
Heparin-Functionalized Adsorbents Eliminate Central Effectors of Immunothrombosis, including Platelet Factor 4, High-Mobility Group Box 1 Protein and Histones

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
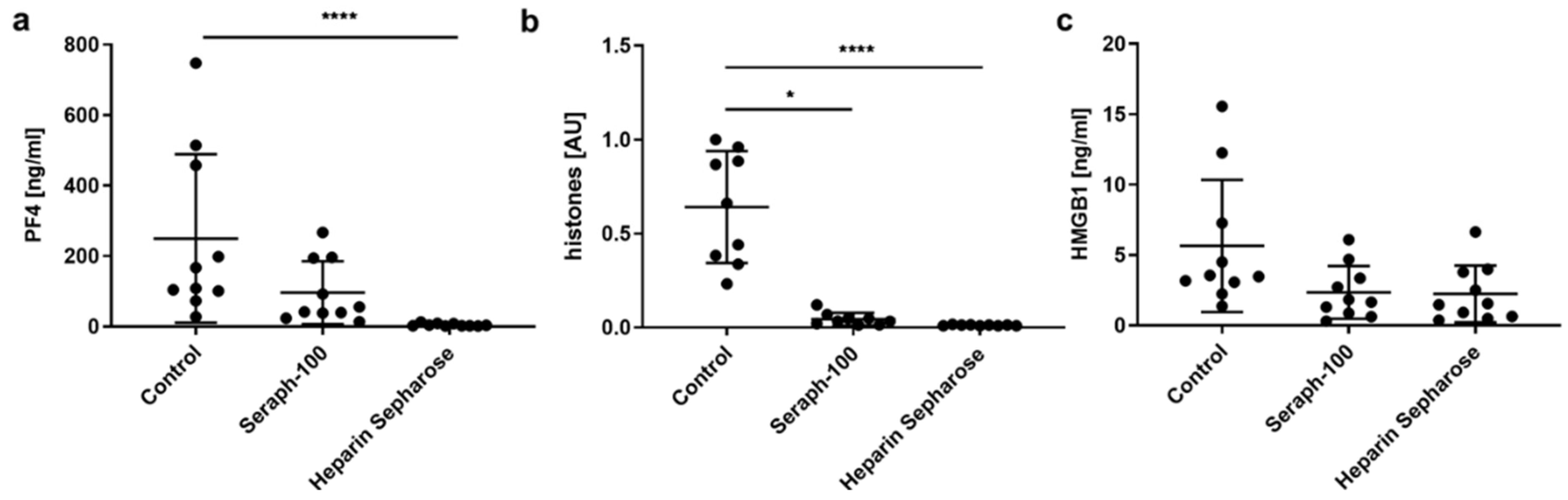
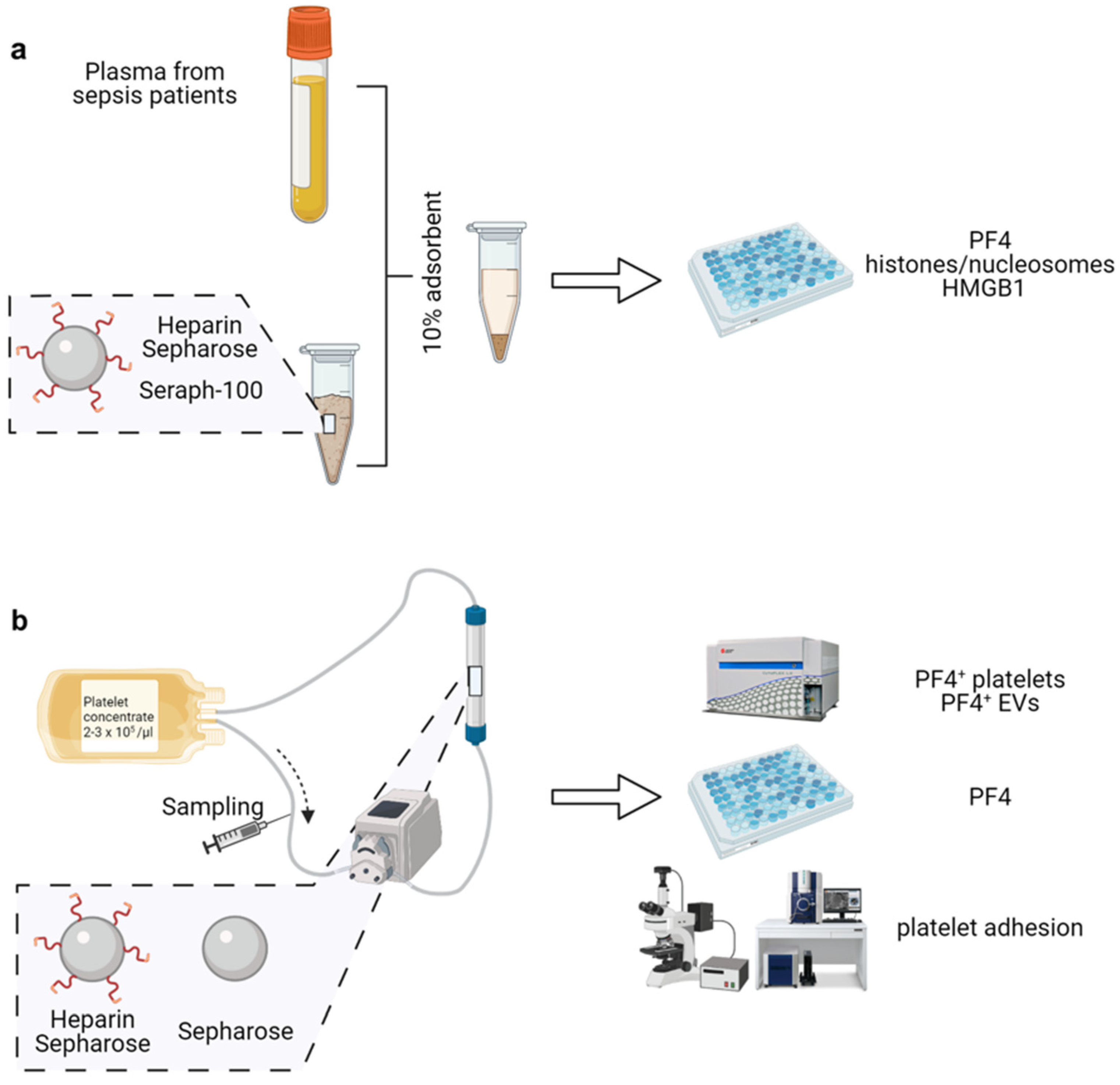
2.1. Heparinized Adsorbents Efficiently Deplete PF4, Histones/Nucleosomes and HMGB1
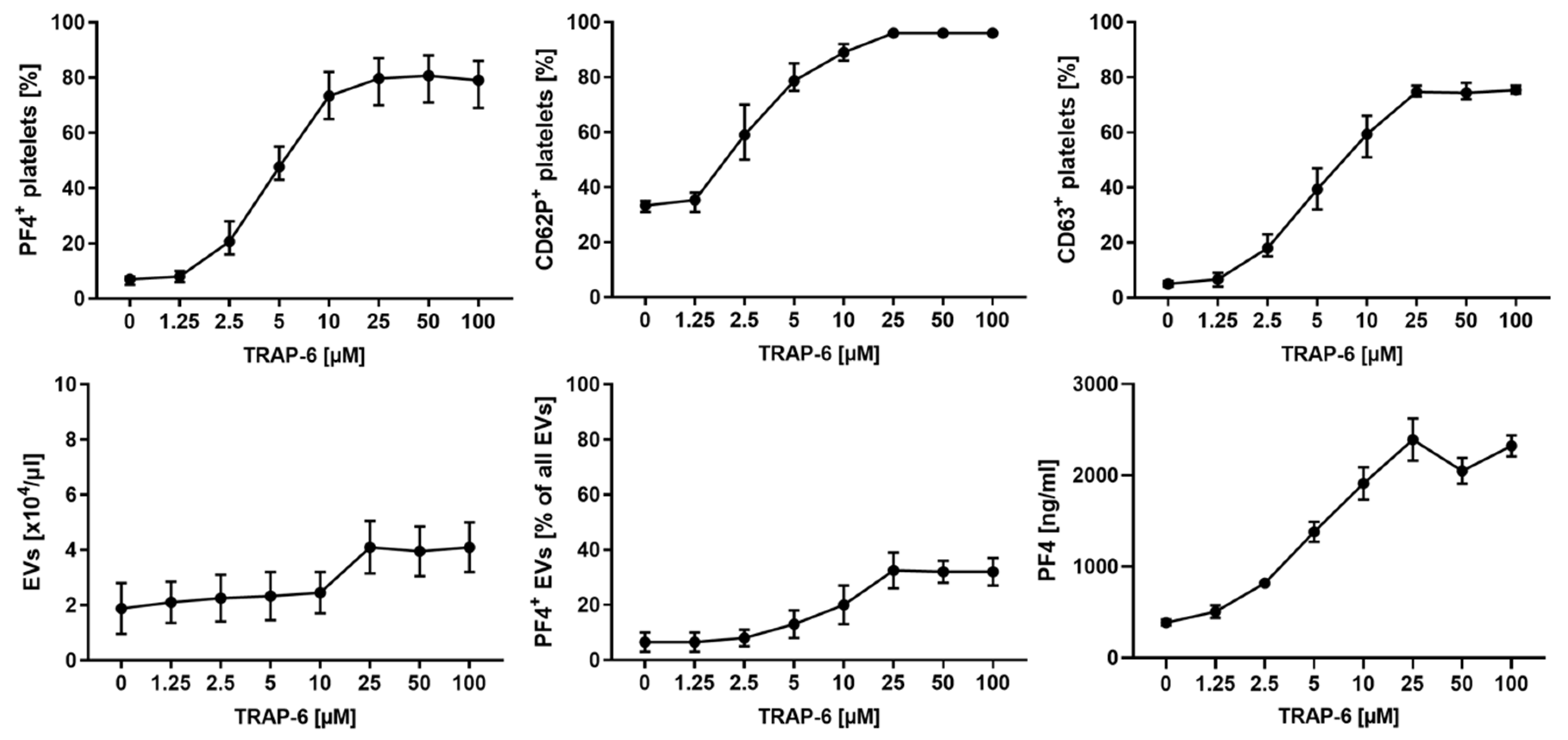
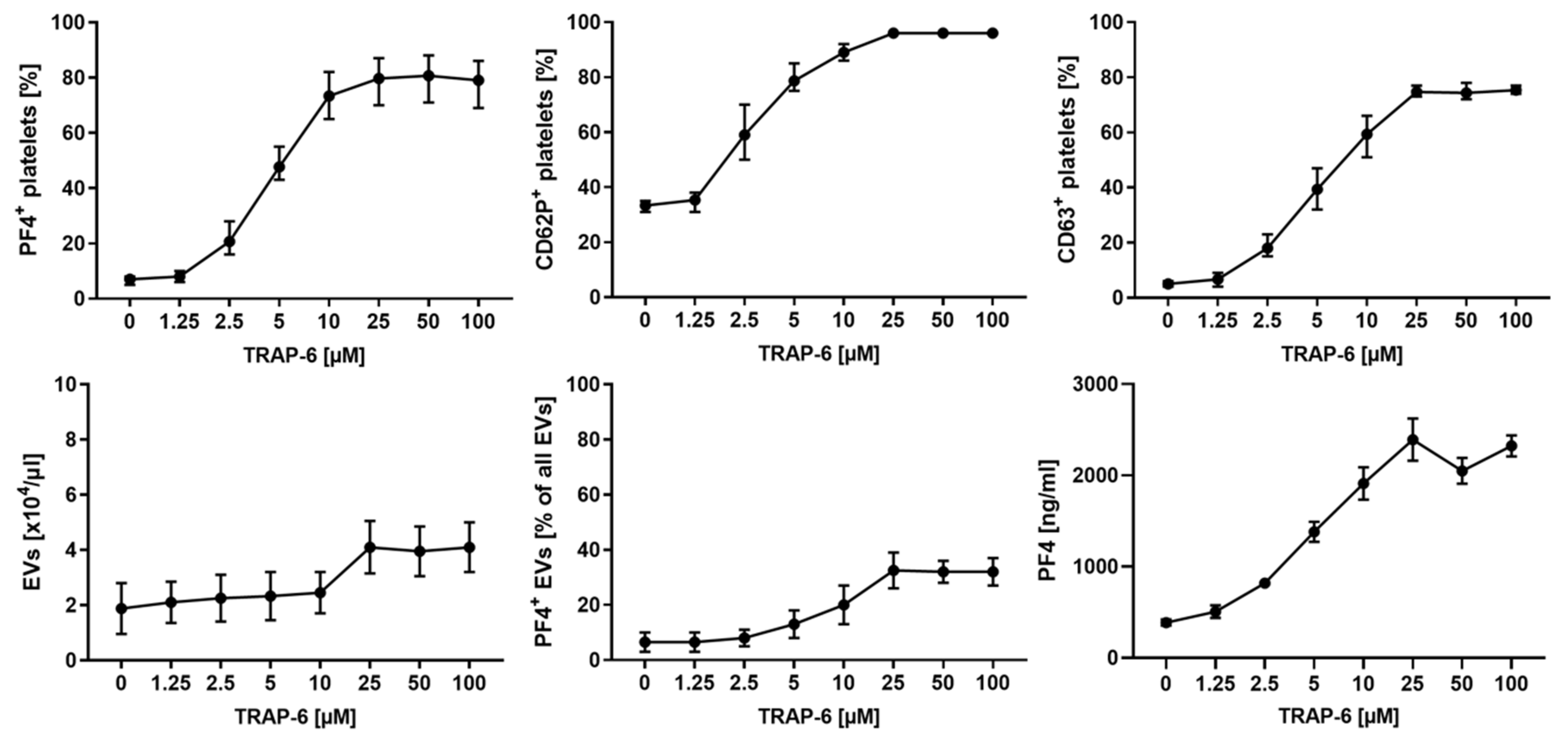
2.2. Platelet Activation and Release of pEVs in Response to TRAP-6 Are Dose-Dependent
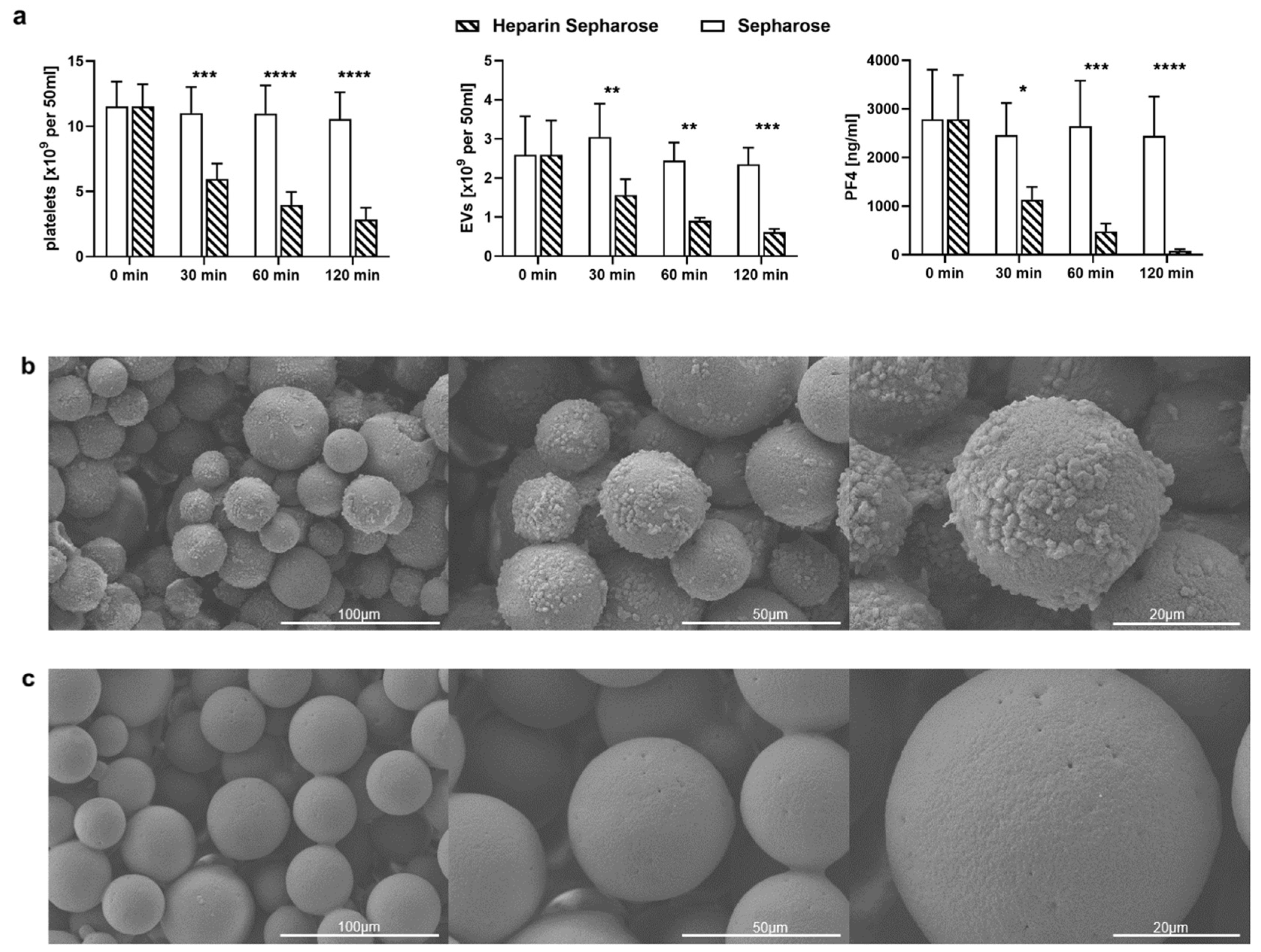
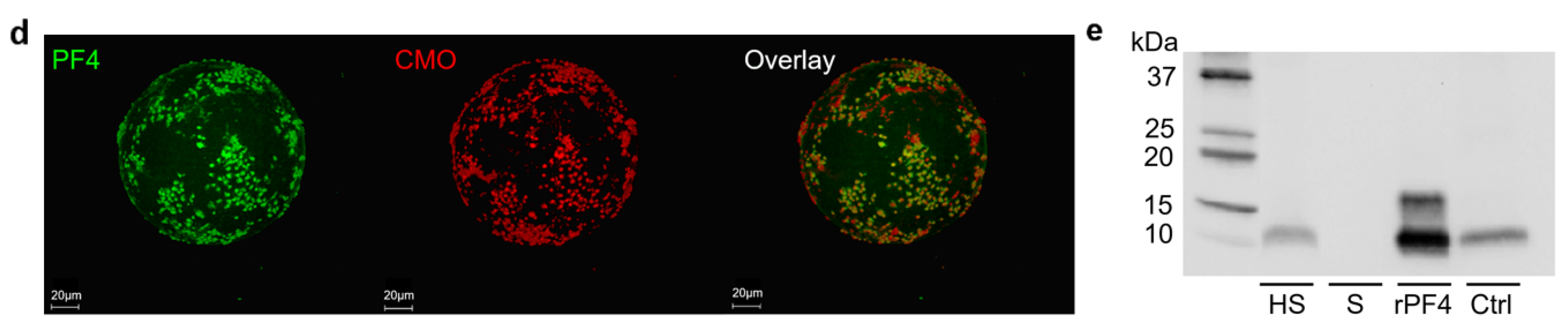
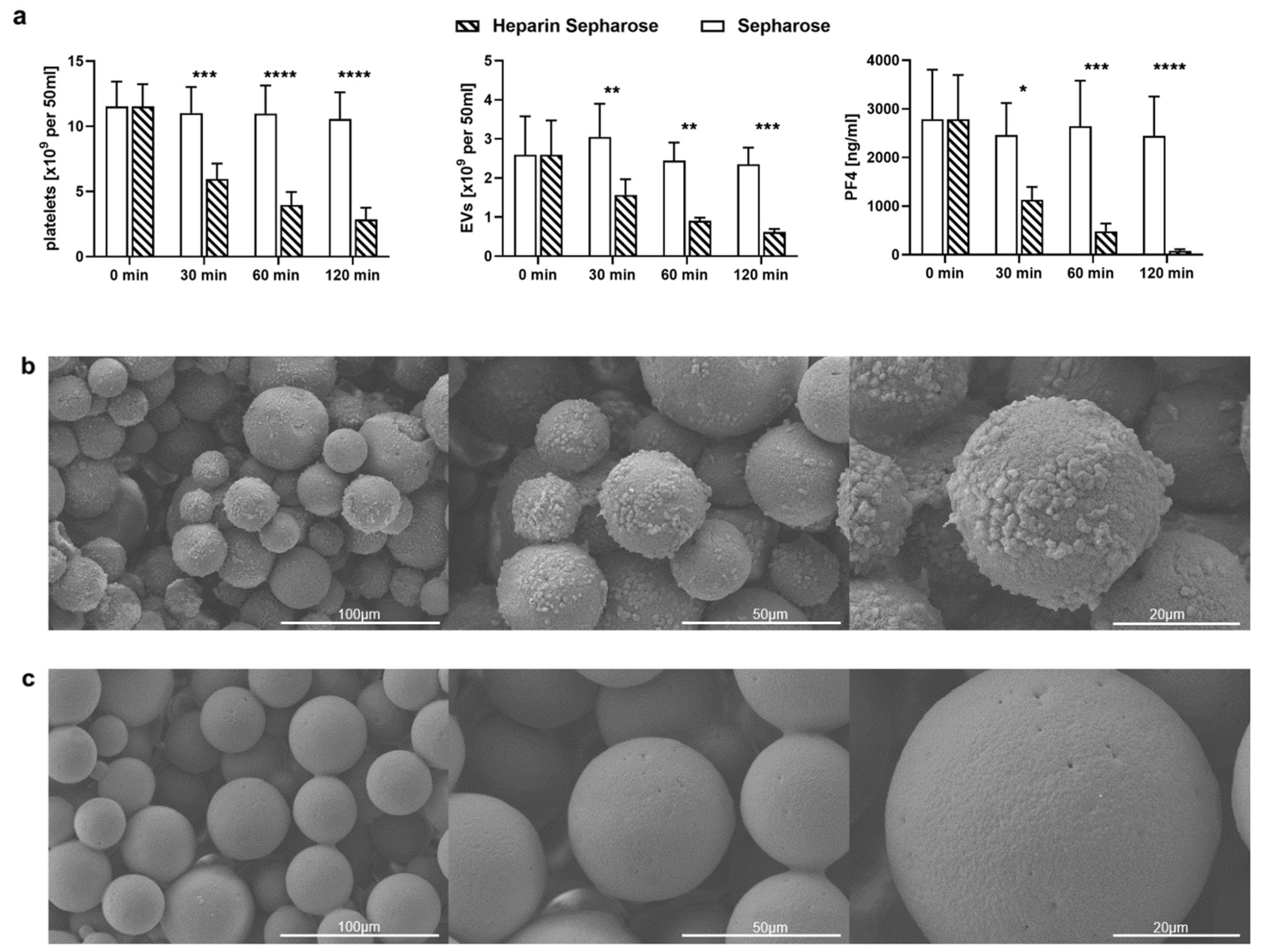
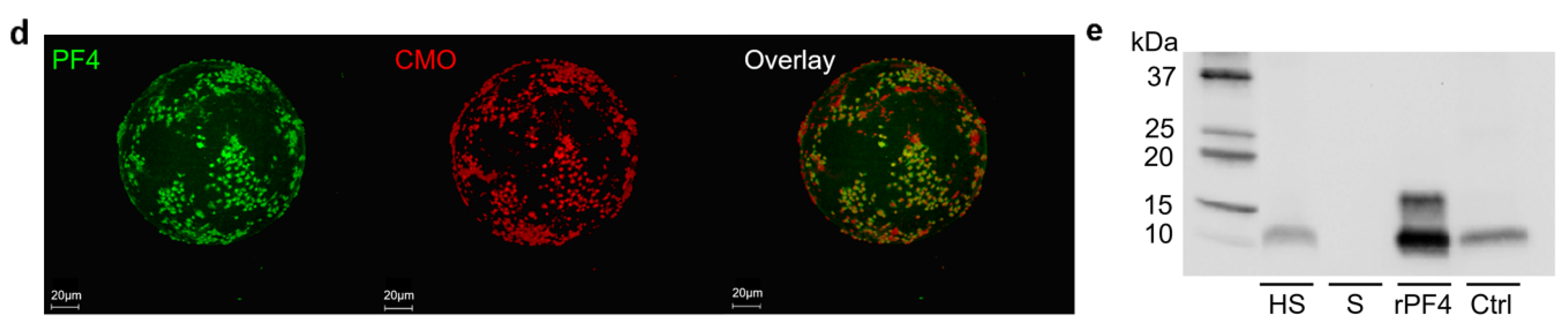
2.3. TRAP-6 Activated Platelets and pEVs Are Bound by Heparin-Functionalized Adsorbents
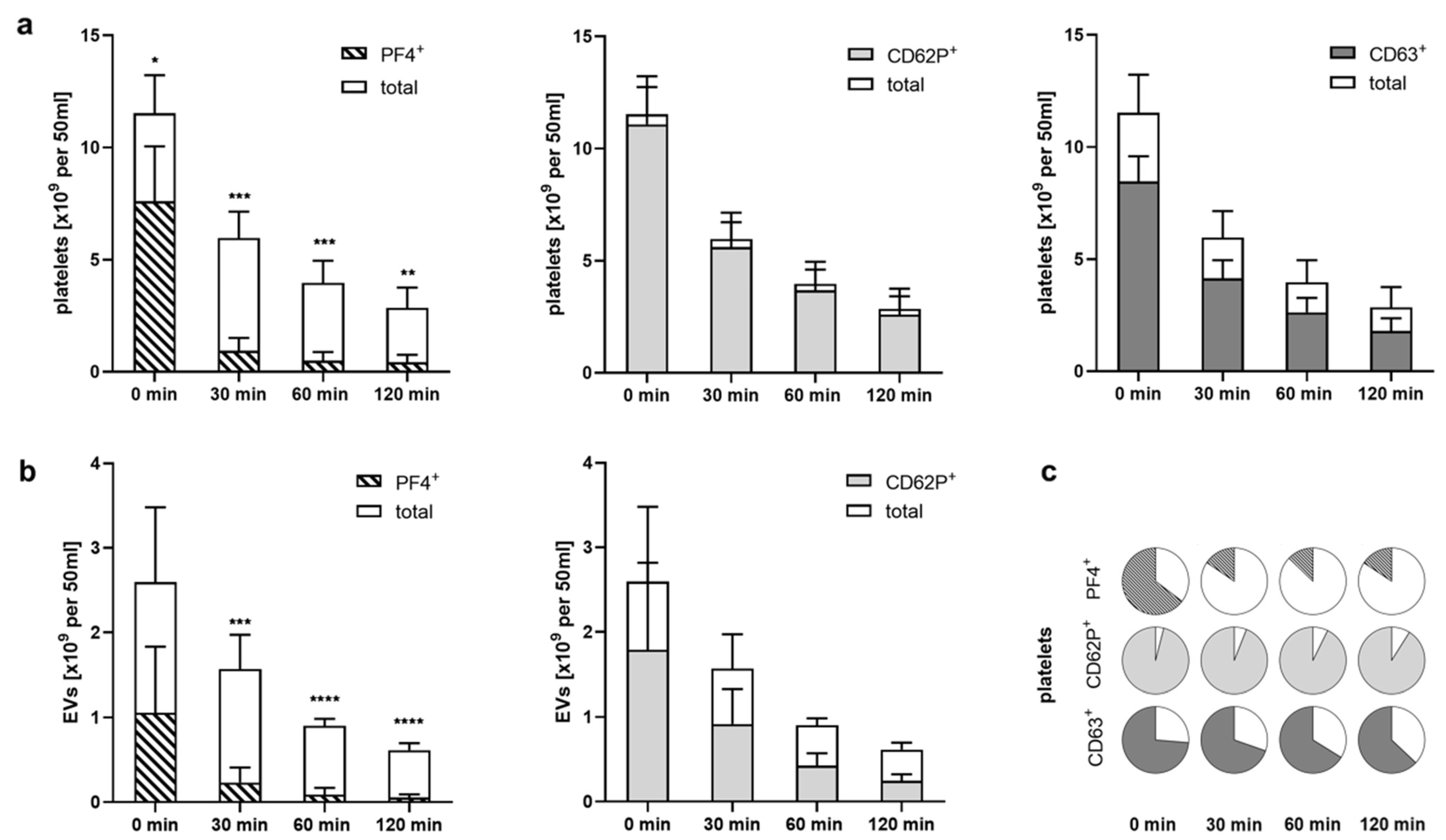
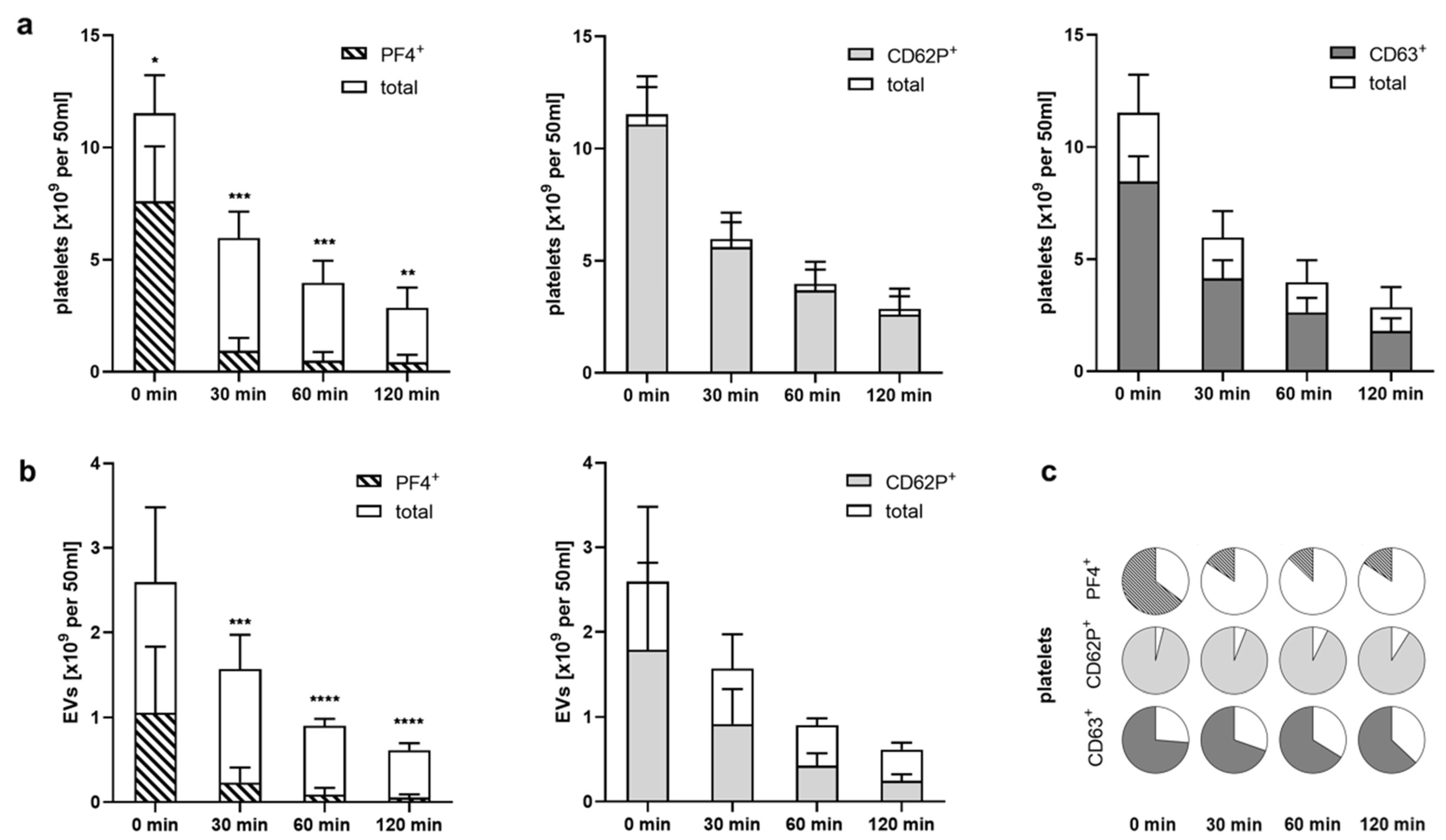
2.4. Heparinized Adsorbents Preferentially Bind PF4+ Platelets and pEVs
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Plasma and Platelet Concentrates
4.3. Adsorbents
4.4. Adsorption of PF4, Histones/Nucleosomes and HMGB1
4.5. Quantification of PF4, Histones/Nucleosomes and HMGB1
4.6. Platelet Activation by TRAP-6
4.7. Flow Cytometric Characterization of Platelets and Platelet-Derived Extracellular Vesicles
4.8. Re-Circulation of Activated Platelets over Adsorbent Columns
4.9. Scanning Electron Microscopy
4.10. Immunofluorescence Staining and Confocal Microscopy
4.11. Gel Electrophoresis and Western Blotting
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Engelmann, B.; Massberg, S. Thrombosis as an Intravascular Effector of Innate Immunity. Nat. Rev. Immunol. 2013, 13, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Hottz, E.D.; Azevedo-Quintanilha, I.G.; Palhinha, L.; Teixeira, L.; Barreto, E.A.; Pão, C.R.R.; Righy, C.; Franco, S.; Souza, T.M.L.; Kurtz, P.; et al. Platelet Activation and Platelet-Monocyte Aggregate Formation Trigger Tissue Factor Expression in Patients with Severe COVID-19. Blood 2020, 136, 1330–1341. [Google Scholar] [CrossRef] [PubMed]
- McFadyen, J.D.; Stevens, H.; Peter, K. The Emerging Threat of (Micro)Thrombosis in COVID-19 and Its Therapeutic Implications. Circ. Res. 2020, 127, 571–587. [Google Scholar] [CrossRef] [PubMed]
- Klok, F.A.; Kruip, M.J.H.A.; van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.A.M.P.J.; Kant, K.M.; Kaptein, F.H.J.; van Paassen, J.; Stals, M.A.M.; Huisman, M.V.; et al. Incidence of Thrombotic Complications in Critically Ill ICU Patients with COVID-19. Thromb. Res. 2020, 191, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yan, X.; Fan, Q.; Liu, H.; Liu, X.; Liu, Z.; Zhang, Z. D-Dimer Levels on Admission to Predict in-Hospital Mortality in Patients with COVID-19. J. Thromb. Haemost. 2020, 18, 1324–1329. [Google Scholar] [CrossRef]
