
Involvement of Mitochondrial Mechanisms and Cyclooxygenase-2 Activation in the Effect of Desethylamiodarone on 4T1 Triple-Negative Breast Cancer Line

 ,
,  ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
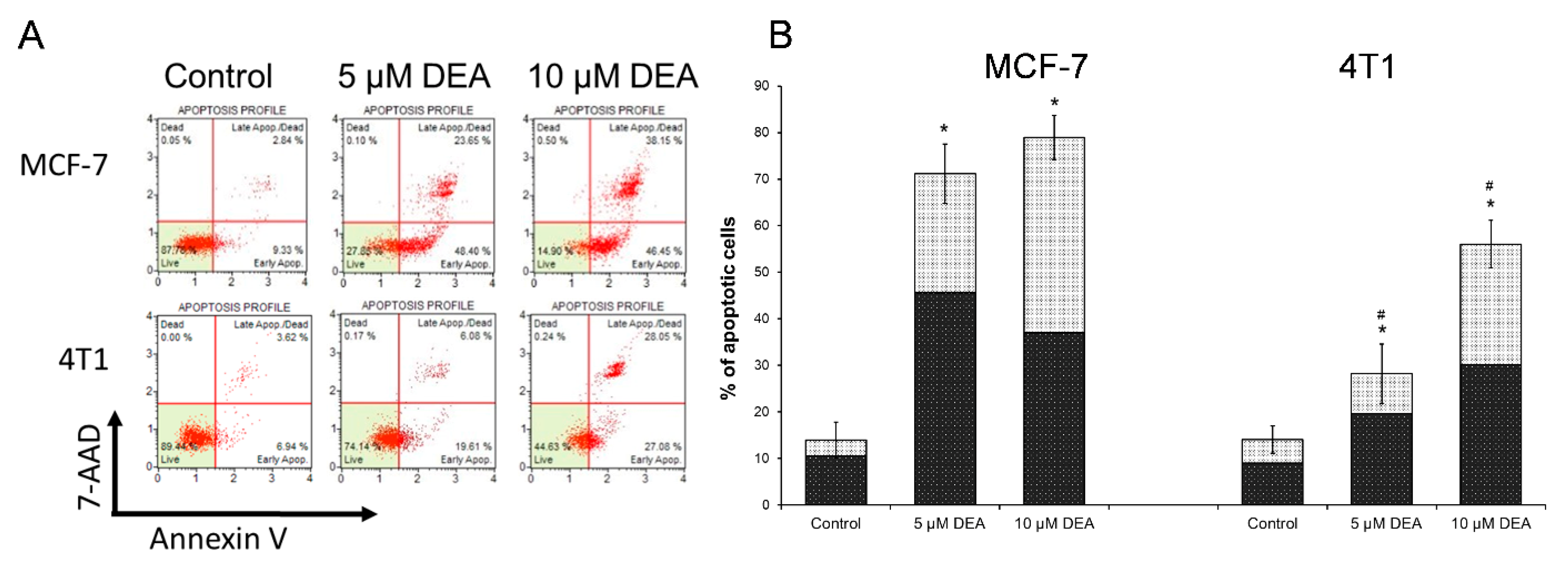
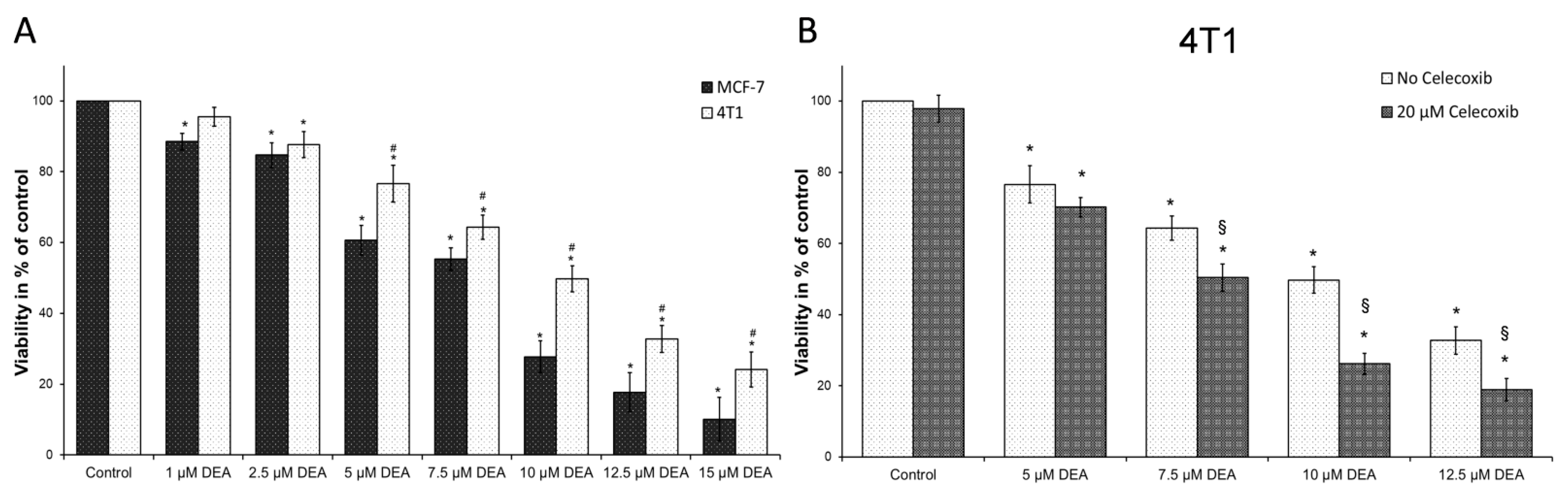
2.1. DEA Induced Apoptotic Cell Death in BC Cell Lines
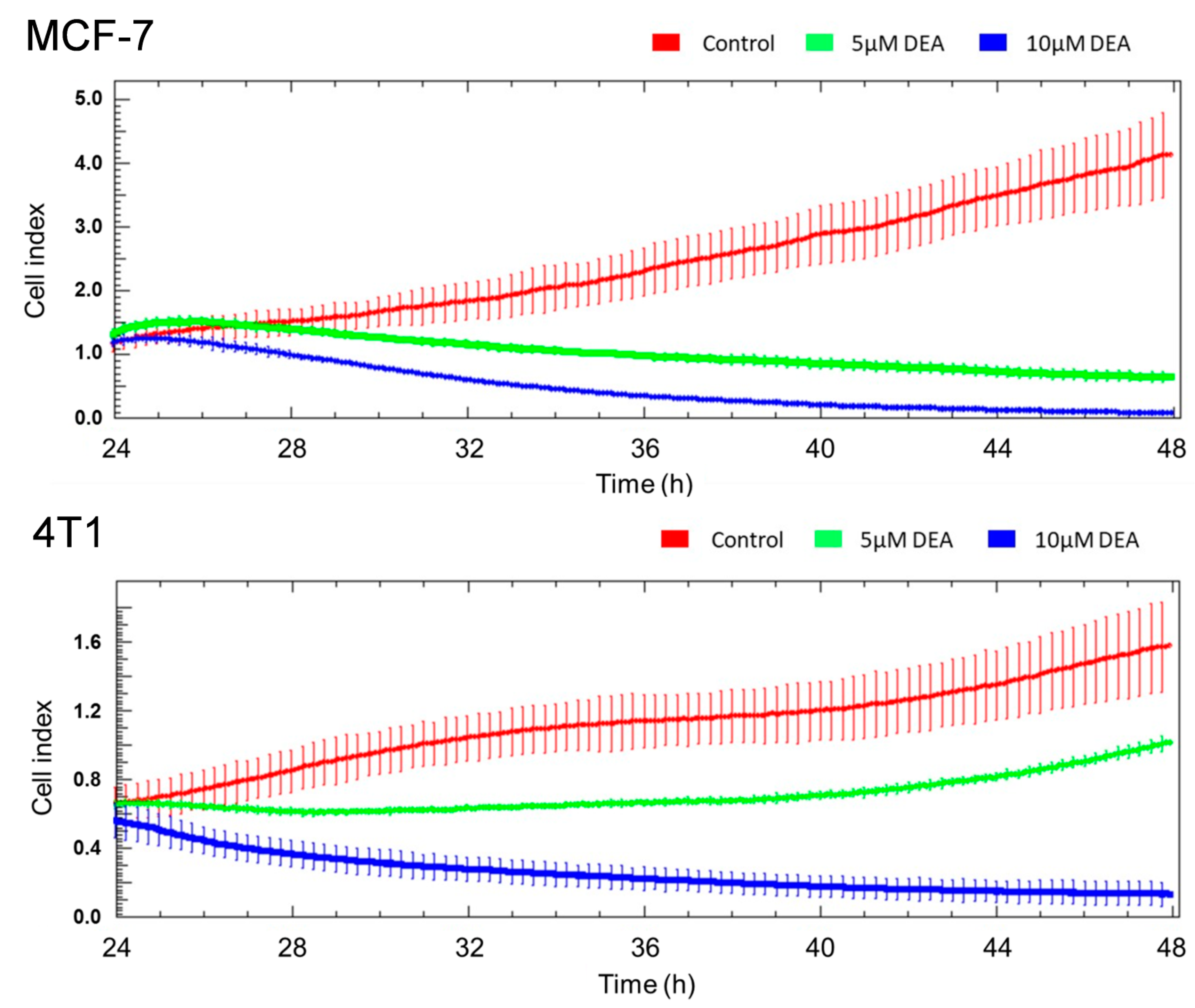
2.2. DEA Mitigated Invasive Growth of BC Cell Lines
2.3. DEA Differentially Modulated Regulators and Markers of the Cell Death Process in the BC Cell Lines
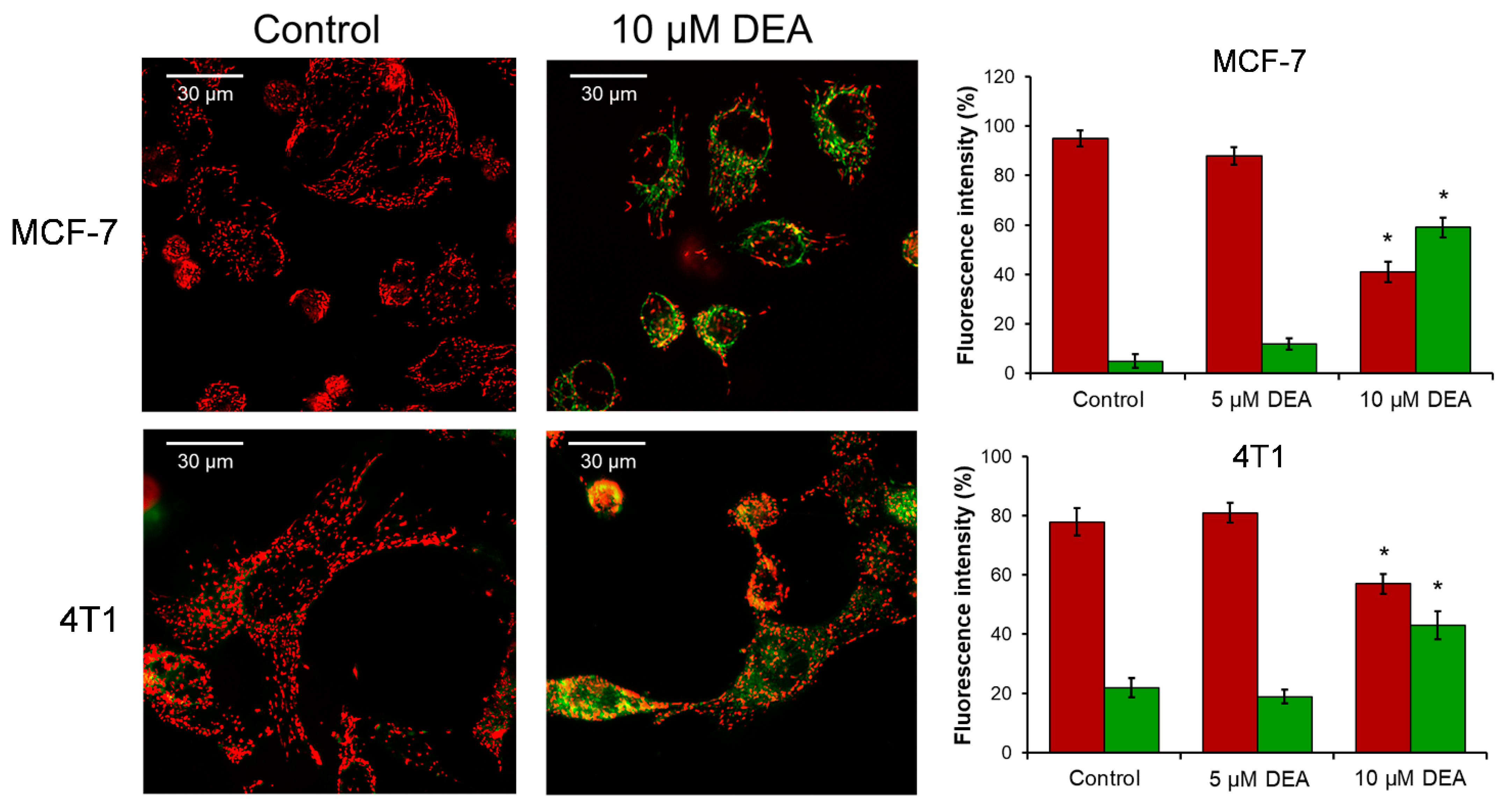
2.4. DEA Caused the Loss of Mitochondrial Membrane Potential (ΔΨm)
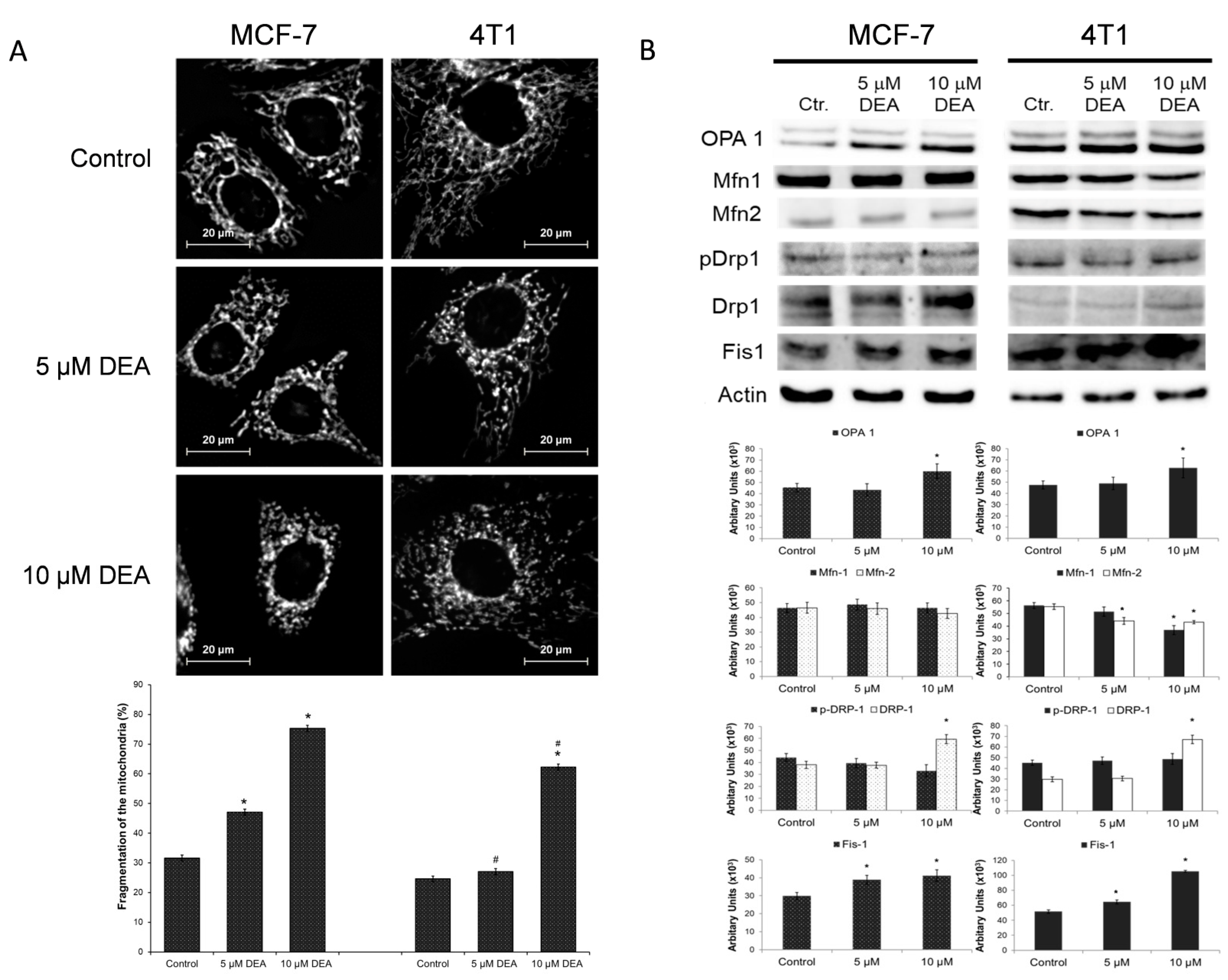
2.5. DEA Induced Mitochondrial Fragmentation in BC Cell Lines
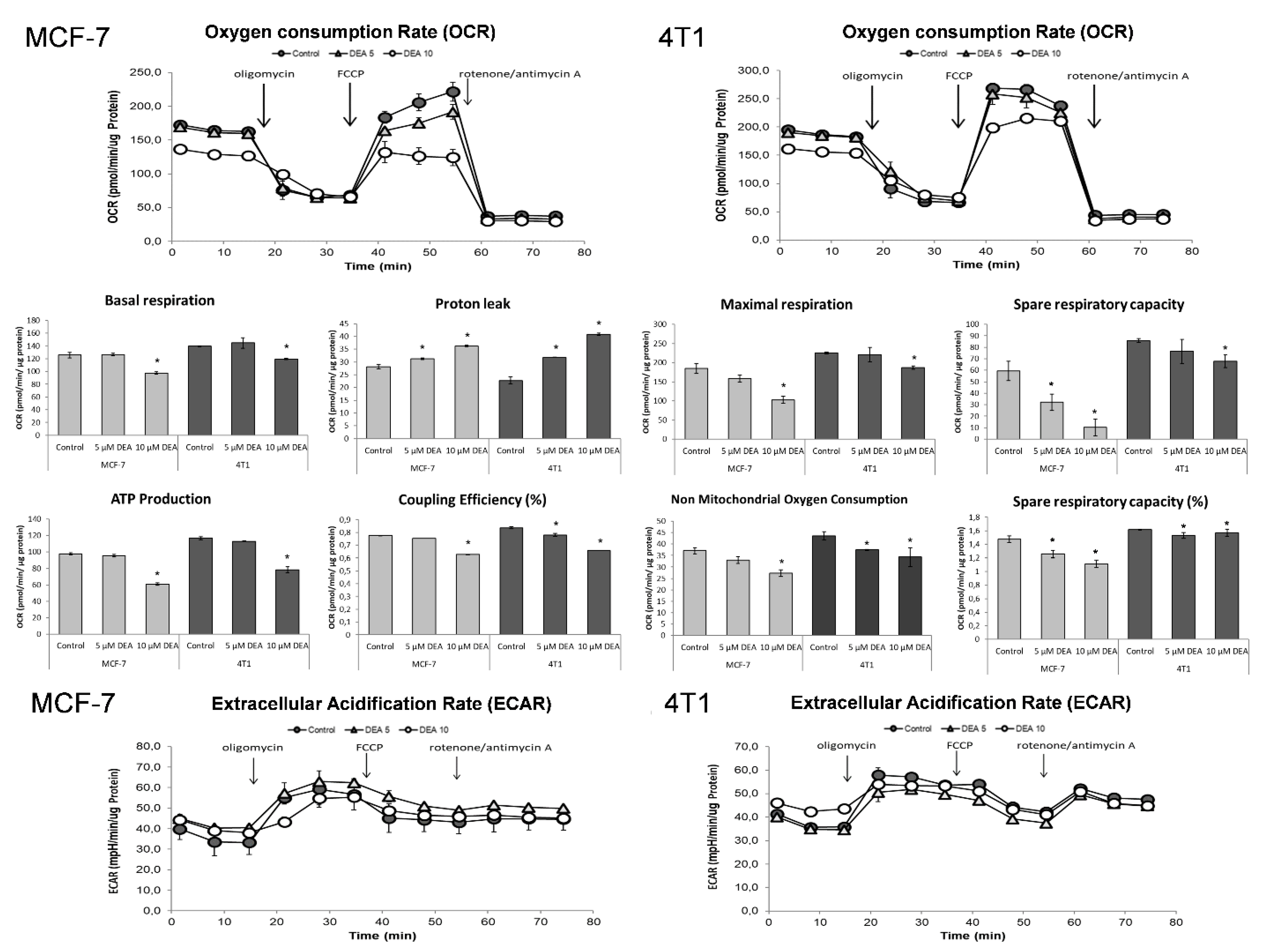
2.6. DEA Impeded Mitochondrial Energy Production in the BC Cell Lines
2.7. COX-2 Inhibition Potentiated DEA’s Anti-Neoplastic Effect in the TN BC Cell Line
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Materials
5.2. Cell Cultures
5.3. Cell Viability Assay
5.4. Apoptosis Assay
5.5. Bioenergetics Assay
5.6. Immunoblot Analysis
5.7. Colony Formation Assay
5.8. Migration Assay
5.9. Measurement of Invasive Growth
5.10. ∆Ψm Assay
5.11. Analysis of Mitochondrial Network Dynamics
5.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Masoud, V.; Pages, G. Targeted therapies in breast cancer: New challenges to fight against resistance. World J. Clin. Oncol 2017, 8, 120–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakrabarty, A.; Chakraborty, S.; Bhattacharya, R.; Chowdhury, G. Senescence-Induced Chemoresistance in Triple Negative Breast Cancer and Evolution-Based Treatment Strategies. Front. Oncol. 2021, 11, 674354. [Google Scholar] [CrossRef] [PubMed]
- Lebert, J.M.; Lester, R.; Powell, E.; Seal, M.; McCarthy, J. Advances in the systemic treatment of triple-negative breast cancer. Curr. Oncol. 2018, 25 (Suppl. 1), S142–S150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Edechi, C.A.; Ikeogu, N.; Uzonna, J.E.; Myal, Y. Regulation of Immunity in Breast Cancer. Cancers 2019, 11, 1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiam-Galvez, K.J.; Allen, B.M.; Spitzer, M.H. Systemic immunity in cancer. Nat. Rev. Cancer 2021, 21, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Pang, L.Y.; Hurst, E.A.; Argyle, D.J. Cyclooxygenase-2: A Role in Cancer Stem Cell Survival and Repopulation of Cancer Cells during Therapy. Stem Cells Int. 2016, 2016, 2048731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bos, P.D.; Zhang, X.H.; Nadal, C.; Shu, W.; Gomis, R.R.; Nguyen, D.X.; Minn, A.J.; van de Vijver, M.J.; Gerald, W.L.; Foekens, J.A.; et al. Genes that mediate breast cancer metastasis to the brain. Nature 2009, 459, 1005–1009. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Fukuda, K.; Xing, F.; Zhang, Y.; Sharma, S.; Liu, Y.; Chan, M.D.; Zhou, X.; Qasem, S.A.; Pochampally, R.; et al. Roles of the cyclooxygenase 2 matrix metalloproteinase 1 pathway in brain metastasis of breast cancer. J. Biol. Chem. 2015, 290, 9842–9854. [Google Scholar] [CrossRef] [Green Version]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007, 11, 69–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, D.L.; Wagers, A.J. No place like home: Anatomy and function of the stem cell niche. Nat. Rev. Mol. Cell. Biol. 2008, 9, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Tian, J.; Hachim, M.Y.; Hachim, I.Y.; Dai, M.; Lo, C.; Raffa, F.A.; Ali, S.; Lebrun, J.J. Cyclooxygenase-2 regulates TGFbeta-induced cancer stemness in triple-negative breast cancer. Sci. Rep. 2017, 7, 40258. [Google Scholar] [CrossRef] [Green Version]
- Tian, J.; Wang, V.; Wang, N.; Khadang, B.; Boudreault, J.; Bakdounes, K.; Ali, S.; Lebrun, J.J. Identification of MFGE8 and KLK5/7 as mediators of breast tumorigenesis and resistance to COX-2 inhibition. Breast Cancer Res. 2021, 23, 23. [Google Scholar] [CrossRef]
- Dong, L.; Neuzil, J. Targeting mitochondria as an anticancer strategy. Cancer Commun. 2019, 39, 63. [Google Scholar] [CrossRef] [Green Version]
- Fontana, F.; Limonta, P. The multifaceted roles of mitochondria at the crossroads of cell life and death in cancer. Free Radic. Biol. Med. 2021, 176, 203–221. [Google Scholar] [CrossRef]
- Hamilton, C.; Fox, J.P.; Longley, D.B.; Higgins, C.A. Therapeutics Targeting the Core Apoptotic Machinery. Cancers 2021, 13, 2618. [Google Scholar] [CrossRef]
- Patra, S.; Elahi, N.; Armorer, A.; Arunachalam, S.; Omala, J.; Hamid, I.; Ashton, A.W.; Joyce, D.; Jiao, X.; Pestell, R.G. Mechanisms Governing Metabolic Heterogeneity in Breast Cancer and Other Tumors. Front. Oncol. 2021, 11, 700629. [Google Scholar] [CrossRef]
