
Expression of a Truncated Form of ODAD1 Associated with an Unusually Mild Primary Ciliary Dyskinesia Phenotype

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Patient Characteristics and Mutation Identification
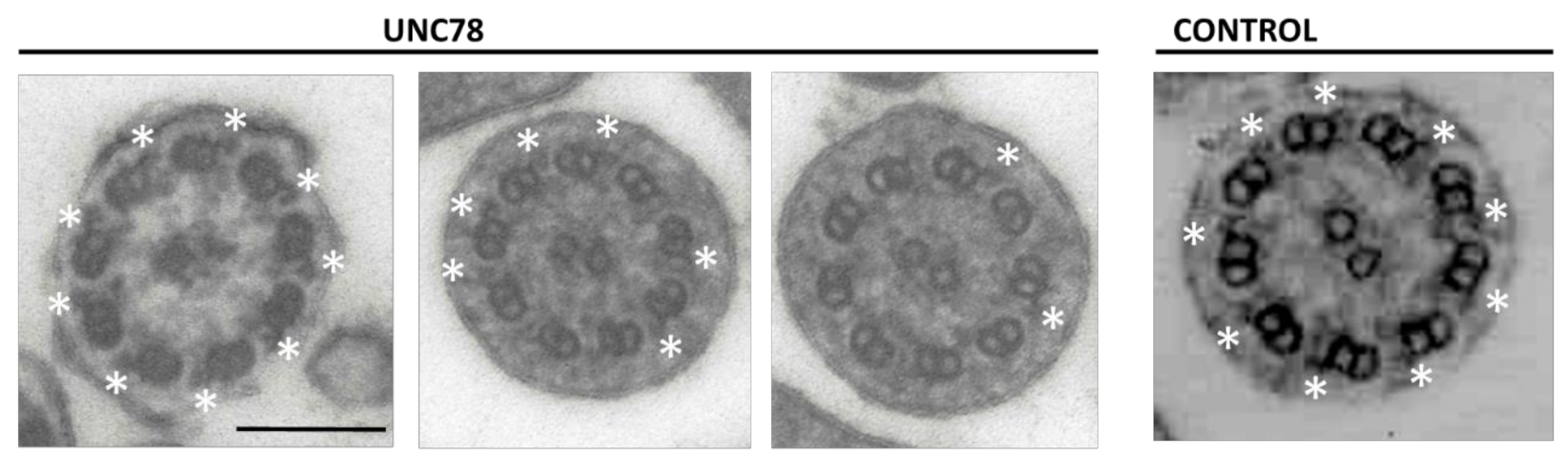
2.2. Quantification of ODA in ODAD1 Subjects
2.3. Ciliary Activity of c.1502+5G>A Mutant Cells in Vitro
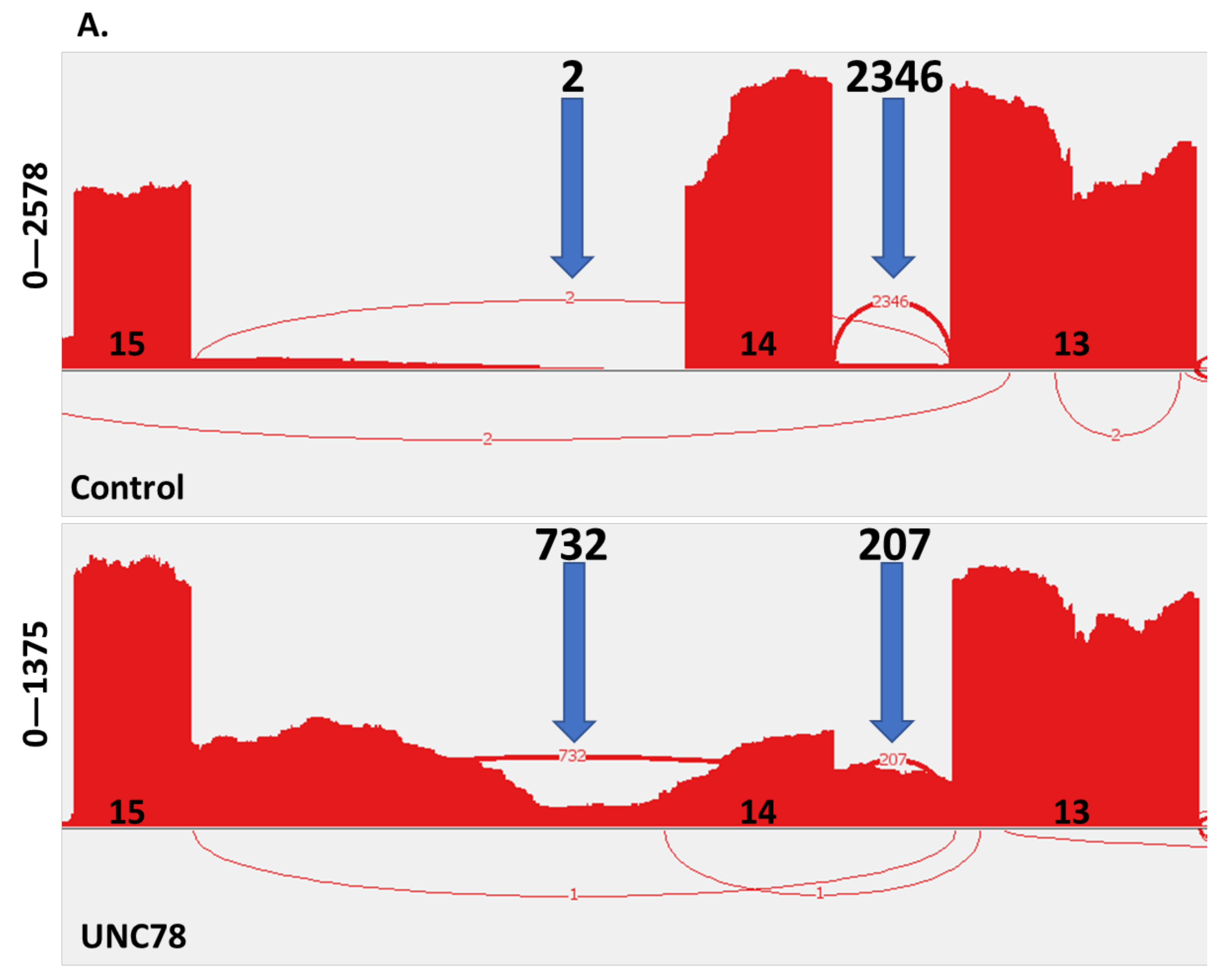
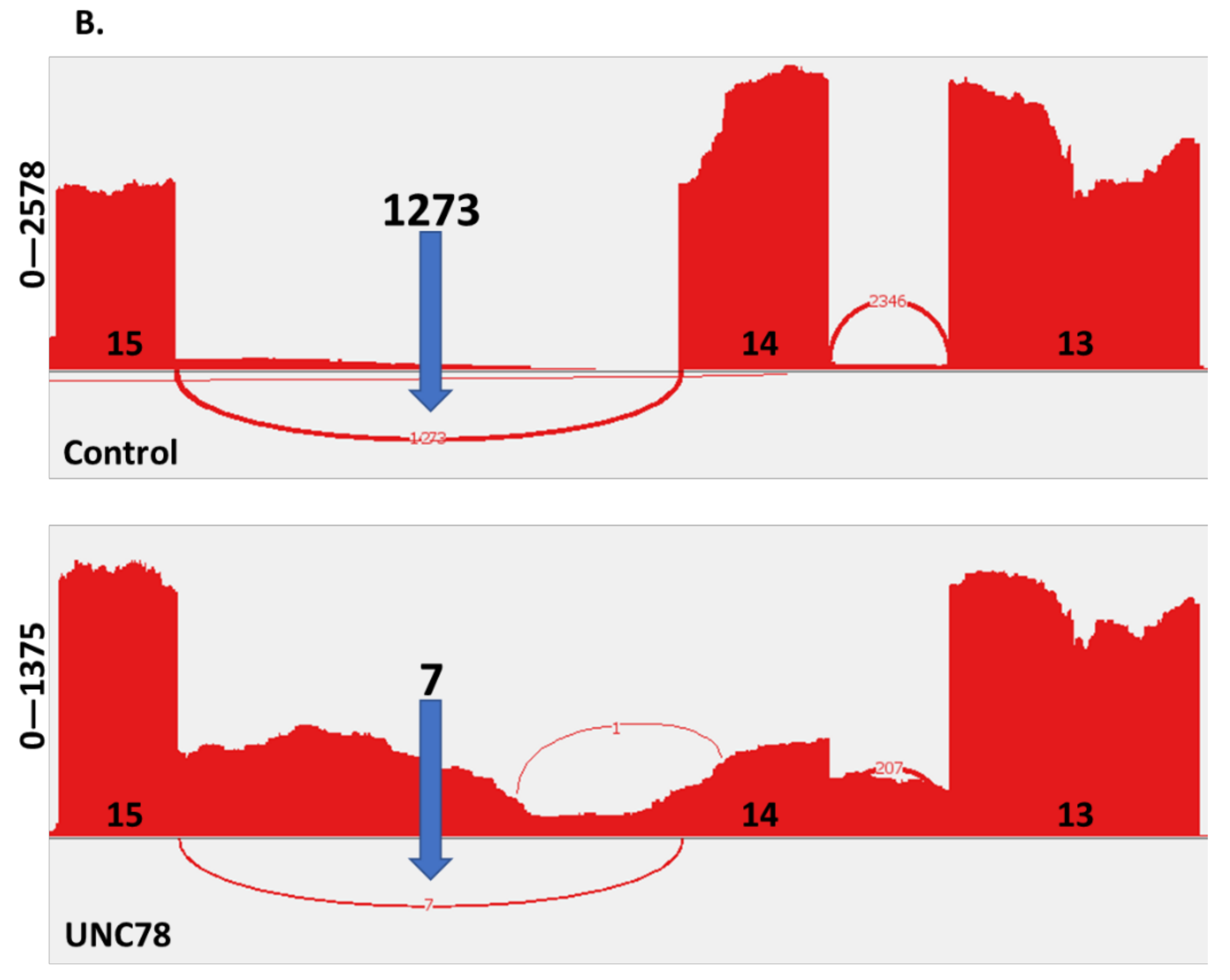
2.4. RNA Expression Analysis
2.5. Expression of a Truncated Form of ODAD1
2.6. Immunofluorescence Localizes Truncated ODAD1 to Cilia
3. Discussion
4. Materials and Methods
4.1. Human Subjects
4.2. Identification of Genetic Variant
4.3. Enumeration of ODA
4.4. Expansion and Culture of Airway Epithelial Cells
4.5. Measurement of Ciliary Activity
4.6. RNA Analysis
4.7. Western Blotting
4.8. Immunofluorescence
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wallmeier, J.; Nielsen, K.G.; Kuehni, C.E.; Lucas, J.S.; Leigh, M.W.; Zariwala, M.A.; Omran, H. Motile ciliopathies. Nat. Rev. Dis. Prim. 2020, 6, 77. [Google Scholar] [CrossRef]
- Zariwala, M.A.; Knowles, M.R.; Leigh, M.W. Primary Ciliary Dyskinesia. In GeneReviews; Ardinger, H.H., Pagon, R.A., Wallace, S.E., Eds.; University of Washington: Seattle, WA, USA, 2015. [Google Scholar]
- Knowles, M.R.; Daniels, L.A.; Davis, S.D.; Zariwala, M.A.; Leigh, M.W. Primary Ciliary Dyskinesia. Recent Advances in Diagnostics, Genetics, and Characterization of Clinical Disease. Am. J. Respir. Crit. Care Med. 2013, 188, 913–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapiro, A.J.; Davis, S.D.; Ferkol, T.; Dell, S.D.; Rosenfeld, M.; Olivier, K.N.; Sagel, S.D.; Milla, C.; Zariwala, M.A.; Wolf, W.; et al. Laterality defects other than situs inversus totalis in primary ciliary dyskinesia: Insights into situs ambiguus and heterotaxy. Chest 2014, 146, 1176–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bustamante-Marin, X.M.; Ostrowski, L.E. Cilia and Mucociliary Clearance. Cold Spring Harb. Perspect. Biol. 2017, 9, a028241. [Google Scholar] [CrossRef] [PubMed]