- Pan, D.; Amison, R.T.; Riffo-Vasquez, Y.; Spina, D.; Cleary, S.J.; Wakelam, M.J.; Page, C.P.; Pitchford, S.C.; Welch, H.C.E. P-Rex and Vav Rac-GEFs in Platelets Control Leukocyte Recruitment to Sites of Inflammation. Blood 2015, 125, 1146–1158. [Google Scholar] [CrossRef]
- Chimen, M.; Evryviadou, A.; Box, C.L.; Harrison, M.J.; Hazeldine, J.; Dib, L.H.; Kuravi, S.J.; Payne, H.; Price, J.M.J.; Kavanagh, D.; et al. Appropriation of GPIbα from Platelet-Derived Extracellular Vesicles Supports Monocyte Recruitment in Systemic Inflammation. Haematologica 2020, 105, 1248–1261. [Google Scholar] [CrossRef]
- Middleton, E.A.; He, X.-Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil Extracellular Traps Contribute to Immunothrombosis in COVID-19 Acute Respiratory Distress Syndrome. Blood 2020, 136, 1169–1179. [Google Scholar] [CrossRef]
- Swystun, L.L.; Liaw, P.C. The Role of Leukocytes in Thrombosis. Blood 2016, 128, 753–762. [Google Scholar] [CrossRef] [Green Version]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 Activates Neutrophil Extracellular Traps to Ensnare Bacteria in Septic Blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef]
- Lindmark, E.; Tenno, T.; Siegbahn, A. Role of Platelet P-Selectin and CD40 Ligand in the Induction of Monocytic Tissue Factor Expression. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2322–2328. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, I.I.; Apta, B.H.R.; Bonna, A.M.; Harper, M.T. Platelet P-Selectin Triggers Rapid Surface Exposure of Tissue Factor in Monocytes. Sci. Rep. 2019, 9, 13397. [Google Scholar] [CrossRef] [PubMed]
- Bakogiannis, C.; Sachse, M.; Stamatelopoulos, K.; Stellos, K. Platelet-Derived Chemokines in Inflammation and Atherosclerosis. Cytokine 2019, 122, 154157. [Google Scholar] [CrossRef] [PubMed]
- Tripisciano, C.; Weiss, R.; Eichhorn, T.; Spittler, A.; Heuser, T.; Fischer, M.B.; Weber, V. Different Potential of Extracellular Vesicles to Support Thrombin Generation: Contributions of Phosphatidylserine, Tissue Factor, and Cellular Origin. Sci. Rep. 2017, 7, 6522. [Google Scholar] [CrossRef] [PubMed]
- Iba, T.; Ogura, H. Role of Extracellular Vesicles in the Development of Sepsis-Induced Coagulopathy. J. Intensive Care 2018, 6, 68. [Google Scholar] [CrossRef] [PubMed]
- Fendl, B. Differential Interaction of Platelet-Derived Extracellular Vesicles With Circulating Immune Cells: Roles of TAM Receptors, CD11b, and Phosphatidylserine. Front. Immunol. 2018, 9, 13. [Google Scholar] [CrossRef] [PubMed]
