J.M.; et al. Reduced Incidence of Stroke in Patients with Gout Using Benzbromarone. J. Pers. Med. 2022, 12, 28. [Google Scholar] [CrossRef] [PubMed]
- Rana, P.; Aleo, M.D.; Wen, X.; Kogut, S. Hepatotoxicity reports in the FDA adverse event reporting system database: A comparison of drugs that cause injury via mitochondrial or other mechanisms. Acta Pharm. Sin. B 2021, 11, 3857–3868. [Google Scholar] [CrossRef]
- Kuriyama, S. Dotinurad: A novel selective urate reabsorption inhibitor as a future therapeutic option for hyperuricemia. Clin. Exp. Nephrol. 2020, 24, 1–5. [Google Scholar] [CrossRef]
- Bove, M.; Cicero, A.F.G.; Veronesi, M.; Borghi, C. An evidence-based review on urate-lowering treatments: Implications for optimal treatment of chronic hyperuricemia. Vasc. Health Risk Manag. 2017, 13, 23–28. [Google Scholar] [CrossRef]
- Ejaz, A.A.; Nakagawa, T.; Kanbay, M.; Kuwabara, M.; Kumar, A.; Garcia Arroyo, F.E.; Roncal-Jimenez, C.; Sasai, F.; Kang, D.H.; Jensen, T.; et al. Hyperuricemia in Kidney Disease: A Major Risk Factor for Cardiovascular Events, Vascular Calcification, and Renal Damage. Semin. Nephrol. 2020, 40, 574–585. [Google Scholar] [CrossRef]
- Bailey, C.J. Uric acid and the cardio-renal effects of SGLT2 inhibitors. Diabetes Obes. Metab. 2019, 21, 1291–1298. [Google Scholar] [CrossRef] [PubMed]
- Takata, T.; Isomoto, H. Pleiotropic Effects of Sodium-Glucose Cotransporter-2 Inhibitors: Renoprotective Mechanisms beyond Glycemic Control. Int. J. Mol. Sci. 2021, 22, 4374. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, C.C.J.; Gansevoort, R.T.; Heerspink, H.J.L. New Diabetes Therapies and Diabetic Kidney Disease Progression: The Role of SGLT-2 Inhibitors. Curr. Diab. Rep. 2018, 18, 27. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, C.C.J.; Petrykiv, S.; Laverman, G.D.; Cherney, D.Z.; Gansevoort, R.T.; Heerspink, H.J.L. Effects of the SGLT-2 inhibitor dapagliflozin on glomerular and tubular injury markers. Diabetes Obes. Metab. 2018, 20, 1988–1993. [Google Scholar] [CrossRef]
- Baer, P.C.; Koch, B.; Freitag, J.; Schubert, R.; Geiger, H. No Cytotoxic and Inflammatory Effects of Empagliflozin and Dapagliflozin on Primary Renal Proximal Tubular Epithelial Cells under Diabetic Conditions In Vitro. Int. J. Mol. Sci. 2020, 21, 391. [Google Scholar] [CrossRef]
- Shaffner, J.; Chen, B.; Malhotra, D.K.; Dworkin, L.D.; Gong, R. Therapeutic Targeting of SGLT2: A New Era in the Treatment of Diabetes and Diabetic Kidney Disease. Front. Endocrinol. 2021, 12, 749010. [Google Scholar] [CrossRef]
- Van Bommel, E.J.M.; Muskiet, M.H.A.; Tonneijck, L.; Kramer, M.H.H.; Nieuwdorp, M.; van Raalte, D.H. SGLT2 Inhibition in the Diabetic Kidney—From Mechanisms to Clinical Outcome. Clin. J. Am. Soc. Nephrol. 2017, 12, 700–710. [Google Scholar] [CrossRef]
- Hu, J.; Xu, W.; Yang, H.; Mu, L. Uric acid participating in female reproductive disorders: A review. Reprod. Biol. Endocrinol. 2021, 19, 65. [Google Scholar] [CrossRef]
- Giandalia, A.; Giuffrida, A.E.; Gembillo, G.; Cucinotta, D.; Squadrito, G.; Santoro, D.; Russo, G.T. Gender differences in diabetic kidney disease: Focus on hormonal, genetic and clinical factors. Int. J. Mol. Sci. 2021, 22, 5808. [Google Scholar] [CrossRef]