- Eden, E.; Choate, R.; Barker, A.; Addrizzo-Harris, D.; Aksamit, T.R.; Daley, C.L.; Daniels, M.L.A.; DiMango, A.; Fennelly, K.; Griffith, D.E.; et al. The Clinical Features of Bronchiectasis Associated with Alpha-1 Antitrypsin Deficiency, Common Variable Immunodeficiency and Primary Ciliary Dyskinesia—Results from the U.S. Bronchiectasis Research Registry. Chronic Obstr. Pulm. Dis. 2019, 6, 145–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrowski, L.E.; Blackburn, K.; Radde, K.M.; Moyer, M.B.; Schlatzer, D.M.; Moseley, A.; Boucher, R.C. A proteomic analysis of human cilia: Identification of novel components. Mol. Cell. Proteom. 2002, 1, 451–465. [Google Scholar] [CrossRef] [Green Version]
- Davis, S.D.; Ferkol, T.W.; Rosenfeld, M.; Lee, H.-S.; Dell, S.D.; Sagel, S.D.; Milla, C.; Zariwala, M.A.; Pittman, J.E.; Shapiro, A.J.; et al. Clinical features of childhood primary ciliary dyskinesia by genotype and ultrastructural phenotype. Am. J. Respir. Crit. Care Med. 2015, 191, 316–324. [Google Scholar] [CrossRef] [Green Version]
- Davis, S.D.; Rosenfeld, M.; Lee, H.-S.; Ferkol, T.W.; Sagel, S.D.; Dell, S.; Milla, C.; Pittman, J.E.; Shapiro, A.; Sullivan, K.M.; et al. Primary Ciliary Dyskinesia: Longitudinal Study of Lung Disease by Ultrastructure Defect and Genotype. Am. J. Respir. Crit. Care Med. 2019, 199, 190–198. [Google Scholar] [CrossRef]
- Pifferi, M.; Bush, A.; Mariani, F.; Piras, M.; Michelucci, A.; Cangiotti, A.; di Cicco, M.; Caligo, M.A.; Miccoli, M.; Boner, A.L.; et al. Lung Function Longitudinal Study by Phenotype and Genotype in Primary Ciliary Dyskinesia. Chest 2020, 158, 117–120. [Google Scholar] [CrossRef]