- Puhm, F.; Boilard, E.; Machlus, K.R. Platelet Extracellular Vesicles: Beyond the Blood. Arterioscler. Thromb. Vasc. Biol. 2020, 41, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Szabó, G.T.; Tarr, B.; Pálóczi, K.; Éder, K.; Lajkó, E.; Kittel, Á.; Tóth, S.; György, B.; Pásztói, M.; Németh, A.; et al. Critical Role of Extracellular Vesicles in Modulating the Cellular Effects of Cytokines. Cell. Mol. Life Sci. 2014, 71, 4055–4067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boilard, E. Thematic Review Series: Exosomes and Microvesicles: Lipids as Key Components of Their Biogenesis and Functions Extracellular Vesicles and Their Content in Bioactive Lipid Mediators: More than a Sack of MicroRNA. J. Lipid Res. 2018, 59, 2037–2046. [Google Scholar] [CrossRef] [Green Version]
- Maugeri, N.; Capobianco, A.; Rovere-Querini, P.; Ramirez, G.A.; Tombetti, E.; Valle, P.D.; Monno, A.; D’Alberti, V.; Gasparri, A.M.; Franchini, S.; et al. Platelet Microparticles Sustain Autophagy-Associated Activation of Neutrophils in Systemic Sclerosis. Sci. Transl. Med. 2018, 10, eaao3089. [Google Scholar] [CrossRef] [Green Version]
- Maharaj, S.; Chang, S. Anti-PF4/Heparin Antibodies Are Increased in Hospitalized Patients with Bacterial Sepsis. Thromb. Res. 2018, 171, 111–113. [Google Scholar] [CrossRef] [PubMed]
- Wegrzyn, G.; Walborn, A.; Rondina, M.; Fareed, J.; Hoppensteadt, D. Biomarkers of Platelet Activation and Their Prognostic Value in Patients With Sepsis-Associated Disseminated Intravascular Coagulopathy. Clin. Appl. Thromb. Hemost. 2021, 27, 1076029620943300. [Google Scholar] [CrossRef] [PubMed]
- Comer, S.P.; Cullivan, S.; Szklanna, P.B.; Weiss, L.; Cullen, S.; Kelliher, S.; Smolenski, A.; Murphy, C.; Altaie, H.; Curran, J.; et al. COVID-19 Induces a Hyperactive Phenotype in Circulating Platelets. PLoS Biol. 2021, 19, e3001109. [Google Scholar] [CrossRef]
- Sachais, B.; Higazi, A.A.-R.; Cines, D.; Poncz, M.; Kowalska, M. Interactions of Platelet Factor 4 with the Vessel Wall. Semin. Thromb. Hemost. 2004, 30, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Petersen, F.; Brandt, E.; Lindahl, U.; Spillmann, D. Characterization of a Neutrophil Cell Surface Glycosaminoglycan That Mediates Binding of Platelet Factor 4. J. Biol. Chem. 1999, 274, 12376–12382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, N.H.; Sørvoll, I.H.; Michelsen, A.E.; Munthe, L.A.; Lund-Johansen, F.; Ahlen, M.T.; Wiedmann, M.; Aamodt, A.-H.; Skattør, T.H.; Tjønnfjord, G.E.; et al. Thrombosis and Thrombocytopenia after ChAdOx1 NCoV-19 Vaccination. N. Engl. J. Med. 2021, 384, 2124–2130. [Google Scholar] [CrossRef] [PubMed]