- Limirio, L.S.; Santos, H.O.; Dos Reis, A.S.; de Oliveira, E.P. Association Between Dietary Intake and Serum Uric Acid Levels in Kidney Transplant Patients. J. Ren. Nutr. 2021, 31, 637–647. [Google Scholar] [CrossRef]
- Ma, J.; Han, R.; Cui, T.; Yang, C.; Wang, S. Effects of high serum uric acid levels on oxidative stress levels and semen parameters in male infertile patients. Medicine 2022, 101, e28442. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.K.; Curhan, G. Coffee, tea, and caffeine consumption and serum uric acid level: The third national health and nutrition examination survey. Arthritis Rheum. 2007, 57, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Guthikonda, S.; Sinkey, C.; Barenz, T.; Haynes, W.G. Xanthine oxidase inhibition reverses endothelial dysfunction in heavy smokers. Circulation 2003, 107, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Furuhashi, M. New insights into purine metabolism in metabolic diseases: Role of xanthine oxidoreductase activity. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E827–E834. [Google Scholar] [CrossRef] [PubMed]
- Bove, M.; Cicero, A.F.G.; Borghi, C. The Effect of Xanthine Oxidase Inhibitors on Blood Pressure and Renal Function. Curr. Hypertens. Rep. 2017, 19, 95. [Google Scholar] [CrossRef]
- Agarwal, V.; Hans, N.; Messerli, F.H. Effect of Allopurinol on Blood Pressure: A Systematic Review and Meta-Analysis. J. Clin. Hypertens. 2013, 15, 435–442. [Google Scholar] [CrossRef]
- Goicoechea, M.; de Vinuesa, S.G.; Verdalles, U.; Ruiz-Caro, C.; Ampuero, J.; Rincón, A.; Arroyo, D.; Luño, J. Effect of allopurinol in chronic kidney disease progression and cardiovascular risk. Clin. J. Am. Soc. Nephrol. 2010, 5, 1388–1393. [Google Scholar] [CrossRef]
- Golmohammadi, S.; Almasi, A.; Manouchehri, M.; Omrani, H.R.; Zandkarimi, M.R. Allopurinol against Progression of Chronic Kidney Disease. Iran. J. Kidney Dis. 2017, 11, 286–293. [Google Scholar]
- Bose, B.; Badve, S.V.; Hiremath, S.S.; Boudville, N.; Brown, F.G.; Cass, A.; de Zoysa, J.R.; Fassett, R.G.; Faull, R.; Harris, D.C.; et al. Effects of uric acid-lowering therapy on renal outcomes: A systematic review and meta-analysis. Nephrol. Dial. Transplant. 2014, 29, 406–413. [Google Scholar] [CrossRef]
- Lee, J.W.; Lee, K.H. Comparison of renoprotective effects of febuxostat and allopurinol in hyperuricemic patients with chronic kidney disease. Int. Urol. Nephrol. 2019, 51, 467–473. [Google Scholar] [CrossRef]
- Zhang, X.; Wan, D.; Yang, G.; Peng, Q.; Wang, X. Febuxostat is superior to allopurinol in delaying the progression of renal impairment in patients with chronic kidney disease and hyperuricemia. Int. Urol. Nephrol. 2019, 51, 2273–2283. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.O.; Wu, I.W.; Chang, S.H.; Lee, C.C.; Tsai, C.Y.; Lin, C.Y.; Lin, W.T.; Huang, Y.T.; Wu, C.Y.; Kuo, G.; et al. Comparative Renoprotective Effect of Febuxostat and Allopurinol in Predialysis Stage 5 Chronic Kidney Disease Patients: A Nationwide Database Analysis. Clin. Pharmacol. Ther. 2020, 107, 1159–1169. [Google Scholar] [CrossRef] [PubMed]