- Boland, M.L.; Chourasia, A.H.; Macleod, K.F. Mitochondrial dysfunction in cancer. Front. Oncol. 2013, 3, 292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, H.; Siddharth, S.; Parida, S.; Wu, X.; Sharma, D. Tumor Microenvironment: Key Players in Triple Negative Breast Cancer Immunomodulation. Cancers 2021, 13, 3357. [Google Scholar] [CrossRef] [PubMed]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef]
- Brien, J.F.; Jimmo, S.; Brennan, F.J.; Ford, S.E.; Armstrong, P.W. Distribution of amiodarone and its metabolite, desethylamiodarone, in human tissues. Can. J. Physiol. Pharmacol. 1987, 65, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Holt, D.W.; Tucker, G.T.; Jackson, P.R.; Storey, G.C. Amiodarone pharmacokinetics. Am. Heart J. 1983, 106 Pt 2, 840–847. [Google Scholar] [CrossRef]
- Colunga Biancatelli, R.M.; Congedo, V.; Calvosa, L.; Ciacciarelli, M.; Polidoro, A.; Iuliano, L. Adverse reactions of Amiodarone. J. Geriatr. Cardiol. 2019, 16, 552–566. [Google Scholar] [PubMed]
- Singh, B.N.; Nademanee, K.; Josephson, M.A.; Ikeda, N.; Venkatesh, N.; Kannan, R. The electrophysiology and pharmacology of verapamil, flecainide, and amiodarone: Correlations with clinical effects and antiarrhythmic actions. Ann. N. Y. Acad. Sci. 1984, 432, 210–235. [Google Scholar] [CrossRef] [PubMed]
- Daniels, J.M.; Brien, J.F.; Massey, T.E. Pulmonary fibrosis induced in the hamster by amiodarone and desethylamiodarone. Toxicol. Appl. Pharmacol. 1989, 100, 350–359. [Google Scholar] [CrossRef]
- Honegger, U.E.; Zuehlke, R.D.; Scuntaro, I.; Schaefer, M.H.; Toplak, H.; Wiesmann, U.N. Cellular accumulation of amiodarone and desethylamiodarone in cultured human cells. Consequences of drug accumulation on cellular lipid metabolism and plasma membrane properties of chronically exposed cells. Biochem. Pharmacol. 1993, 45, 349–356. [Google Scholar] [CrossRef]
- Morvay, N.; Baczko, I.; Sztojkov-Ivanov, A.; Falkay, G.; Papp, J.G.; Varro, A.; Lepran, I. Long-term pretreatment with desethylamiodarone (DEA) or amiodarone (AMIO) protects against coronary artery occlusion induced ventricular arrhythmias in conscious rats. Can. J. Physiol. Pharmacol. 2015, 93, 773–777. [Google Scholar] [CrossRef] [Green Version]