- Greinacher, A.; Thiele, T.; Warkentin, T.E.; Weisser, K.; Kyrle, P.A.; Eichinger, S. Thrombotic Thrombocytopenia after ChAdOx1 NCov-19 Vaccination. N. Engl. J. Med. 2021, 384, 2092–2101. [Google Scholar] [CrossRef]
- Maugeri, N.; Franchini, S.; Campana, L.; Baldini, M.; Ramirez, G.A.; Sabbadini, M.G.; Rovere-Querini, P.; Manfredi, A.A. Circulating Platelets as a Source of the Damage-Associated Molecular Pattern HMGB1 in Patients with Systemic Sclerosis. Autoimmunity 2012, 45, 584–587. [Google Scholar] [CrossRef]
- Rouhiainen, A.; Imai, S.; Rauvala, H.; Parkkinen, J. Occurrence of Amphoterin (HMG1) as an Endogenous Protein of Human Platelets That Is Exported to the Cell Surface upon Platelet Activation. Thromb. Haemost. 2000, 84, 1087–1094. [Google Scholar] [CrossRef]
- Eichhorn, T.; Linsberger, I.; Lauková, L.; Tripisciano, C.; Fendl, B.; Weiss, R.; König, F.; Valicek, G.; Miestinger, G.; Hörmann, C.; et al. Analysis of Inflammatory Mediator Profiles in Sepsis Patients Reveals That Extracellular Histones Are Strongly Elevated in Nonsurvivors. Mediators Inflamm. 2021, 2021, 8395048. [Google Scholar] [CrossRef]
- Chen, R.; Huang, Y.; Quan, J.; Liu, J.; Wang, H.; Billiar, T.R.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. HMGB1 as a Potential Biomarker and Therapeutic Target for Severe COVID-19. Heliyon 2020, 6, e05672. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, N.; Campana, L.; Gavina, M.; Covino, C.; De Metrio, M.; Panciroli, C.; Maiuri, L.; Maseri, A.; D’Angelo, A.; Bianchi, M.E.; et al. Activated Platelets Present High Mobility Group Box 1 to Neutrophils, Inducing Autophagy and Promoting the Extrusion of Neutrophil Extracellular Traps. J. Thromb. Haemost. 2014, 12, 2074–2088. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Kawahara, K.; Nakamura, T.; Yamada, S.; Nakamura, T.; Abeyama, K.; Hashiguchi, T.; Maruyama, I. High-Mobility Group Box 1 Protein Promotes Development of Microvascular Thrombosis in Rats: Prothrombotic Effects of HMGB1. J. Thromb. Haemost. 2007, 5, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Vogel, S.; Bodenstein, R.; Chen, Q.; Feil, S.; Feil, R.; Rheinlaender, J.; Schäffer, T.E.; Bohn, E.; Frick, J.-S.; Borst, O.; et al. Platelet-Derived HMGB1 Is a Critical Mediator of Thrombosis. J. Clin. Invest. 2015, 125, 4638–4654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, Y.; Yang, Z.-Y.; Yin, T.; Li, L.; Yuan, W.-W.; Wu, H.-S.; Wang, C.-Y. Heparin Changes the Conformation of High-Mobility Group Protein 1 and Decreases Its Affinity toward Receptor for Advanced Glycation Endproducts in Vitro. Int. Immunopharmacol. 2011, 11, 187–193. [Google Scholar] [CrossRef]