- Pifferi, M.; Bush, A.; Mulé, G.; Gracci, S.; Fonnesu, R.; Michelucci, A.; Cangiotti, A.; Caligo, M.A.; Miccoli, M.; Boner, A.L.; et al. Longitudinal Lung Volume Changes by Ultrastructure and Genotype in Primary Ciliary Dyskinesia. Ann. Am. Thorac. Soc. 2021, 18, 963–970. [Google Scholar] [CrossRef]
- Knowles, M.R.; Ostrowski, L.E.; Leigh, M.W.; Sears, P.R.; Davis, S.D.; Wolf, W.E.; Hazucha, M.J.; Carson, J.L.; Olivier, K.N.; Sagel, S.D.; et al. Mutations in RSPH1 cause primary ciliary dyskinesia with a unique clinical and ciliary phenotype. Am. J. Respir. Crit. Care Med. 2014, 189, 707–717. [Google Scholar] [CrossRef] [Green Version]
- Yin, W.; Livraghi-Butrico, A.; Sears, P.R.; Rogers, T.D.; Burns, K.A.; Grubb, B.R.; Ostrowski, L.E. Mice with a Deletion of Rsph1 Exhibit a Low Level of Mucociliary Clearance and Develop a Primary Ciliary Dyskinesia Phenotype. Am. J. Respir. Cell Mol. Biol. 2019, 61, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Hornef, N.; Olbrich, H.; Horvath, J.; Zariwala, M.A.; Fliegauf, M.; Loges, N.T.; Wildhaber, J.; Noone, P.G.; Kennedy, M.; Antonarakis, S.E.; et al. DNAH5 Mutations Are a Common Cause of Primary Ciliary Dyskinesia with Outer Dynein Arm Defects. Am. J. Respir. Crit. Care Med. 2006, 174, 120–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kispert, A.; Petry, M.; Olbrich, H.; Volz, A.; Ketelsen, U.-P.; Horvath, J.; Melkaoui, R.; Omran, H.; Zariwala, M.; Noone, P.G.; et al. Genotype-phenotype correlations in PCD patients carrying DNAH5 mutations. Thorax 2003, 58, 552–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takada, S.; Wilkerson, C.G.; Wakabayashi, K.-I.; Kamiya, R.; Witman, G.B. The Outer Dynein Arm-Docking Complex: Composition and Characterization of a Subunit (Oda1) Necessary for Outer Arm Assembly. Mol. Biol. Cell 2002, 13, 1015–1029. [Google Scholar] [CrossRef] [Green Version]
- Knowles, M.R.; Leigh, M.W.; Ostrowski, L.E.; Huang, L.; Carson, J.L.; Hazucha, M.J.; Yin, W.; Berg, J.S.; Davis, S.D.; Dell, S.D.; et al. Exome sequencing identifies mutations in CCDC114 as a cause of primary ciliary dyskinesia. Am. J. Hum. Genet. 2013, 92, 99–106. [Google Scholar] [CrossRef] [Green Version]