- Bognar, Z.; Fekete, K.; Antus, C.; Hocsak, E.; Bognar, R.; Tapodi, A.; Boronkai, A.; Farkas, N.; Gallyas, F., Jr.; Sumegi, B.; et al. Desethylamiodarone-A metabolite of amiodarone-Induces apoptosis on T24 human bladder cancer cells via multiple pathways. PLoS ONE 2017, 12, e0189470. [Google Scholar] [CrossRef] [PubMed]
- Bognar, Z.; Fekete, K.; Bognar, R.; Szabo, A.; Vass, R.A.; Sumegi, B. Amiodarone’s major metabolite, desethylamiodarone, induces apoptosis in human cervical cancer cells. Can. J. Physiol. Pharmacol. 2018, 96, 1004–1011. [Google Scholar] [CrossRef] [PubMed]
- Bognar, Z.; Cseh, A.M.; Fekete, K.; Antus, C.; Bognar, R.; Tapodi, A.; Ramadan, F.H.J.; Sumegi, B.; Gallyas, F., Jr. Amiodarone’s major metabolite, desethylamiodarone inhibits proliferation of B16-F10 melanoma cells and limits lung metastasis formation in an in vivo experimental model. PLoS ONE 2020, 15, e0239088. [Google Scholar] [CrossRef] [PubMed]
- Ramadan, F.H.J.; Szabo, A.; Kovacs, D.; Takatsy, A.; Bognar, R.; Gallyas, F., Jr.; Bognar, Z. Involvement of Mitochondrial Mechanisms in the Cytostatic Effect of Desethylamiodarone in B16F10 Melanoma Cells. Int. J. Mol. Sci. 2020, 21, 7346. [Google Scholar] [CrossRef] [PubMed]
- Varbiro, G.; Toth, A.; Tapodi, A.; Bognar, Z.; Veres, B.; Sumegi, B.; Gallyas, F., Jr. Protective effect of amiodarone but not N-desethylamiodarone on postischemic hearts through the inhibition of mitochondrial permeability transition. J. Pharmacol. Exp. Ther. 2003, 307, 615–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ensenyat-Mendez, M.; Llinas-Arias, P.; Orozco, J.I.J.; Iniguez-Munoz, S.; Salomon, M.P.; Sese, B.; DiNome, M.L.; Marzese, D.M. Current Triple-Negative Breast Cancer Subtypes: Dissecting the Most Aggressive Form of Breast Cancer. Front. Oncol. 2021, 11, 681476. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.; Werner, S. Cancer as an overhealing wound: An old hypothesis revisited. Nat. Rev. Mol. Cell. Biol. 2008, 9, 628–638. [Google Scholar] [CrossRef]
- Karamanou, K.; Franchi, M.; Vynios, D.; Brezillon, S. Epithelial-to-mesenchymal transition and invadopodia markers in breast cancer: Lumican a key regulator. Semin. Cancer Biol. 2020, 62, 125–133. [Google Scholar] [CrossRef]