- Ekaney, M.L.; Otto, G.P.; Sossdorf, M.; Sponholz, C.; Boehringer, M.; Loesche, W.; Rittirsch, D.; Wilharm, A.; Kurzai, O.; Bauer, M.; et al. Impact of Plasma Histones in Human Sepsis and Their Contribution to Cellular Injury and Inflammation. Crit. Care 2014, 18, 543. [Google Scholar] [CrossRef] [Green Version]
- Veras, F.P.; Pontelli, M.C.; Silva, C.M.; Toller-Kawahisa, J.E.; de Lima, M.; Nascimento, D.C.; Schneider, A.H.; Caetité, D.; Tavares, L.A.; Paiva, I.M.; et al. SARS-CoV-2–Triggered Neutrophil Extracellular Traps Mediate COVID-19 Pathology. J. Exp. Med. 2020, 217, e20201129. [Google Scholar] [CrossRef]
- Barnes, B.J.; Adrover, J.M.; Baxter-Stoltzfus, A.; Borczuk, A.; Cools-Lartigue, J.; Crawford, J.M.; Daßler-Plenker, J.; Guerci, P.; Huynh, C.; Knight, J.S.; et al. Targeting Potential Drivers of COVID-19: Neutrophil Extracellular Traps. J. Exp. Med. 2020, 217, e20200652. [Google Scholar] [CrossRef]
- Traby, L.; Kollars, M.; Kussmann, M.; Karer, M.; Šinkovec, H.; Lobmeyr, E.; Hermann, A.; Staudinger, T.; Schellongowski, P.; Rössler, B.; et al. Extracellular Vesicles and Citrullinated Histone H3 in Coronavirus Disease 2019 Patients. Thromb. Haemost. 2021, 113–122. [Google Scholar] [CrossRef]
- Shaw, R.J.; Austin, J.; Taylor, J.; Dutt, T.; Wang, G.; Abrams, S.T.; Toh, C.H. Circulating Histone Levels Correlate with the Severity of COVID-19 and the Extent of Coagulation Activation and Inflammation. Blood 2020, 136, 19. [Google Scholar] [CrossRef]
- Rock, G.; Weber, V.; Stegmayr, B. Therapeutic Plasma Exchange (TPE) as a Plausible Rescue Therapy in Severe Vaccine-Induced Immune Thrombotic Thrombocytopenia. Transfus. Apher. Sci. 2021, 60, 103174. [Google Scholar] [CrossRef] [PubMed]
- Ronco, C.; Bagshaw, S.M.; Bellomo, R.; Clark, W.R.; Husain-Syed, F.; Kellum, J.A.; Ricci, Z.; Rimmelé, T.; Reis, T.; Ostermann, M. Extracorporeal Blood Purification and Organ Support in the Critically Ill Patient during COVID-19 Pandemic: Expert Review and Recommendation. Blood Purif. 2021, 50, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Yin, Z.; Hu, Y.; Mei, H. Controlling Cytokine Storm Is Vital in COVID-19. Front. Immunol. 2020, 11, 570993. [Google Scholar] [CrossRef] [PubMed]