- Onoufriadis, A.; Paff, T.; Antony, D.; Shoemark, A.; Micha, D.; Kuyt, B.; Schmidts, M.; Petridi, S.; Dankert-Roelse, J.E.; Haarman, E.G.; et al. Splice-Site Mutations in the Axonemal Outer Dynein Arm Docking Complex Gene CCDC114 Cause Primary Ciliary Dyskinesia. Am. J. Hum. Genet. 2013, 92, 88–98. [Google Scholar] [CrossRef] [Green Version]
- Halbritter, J.; Diaz, K.; Chaki, M.; Porath, J.D.; Tarrier, B.; Fu, C.; Innis, J.L.; Allen, S.J.; Lyons, R.H.; Stefanidis, C.J.; et al. High-throughput mutation analysis in patients with a nephronophthisis-associated ciliopathy applying multiplexed barcoded array-based PCR amplification and next-generation sequencing. J. Med. Genet. 2012, 49, 756–767. [Google Scholar] [CrossRef]
- Halbritter, J.; Porath, J.D.; Diaz, K.A.; Braun, D.A.; Kohl, S.; Chaki, M.; Allen, S.J.; Soliman, N.; Hildebrandt, F.; Otto, E.A.; et al. Identification of 99 novel mutations in a worldwide cohort of 1056 patients with a nephronophthisis-related ciliopathy. Hum. Genet. 2013, 132, 865–884. [Google Scholar] [CrossRef] [Green Version]
- Leigh, M.W.; Hazucha, M.J.; Chawla, K.K.; Baker, B.R.; Shapiro, A.; Brown, D.E.; LaVange, L.M.; Horton, B.J.; Qaqish, B.; Carson, J.L.; et al. Standardizing Nasal Nitric Oxide Measurement as a Test for Primary Ciliary Dyskinesia. Ann. Am. Thorac. Soc. 2013, 10, 574–581. [Google Scholar] [CrossRef] [Green Version]
- Mullowney, T.; Manson, D.; Kim, R.; Stephens, D.; Shah, V.; Dell, S. Primary Ciliary Dyskinesia and Neonatal Respiratory Distress. Pediatrics 2014, 134, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Vanaken, G.-J.; Bassinet, L.; Boon, M.; Mani, R.; Honoré, I.; Papon, J.F.; Cuppens, H.; Jaspers, M.; Lorent, N.; Coste, A.; et al. Infertility in an adult cohort with primary ciliary dyskinesia: Phenotype–gene association. Eur. Respir. J. 2017, 50, 1750314. [Google Scholar] [CrossRef] [PubMed]