- Steenbrugge, J.; Vander Elst, N.; Demeyere, K.; De Wever, O.; Sanders, N.N.; Van Den Broeck, W.; Dirix, L.; Van Laere, S.; Meyer, E. Comparative Profiling of Metastatic 4T1- vs. Non-metastatic Py230-Based Mammary Tumors in an Intraductal Model for Triple-Negative Breast Cancer. Front. Immunol. 2019, 10, 2928. [Google Scholar] [CrossRef]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef]
- Del Campo, A.; Parra, V.; Vasquez-Trincado, C.; Gutierrez, T.; Morales, P.E.; Lopez-Crisosto, C.; Bravo-Sagua, R.; Navarro-Marquez, M.F.; Verdejo, H.E.; Contreras-Ferrat, A.; et al. Mitochondrial fragmentation impairs insulin-dependent glucose uptake by modulating Akt activity through mitochondrial Ca2+ uptake. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E1–E13. [Google Scholar] [CrossRef] [PubMed]
- Zhan, L.; Cao, H.; Wang, G.; Lyu, Y.; Sun, X.; An, J.; Wu, Z.; Huang, Q.; Liu, B.; Xing, J. Drp1-mediated mitochondrial fission promotes cell proliferation through crosstalk of p53 and NF-kappaB pathways in hepatocellular carcinoma. Oncotarget 2016, 7, 65001–65011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adebayo, M.; Singh, S.; Singh, A.P.; Dasgupta, S. Mitochondrial fusion and fission: The fine-tune balance for cellular homeostasis. FASEB J. 2021, 35, e21620. [Google Scholar] [CrossRef] [PubMed]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef] [PubMed]
- Moindjie, H.; Rodrigues-Ferreira, S.; Nahmias, C. Mitochondrial Metabolism in Carcinogenesis and Cancer Therapy. Cancers 2021, 13, 3311. [Google Scholar] [CrossRef]
- Hirpara, J.; Eu, J.Q.; Tan, J.K.M.; Wong, A.L.; Clement, M.V.; Kong, L.R.; Ohi, N.; Tsunoda, T.; Qu, J.; Goh, B.C.; et al. Metabolic reprogramming of oncogene-addicted cancer cells to OXPHOS as a mechanism of drug resistance. Redox Biol. 2019, 25, 101076. [Google Scholar] [CrossRef]
- Zhang, G.; Frederick, D.T.; Wu, L.; Wei, Z.; Krepler, C.; Srinivasan, S.; Chae, Y.C.; Xu, X.; Choi, H.; Dimwamwa, E.; et al. Targeting mitochondrial biogenesis to overcome drug resistance to MAPK inhibitors. J. Clin. Invest. 2016, 126, 1834–1856. [Google Scholar] [CrossRef]
- LeBleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.; Wikman, H.; Pantel, K.; Haigis, M.C.; de Carvalho, F.M.; Damascena, A.; Domingos Chinen, L.T.; Rocha, R.M.; et al. PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell. Biol. 2014, 16, 992–1003. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.S.; Liu, L.T.; Ou, L.H.; Pan, S.C.; Lin, C.I.; Wei, Y.H. Role of mitochondrial function in the invasiveness of human colon cancer cells. Oncol. Rep. 2018, 39, 316–330. [Google Scholar] [CrossRef] [Green Version]
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef] [Green Version]
- Guerra, F.; Arbini, A.A.; Moro, L. Mitochondria and cancer chemoresistance. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 686–699. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, S.E.; Chandel, N.S. Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 2015, 11, 9–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, P.J. Mitochondria, Bioenergetics and Apoptosis in Cancer. Trends Cancer 2017, 3, 857–870. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Zheng, S.; Yang, A.; Zhang, X.; Zou, Y.; Tang, H.; Xie, X. Breast cancer subtypes and the risk of distant metastasis at initial diagnosis: A population-based study. Cancer Manag. Res. 2018, 10, 5329–5338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashley, N.; Poulton, J. Mitochondrial DNA is a direct target of anti-cancer anthracycline drugs. Biochem. Biophys. Res. Commun. 2009, 378, 450–455. [Google Scholar] [CrossRef]
- Srinivasan, S.; Guha, M.; Kashina, A.; Avadhani, N.G. Mitochondrial dysfunction and mitochondrial dynamics-The cancer connection. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 602–614. [Google Scholar] [CrossRef]
- Mishra, P.; Chan, D.C. Metabolic regulation of mitochondrial dynamics. J. Cell Biol. 2016, 212, 379–387. [Google Scholar] [CrossRef] [Green Version]
- Rehman, J.; Zhang, H.J.; Toth, P.T.; Zhang, Y.; Marsboom, G.; Hong, Z.; Salgia, R.; Husain, A.N.; Wietholt, C.; Archer, S.L. Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. FASEB J. 2012, 26, 2175–2186. [Google Scholar] [CrossRef] [Green Version]
- Inoue-Yamauchi, A.; Oda, H. Depletion of mitochondrial fission factor DRP1 causes increased apoptosis in human colon cancer cells. Biochem. Biophys. Res. Commun. 2012, 421, 81–85. [Google Scholar] [CrossRef]