- Seffer, M.-T.; Cottam, D.; Forni, L.G.; Kielstein, J.T. Heparin 2.0: A New Approach to the Infection Crisis. Blood Purif. 2021, 50, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Connors, J.M.; Levy, J.H. COVID-19 and Its Implications for Thrombosis and Anticoagulation. Blood 2020, 135, 2033–2040. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in COVID-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Skendros, P.; Mitsios, A.; Chrysanthopoulou, A.; Mastellos, D.C.; Metallidis, S.; Rafailidis, P.; Ntinopoulou, M.; Sertaridou, E.; Tsironidou, V.; Tsigalou, C.; et al. Complement and Tissue Factor–Enriched Neutrophil Extracellular Traps Are Key Drivers in COVID-19 Immunothrombosis. J. Clin. Investig. 2020, 130, 6151–6157. [Google Scholar] [CrossRef]
- Kapur, R.; Zufferey, A.; Boilard, E.; Semple, J.W. Nouvelle Cuisine: Platelets Served with Inflammation. J. Immunol. 2015, 194, 5579–5587. [Google Scholar] [CrossRef] [Green Version]
- Passacquale, G.; Vamadevan, P.; Pereira, L.; Hamid, C.; Corrigall, V.; Ferro, A. Monocyte-Platelet Interaction Induces a Pro-Inflammatory Phenotype in Circulating Monocytes. PLoS ONE 2011, 6, e25595. [Google Scholar] [CrossRef] [Green Version]
- Fendl, B.; Weiss, R.; Eichhorn, T.; Spittler, A.; Fischer, M.B.; Weber, V. Storage of Human Whole Blood, but Not Isolated Monocytes, Preserves the Distribution of Monocyte Subsets. Biochem. Biophys. Res. Commun. 2019, 517, 709–714. [Google Scholar] [CrossRef]
- Lee, S.J.; Yoon, B.R.; Kim, H.Y.; Yoo, S.-J.; Kang, S.W.; Lee, W.-W. Activated Platelets Convert CD14+CD16- Into CD14+CD16+ Monocytes With Enhanced FcγR-Mediated Phagocytosis and Skewed M2 Polarization. Front. Immunol. 2021, 11, 611133. [Google Scholar] [CrossRef] [PubMed]
- Payen, D.; Cravat, M.; Maadadi, H.; Didelot, C.; Prosic, L.; Dupuis, C.; Losser, M.-R.; De Carvalho Bittencourt, M. A Longitudinal Study of Immune Cells in Severe COVID-19 Patients. Front. Immunol. 2020, 11, 580250. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Young, J.; Song, D.; Esko, J.D. Heparan Sulfate Is Essential for High Mobility Group Protein 1 (HMGB1) Signaling by the Receptor for Advanced Glycation End Products (RAGE). J. Biol. Chem. 2011, 286, 41736–41744. [Google Scholar] [CrossRef] [Green Version]
- McCrea, K.; Ward, R.; LaRosa, S.P. Removal of Carbapenem-Resistant Enterobacteriaceae (CRE) from Blood by Heparin-Functional Hemoperfusion Media. PLoS ONE 2014, 9, e114242. [Google Scholar] [CrossRef] [PubMed]
- Seffer, M.-T.; Eden, G.; Engelmann, S.; Kielstein, J.T. Elimination of Staphylococcus Aureus from the Bloodstream Using a Novel Biomimetic Sorbent Haemoperfusion Device. BMJ Case Rep. 2020, 13, e235262. [Google Scholar] [CrossRef]
- Fajnzylber, J.; Regan, J.; Coxen, K.; Corry, H.; Wong, C.; Rosenthal, A.; Worrall, D.; Giguel, F.; Piechocka-Trocha, A.; Atyeo, C.; et al. SARS-CoV-2 Viral Load Is Associated with Increased Disease Severity and Mortality. Nat. Commun. 2020, 11, 5493. [Google Scholar] [CrossRef]