- Mauer, M.; Doria, A. Uric acid and risk of diabetic kidney disease. J. Nephrol. 2020, 33, 995–999. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhai, T.; Ma, R.; Luo, C.; Wang, H.; Liu, L. Effects of uric acid-lowering therapy on the progression of chronic kidney disease: A systematic review and meta-analysis. Ren. Fail. 2018, 40, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Sampson, A.L.; Singer, R.F.; Walters, G.D. Uric acid lowering therapies for preventing or delaying the progression of chronic kidney disease. Cochrane Database Syst. Rev. 2017, 10, CD009460. [Google Scholar] [CrossRef]
- Su, X.; Xu, B.; Yan, B.; Qiao, X.; Wang, L. Effects of uric acid-lowering therapy in patients with chronic kidney disease: A meta-analysis. PLoS ONE 2017, 12, e0187550. [Google Scholar] [CrossRef]
- Chen, Q.; Wang, Z.; Zhou, J.; Chen, Z.; Li, Y.; Li, S.; Zhao, H.; Badve, S.V.; Lv, J. Effect of Urate-Lowering Therapy on Cardiovascular and Kidney Outcomes: A Systematic Review and Meta-Analysis. Clin. J. Am. Soc. Nephrol. 2020, 15, 1576–1586. [Google Scholar] [CrossRef]
- Li, X.; Meng, X.; Timofeeva, M.; Tzoulaki, I.; Tsilidis, K.K.; Ioannidis, J.P.; Campbell, H.; Theodoratou, E. Serum uric acid levels and multiple health outcomes: Umbrella review of evidence from observational studies, randomised controlled trials, and Mendelian randomisation studies. BMJ 2017, 357, j2376. [Google Scholar] [CrossRef]
- Lin, T.C.; Hung, L.Y.; Chen, Y.C.; Lo, W.C.; Lin, C.H.; Tam, K.W.; Wu, M.Y. Effects of febuxostat on renal function in patients with chronic kidney disease: A systematic review and meta-analysis. Medicine 2019, 98, e16311. [Google Scholar] [CrossRef]
- FitzGerald, J.D.; Dalbeth, N.; Mikuls, T.; Brignardello-Petersen, R.; Guyatt, G.; Abeles, A.M.; Gelber, A.C.; Harrold, L.R.; Khanna, D.; King, C.; et al. 2020 American College of Rheumatology Guideline for the Management of Gout. Arthritis Care Res. 2020, 72, 744–760. [Google Scholar] [CrossRef]
- Yamanaka, H. Japanese Society of Gout and Nucleic Acid Metabolism. Japanese guideline for the management of hyperuricemia and gout: Second edition. Nucleosides Nucleotides Nucleic Acids 2011, 30, 1018–1029. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.A.; Edwards, N.L. EULAR gout treatment guidelines by Richette et al.: Uric acid and neurocognition. Ann. Rheum. Dis. 2018, 77, e20. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; Pletinck, A.; Schepers, E.; Glorieux, G. Biochemical and Clinical Impact of Organic Uremic Retention Solutes: A Comprehensive Update. Toxins 2018, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Viggiano, D.; Gigliotti, G.; Vallone, G.; Giammarino, A.; Nigro, M.; Capasso, G. Urate-Lowering Agents in Asymptomatic Hyperuricemia: Role of Urine Sediment Analysis and Musculoskeletal Ultrasound. Kidney Blood Press. Res. 2018, 43, 606–615. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Study | Studied Medication | Included Subjects | Results |
|---|---|---|---|
| Agarwal et al. (2012, meta-analysis) [107] | Allopurinol vs. placebo 1:1 | 10 clinical trials with n = 738 subjects with eGFR < 60 mL/min | 3.3 mmHg BP reduction in placebo group |
| Goicoechea et al. (2010) [108] | Allopurinol vs. placebo 1:1 | n = 113 with eGFR < 60 mL/min | Significant reduction of CRP level eGFR 1.3 mL/min/24 months increment |