- Grabacka, M.; Plonka, P.M.; Urbanska, K.; Reiss, K. Peroxisome proliferator-activated receptor alpha activation decreases metastatic potential of melanoma cells in vitro via down-regulation of Akt. Clin. Cancer Res. 2006, 12, 3028–3036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, K.M.; Anderson, N.G. The protein kinase B/Akt signalling pathway in human malignancy. Cell. Signal. 2002, 14, 381–395. [Google Scholar] [CrossRef]
- Pascual, J.; Turner, N.C. Targeting the PI3-kinase pathway in triple-negative breast cancer. Ann. Oncol 2019, 30, 1051–1060. [Google Scholar] [CrossRef] [Green Version]
- Fidler, I.J. Selection of successive tumour lines for metastasis. Nat. New Biol. 1973, 242, 148–149. [Google Scholar] [CrossRef]
- Gallo, O.; Masini, E.; Bianchi, B.; Bruschini, L.; Paglierani, M.; Franchi, A. Prognostic significance of cyclooxygenase-2 pathway and angiogenesis in head and neck squamous cell carcinoma. Hum. Pathol. 2002, 33, 708–714. [Google Scholar] [CrossRef]
- Jiao, G.; Ren, T.; Lu, Q.; Sun, Y.; Lou, Z.; Peng, X.; Liang, W.; Guo, W. Prognostic significance of cyclooxygenase-2 in osteosarcoma: A meta-analysis. Tumour Biol. 2013, 34, 2489–2495. [Google Scholar] [CrossRef]
- Sicking, I.; Rommens, K.; Battista, M.J.; Bohm, D.; Gebhard, S.; Lebrecht, A.; Cotarelo, C.; Hoffmann, G.; Hengstler, J.G.; Schmidt, M. Prognostic influence of cyclooxygenase-2 protein and mRNA expression in node-negative breast cancer patients. BMC Cancer 2014, 14, 952. [Google Scholar] [CrossRef] [Green Version]
- Falandry, C.; Canney, P.A.; Freyer, G.; Dirix, L.Y. Role of combination therapy with aromatase and cyclooxygenase-2 inhibitors in patients with metastatic breast cancer. Ann. Oncol. 2009, 20, 615–620. [Google Scholar] [CrossRef]
- Hida, T.; Yatabe, Y.; Achiwa, H.; Muramatsu, H.; Kozaki, K.; Nakamura, S.; Ogawa, M.; Mitsudomi, T.; Sugiura, T.; Takahashi, T. Increased expression of cyclooxygenase 2 occurs frequently in human lung cancers, specifically in adenocarcinomas. Cancer Res. 1998, 58, 3761–3764. [Google Scholar]
- Hwang, D.; Scollard, D.; Byrne, J.; Levine, E. Expression of cyclooxygenase-1 and cyclooxygenase-2 in human breast cancer. J. Natl. Cancer Inst. 1998, 90, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Soslow, R.A.; Dannenberg, A.J.; Rush, D.; Woerner, B.M.; Khan, K.N.; Masferrer, J.; Koki, A.T. COX-2 is expressed in human pulmonary, colonic, and mammary tumors. Cancer 2000, 89, 2637–2645. [Google Scholar] [CrossRef]