- Sears, P.R.; Yin, W.-N.; Ostrowski, L.E. Continuous mucociliary transport by primary human airway epithelial cells in vitro. Am. J. Physiol. Cell. Mol. Physiol. 2015, 309, L99–L108. [Google Scholar] [CrossRef] [Green Version]
- Hjeij, R.; Onoufriadis, A.; Watson, C.M.; Slagle, C.E.; Klena, N.T.; Dougherty, G.W.; Kurkowiak, M.; Loges, N.T.; Diggle, C.P.; Morante, N.F.C.; et al. CCDC151 mutations cause primary ciliary dyskinesia by disruption of the outer dynein arm docking complex formation. Am. J. Hum. Genet. 2014, 95, 257–274. [Google Scholar] [CrossRef] [Green Version]
- Button, B.; Okada, S.F.; Frederick, C.B.; Thelin, W.R.; Boucher, R.C. Mechanosensitive ATP Release Maintains Proper Mucus Hydration of Airways. Sci. Signal. 2013, 6, ra46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, W.T.; Jackson, C.L.; Lackie, P.M.; Hogg, C.; Lucas, J.S. Nitric oxide in primary ciliary dyskinesia. Eur. Respir. J. 2012, 40, 1024–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniels, M.L.A.; Leigh, M.W.; Davis, S.D.; Armstrong, M.C.; Carson, J.L.; Hazucha, M.; Dell, S.D.; Eriksson, M.; Collins, F.S.; Knowles, M.R.; et al. Founder mutation in RSPH4A identified in patients of Hispanic descent with primary ciliary dyskinesia. Hum. Mutat. 2013, 34, 1352–1356. [Google Scholar] [CrossRef] [Green Version]
- Marin, X.B.; Yin, W.-N.; Sears, P.R.; Werner, M.E.; Brotslaw, E.; Mitchell, B.J.; Jania, C.M.; Zeman, K.L.; Rogers, T.D.; Herring, L.E.; et al. Lack of GAS2L2 Causes PCD by Impairing Cilia Orientation and Mucociliary Clearance. Am. J. Hum. Genet. 2019, 104, 229–245. [Google Scholar] [CrossRef] [Green Version]
- Suprynowicz, F.A.; Upadhyay, G.; Krawczyk, E.; Kramer, S.C.; Hebert, J.D.; Liu, X.; Yuan, H.; Cheluvaraju, C.; Clapp, P.W.; Boucher, R.C., Jr.; et al. Conditionally reprogrammed cells represent a stem-like state of adult epithelial cells. Proc. Natl. Acad. Sci. USA 2012, 109, 20035–20040. [Google Scholar] [CrossRef] [Green Version]