- Tandon, R.; Sharp, J.S.; Zhang, F.; Pomin, V.H.; Ashpole, N.M.; Mitra, D.; McCandless, M.G.; Jin, W.; Liu, H.; Sharma, P.; et al. Effective Inhibition of SARS-CoV-2 Entry by Heparin and Enoxaparin Derivatives. J. Virol. 2021, 95, e01987-20. [Google Scholar] [CrossRef]
- Kalra, R.S.; Kandimalla, R. Engaging the Spikes: Heparan Sulfate Facilitates SARS-CoV-2 Spike Protein Binding to ACE2 and Potentiates Viral Infection. Signal Transduct. Target. Ther. 2021, 6, 39. [Google Scholar] [CrossRef]
- Rifkin, B.S.; Stewart, I.J. Seraph-100 Hemoperfusion in SARS-CoV-2-Infected Patients Early in Critical Illness: A Case Series. Blood Purif. 2021, 1–4. [Google Scholar] [CrossRef]
- Kielstein, J.T.; Borchina, D.-N.; Fühner, T.; Hwang, S.; Mattoon, D.; Ball, A.J. Hemofiltration with the Seraph® 100 Microbind® Affinity Filter Decreases SARS-CoV-2 Nucleocapsid Protein in Critically Ill COVID-19 Patients. Crit. Care 2021, 25, 190. [Google Scholar] [CrossRef]
- Lamy, B.; Dargère, S.; Arendrup, M.C.; Parienti, J.-J.; Tattevin, P. How to Optimize the Use of Blood Cultures for the Diagnosis of Bloodstream Infections? A State-of-the Art. Front. Microbiol. 2016, 7, 697. [Google Scholar] [CrossRef]
- Eisenreich, W.; Rudel, T.; Heesemann, J.; Goebel, W. How Viral and Intracellular Bacterial Pathogens Reprogram the Metabolism of Host Cells to Allow Their Intracellular Replication. Front. Cell. Infect. Microbiol. 2019, 9, 42. [Google Scholar] [CrossRef] [Green Version]
- Percivalle, E.; Sammartino, J.C.; Cassaniti, I.; Arbustini, E.; Urtis, M.; Smirnova, A.; Concardi, M.; Belgiovine, C.; Ferrari, A.; Lilleri, D.; et al. Macrophages and Monocytes: “Trojan Horses” in COVID-19. Viruses 2021, 13, 2178. [Google Scholar] [CrossRef]
- Knoll, R.; Schultze, J.L.; Schulte-Schrepping, J. Monocytes and Macrophages in COVID-19. Front. Immunol. 2021, 12, 720109. [Google Scholar] [CrossRef]
- Carestia, A.; Kaufman, T.; Schattner, M. Platelets: New Bricks in the Building of Neutrophil Extracellular Traps. Front. Immunol. 2016, 7, 271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carestia, A.; Kaufman, T.; Rivadeneyra, L.; Landoni, V.I.; Pozner, R.G.; Negrotto, S.; D’Atri, L.P.; Gómez, R.M.; Schattner, M. Mediators and Molecular Pathways Involved in the Regulation of Neutrophil Extracellular Trap Formation Mediated by Activated Platelets. J. Leukoc. Biol. 2016, 99, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Gollomp, K.; Kim, M.; Johnston, I.; Hayes, V.; Welsh, J.; Arepally, G.M.; Kahn, M.; Lambert, M.P.; Cuker, A.; Cines, D.B.; et al. Neutrophil Accumulation and NET Release Contribute to Thrombosis in HIT. JCI Insight 2018, 3, e99445. [Google Scholar] [CrossRef] [Green Version]