- Subbaramaiah, K.; Telang, N.; Ramonetti, J.T.; Araki, R.; DeVito, B.; Weksler, B.B.; Dannenberg, A.J. Transcription of cyclooxygenase-2 is enhanced in transformed mammary epithelial cells. Cancer Res. 1996, 56, 4424–4429. [Google Scholar] [PubMed]
- Claria, J. Cyclooxygenase-2 biology. Curr. Pharm. Des. 2003, 9, 2177–2190. [Google Scholar] [CrossRef] [Green Version]
- Allen, C.P.; Tinganelli, W.; Sharma, N.; Nie, J.; Sicard, C.; Natale, F.; King, M., 3rd; Keysar, S.B.; Jimeno, A.; Furusawa, Y.; et al. DNA Damage Response Proteins and Oxygen Modulate Prostaglandin E2 Growth Factor Release in Response to Low and High LET Ionizing Radiation. Front. Oncol. 2015, 5, 260. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gallyas, F., Jr.; Ramadan, F.H.J.; Andreidesz, K.; Hocsak, E.; Szabo, A.; Tapodi, A.; Kiss, G.N.; Fekete, K.; Bognar, R.; Szanto, A.; et al. Involvement of Mitochondrial Mechanisms and Cyclooxygenase-2 Activation in the Effect of Desethylamiodarone on 4T1 Triple-Negative Breast Cancer Line. Int. J. Mol. Sci. 2022, 23, 1544. https://doi.org/10.3390/ijms23031544
Gallyas F Jr., Ramadan FHJ, Andreidesz K, Hocsak E, Szabo A, Tapodi A, Kiss GN, Fekete K, Bognar R, Szanto A, et al. Involvement of Mitochondrial Mechanisms and Cyclooxygenase-2 Activation in the Effect of Desethylamiodarone on 4T1 Triple-Negative Breast Cancer Line. International Journal of Molecular Sciences. 2022; 23(3):1544. https://doi.org/10.3390/ijms23031544
Chicago/Turabian StyleGallyas, Ferenc, Jr., Fadi H. J. Ramadan, Kitti Andreidesz, Eniko Hocsak, Aliz Szabo, Antal Tapodi, Gyongyi N. Kiss, Katalin Fekete, Rita Bognar, Arpad Szanto, and et al. 2022. "Involvement of Mitochondrial Mechanisms and Cyclooxygenase-2 Activation in the Effect of Desethylamiodarone on 4T1 Triple-Negative Breast Cancer Line" International Journal of Molecular Sciences 23, no. 3: 1544. https://doi.org/10.3390/ijms23031544
APA StyleGallyas, F., Jr., Ramadan, F. H. J., Andreidesz, K., Hocsak, E., Szabo, A., Tapodi, A., Kiss, G. N., Fekete, K., Bognar, R., Szanto, A., & Bognar, Z. (2022). Involvement of Mitochondrial Mechanisms and Cyclooxygenase-2 Activation in the Effect of Desethylamiodarone on 4T1 Triple-Negative Breast Cancer Line. International Journal of Molecular Sciences, 23(3), 1544. https://doi.org/10.3390/ijms23031544