- Fulcher, M.L.; Randell, S.H. Human Nasal and Tracheo-Bronchial Respiratory Epithelial Cell Culture. Methods Mol. Biol. 2013, 945, 109–121. [Google Scholar]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackburn, K.; Bustamante-Marin, X.; Yin, W.; Goshe, M.B.; Ostrowski, L.E. Quantitative Proteomic Analysis of Human Airway Cilia Identifies Previously Uncharacterized Proteins of High Abundance. J. Proteome Res. 2017, 16, 1579–1592. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Allele 1 | Allele 2 | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| UNC # | Study | Sex | Age | Ethnicity | nNO nl/min a | Situs Status | Neo RDS | Bxsis | Sinusitis | Otitis Media | Pathogens b | FEV1 % Pred | Exon/Intron | Base Change | Amino Acid Change | Exon/Intron | Base Change | Amino Acid Change |
| Homozygous Mutations | ||||||||||||||||||
| 78 | Current | F | 34 | white c | 205/188 d | SI | no | no | no | no | none | 95 e | Int 14 | c.1502+5G>A | p.Ser469Argfs*7 | Int 14 | c.1502+5G>A | p.Ser469Argfs*7 |
| 359 | ref [17] | F | 39 | white | 36.0 | SS | yes | yes | yes | yes | Ps a | 84 | Ex 9 | c.853G>A f | p.Ala285Serfs*52 | Ex 9 | c.853G>A f | p.Ala285Serfs*52 |
| 360 | ref [17] | F | 33 | white | 31.5 | SS | yes | yes | yes | yes | Ps a; St p | 92 | Ex 9 | c.853G>A f | p.Ala285Serfs*52 | Ex 9 | c.853G>A f | p.Ala285Serfs*52 |
| 961 | Current | F | 18 | Asian/Indian | 18.9 | SI | no | yes | yes | yes | Ps a | 42 | Ex 6 | c.448C>T | p.Arg150* | Ex 6 | c.448C>T | p.Arg150* |
| Comound Heterozygous Mutations | ||||||||||||||||||
| 891 | ref [17] | F | 59 | white | 32.4 | SS | yes | yes | yes | yes | Ps a | 57 | Ex 9 | c.853G>A f | p.Ala285Serfs*52 | Int 7 | c.598-2A>G | p.Glu200Glyfs*60, p.Glu200_Val221delins g |