| Golmohammadi et al. (2017) [109] | Allopurinol vs. placebo 1:1 | n = 216 with eGFR 15–60 mL/min | Reduction of CKD decline with a mean difference of 1 mL/min/year |
| Bose et al. (2014, meta-analysis) [110] | Allopurinol vs. placebo 1:1 | 8 clinical trials with n = 476 with eGFR < 60 mL/min | Mean eGFR retarded by 3.3 mL/min/year in 5 from 8 studies |
| Lee et al. (2019) [111] | Allopurinol vs. Febuxostat vs placebo ½:½:1 | n = 141, mean eGFR baseline = 42.1 mL/min | Febuxostat significant decreased UA level and maintain eGFR significant higher for 4 years in contrast to Allopurinol or placebo |
| Zhang et al. (2019) [112] | Febuxostat vs. Allopurinol 1:1 | n = 152 with CKD stage 2–3 | Febuxostat showed superiority in eGFR decline but not in proteinuria or uric acid control |
| Hsu et al. (2020, meta-analysis) [113] | Febuxostat vs. Allopurinol 1:1 | n = 6057 with CKD stage 5 | Lower risk of progression to dialysis on febuxostat group |
| Liu et al. (2018, meta-analysis) [115] | Uric-acid-lowering therapy | 12 clinical trials with 832 CKD subjects | 8 trials showed a mean serum creatinine reduction by −0.63 |
| Sampson et al. (2017, meta-analysis) [116] | Uric-acid-lowering therapy | 12 clinical trials with 1187 CKD subjects | Conflicting evidence—no apparent benefits in eGFR, blood pressure, or proteinuria control |
| Su et al. (2017, meta-analysis) [117] | Uric-acid-lowering therapy | 16 clinical trial with 1211 CKD subjects | eGFR progression retarded by 4.1 mL/min/year 55% reduced risk of AKI 60% reduced risk in cardiovascular events |
| Chen et al. (2020, meta-analysis) [118] | Uric-acid-lowering therapy | 28 trials with 6458 CKD subjects | No benefits in kidney failure or cardiovascular events |
| Lin et al. (2019, meta-analysis) [120] | Febuxostat vs. placebo | 11 trials with 1317 CKD stage 3–4 subjects | Reno-protective effects with a mean difference in eGFR of 3.6 mL/min |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gherghina, M.-E.; Peride, I.; Tiglis, M.; Neagu, T.P.; Niculae, A.; Checherita, I.A. Uric Acid and Oxidative Stress—Relationship with Cardiovascular, Metabolic, and Renal Impairment. Int. J. Mol. Sci. 2022, 23, 3188. https://doi.org/10.3390/ijms23063188
Gherghina M-E, Peride I, Tiglis M, Neagu TP, Niculae A, Checherita IA. Uric Acid and Oxidative Stress—Relationship with Cardiovascular, Metabolic, and Renal Impairment. International Journal of Molecular Sciences. 2022; 23(6):3188. https://doi.org/10.3390/ijms23063188
Chicago/Turabian StyleGherghina, Mihai-Emil, Ileana Peride, Mirela Tiglis, Tiberiu Paul Neagu, Andrei Niculae, and Ionel Alexandru Checherita. 2022. "Uric Acid and Oxidative Stress—Relationship with Cardiovascular, Metabolic, and Renal Impairment" International Journal of Molecular Sciences 23, no. 6: 3188. https://doi.org/10.3390/ijms23063188
APA StyleGherghina, M.-E., Peride, I., Tiglis, M., Neagu, T. P., Niculae, A., & Checherita, I. A. (2022). Uric Acid and Oxidative Stress—Relationship with Cardiovascular, Metabolic, and Renal Impairment. International Journal of Molecular Sciences, 23(6), 3188. https://doi.org/10.3390/ijms23063188