| 897 | ref [17] | M | 10 | white | 9.6 | SS | yes | no | no | yes | none | 94 | Int 14 | c.1502+5G>A | p.Ser469Argfs*7 | Ex 9 | c.853G>A f | p.Ala285Serfs*52 |
| 1107 | ref [17] | F | 34 | white | 6 | SS | no | yes | yes | yes | Ps a; Kl p | 46 | Ex 9 | c.853G>A f | p.Ala285Serfs*52 | Ex 11 | c.1050delT | p.His350Glnfs*14 |
| Subject # | Genotype | # of Cilia Examined | # of Cilia w ODA (%) | # of ODA | # of ODA/# Cilia with ODA |
|---|---|---|---|---|---|
| 78 | c.1502+5G>A c.1502+5G>A | 63 | 18 (28) | 92 | 5.1 |
| 897 | c.1502+5G>A c.853G>A | 43 | 12 (28) | 65 | 5.4 |
| 359 | c.853G>A c.853G>A | 36 | 4 (11) | 18 | 4.5 |
| 360 | c.853G>A c.853G>A | 35 | 5 (14) | 8 | 1.6 |
| 891 | c.853G>A c.598-2A>G | 55 | 0 | 0 | 0 |
| 961 | p.Arg150* p.Arg150* | 34 | 0 | 0 | 0 |
| 1107 | c.853G>A c.1050delT | 53 | 0 | 0 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ostrowski, L.E.; Yin, W.; Smith, A.J.; Sears, P.R.; Bustamante-Marin, X.M.; Dang, H.; Hildebrandt, F.; Daniels, L.A.; Capps, N.A.; Sullivan, K.M.; et al. Expression of a Truncated Form of ODAD1 Associated with an Unusually Mild Primary Ciliary Dyskinesia Phenotype. Int. J. Mol. Sci. 2022, 23, 1753. https://doi.org/10.3390/ijms23031753
Ostrowski LE, Yin W, Smith AJ, Sears PR, Bustamante-Marin XM, Dang H, Hildebrandt F, Daniels LA, Capps NA, Sullivan KM, et al. Expression of a Truncated Form of ODAD1 Associated with an Unusually Mild Primary Ciliary Dyskinesia Phenotype. International Journal of Molecular Sciences. 2022; 23(3):1753. https://doi.org/10.3390/ijms23031753
Chicago/Turabian StyleOstrowski, Lawrence E., Weining Yin, Amanda J. Smith, Patrick R. Sears, Ximena M. Bustamante-Marin, Hong Dang, Friedhelm Hildebrandt, Leigh Anne Daniels, Nicole A. Capps, Kelli M. Sullivan, and et al. 2022. "Expression of a Truncated Form of ODAD1 Associated with an Unusually Mild Primary Ciliary Dyskinesia Phenotype" International Journal of Molecular Sciences 23, no. 3: 1753. https://doi.org/10.3390/ijms23031753