- McCrea, K.R.; Ward, R.S. An Affinity Adsorption Media That Mimics Heparan Sulfate Proteoglycans for the Treatment of Drug-Resistant Bacteremia. Surf. Sci. 2016, 648, 42–46. [Google Scholar] [CrossRef]
- Weiss, R.; Gröger, M.; Rauscher, S.; Fendl, B.; Eichhorn, T.; Fischer, M.B.; Spittler, A.; Weber, V. Differential Interaction of Platelet-Derived Extracellular Vesicles with Leukocyte Subsets in Human Whole Blood. Sci. Rep. 2018, 8, 6598. [Google Scholar] [CrossRef] [PubMed]
- George, S.K.; Lauková, L.; Weiss, R.; Semak, V.; Fendl, B.; Weiss, V.U.; Steinberger, S.; Allmaier, G.; Tripisciano, C.; Weber, V. Comparative Analysis of Platelet-Derived Extracellular Vesicles Using Flow Cytometry and Nanoparticle Tracking Analysis. Int. J. Mol. Sci. 2021, 22, 3839. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Adsorbent | Polymer | Mean Particle Size [µm] | Ligand Density | Supplier |
|---|---|---|---|---|
| Seraph-100 | Polyethylene | 370 | Porcine heparin 2 mg/g beads | ExThera Medical, Martinez, CA, USA |
| Heparin Sepharose | Cross-linked agarose | 90 | Porcine heparin 2 mg/mL beads | Cytiva, Uppsala, Sweden |
| Sepharose | Cross-linked agarose | 90 | none | Cytiva, Uppsala, Sweden |
| Flow Cytometry | ||||||
| Antigen | Origin | Clone | Marker for | Fluorochrome | Abbreviation | Supplier |
| CD41 | Mouse | P2 | Platelets | Phycoerythrin Cyanin 7 | PC7 | Beckman Coulter |
| CD62P | Mouse | CLB Thromb6 | Activated platelets | Fluorescein Isothiocyanate | FITC | Beckman Coulter |
| CD63 | Mouse | H5C6 | Activated platelets | Alexa Fluor 647 | AF647 | BioLegend |
| PF4 | Mouse | # 170138 | Activated platelets | Phycoerythrin | PE | R&D Systems |
| Anx5 | - | - | Phosphatidyl-serine | Allophyco-cyanin | APC | BD Biosciences |
| Confocal Microscopy | ||||||
| Antigen | Origin | Clone | Marker for | Fluorochrome | Abbreviation | Supplier |
| PF4 | Mouse | D7 | Activated platelets | - | - | Santa Cruz Biotechnology |
| Mouse IgG | Goat | Polyclonal | - | AlexaFluor 488 | AF488 | Jackson Immunoresearch |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ebeyer-Masotta, M.; Eichhorn, T.; Weiss, R.; Semak, V.; Lauková, L.; Fischer, M.B.; Weber, V. Heparin-Functionalized Adsorbents Eliminate Central Effectors of Immunothrombosis, including Platelet Factor 4, High-Mobility Group Box 1 Protein and Histones. Int. J. Mol. Sci. 2022, 23, 1823. https://doi.org/10.3390/ijms23031823
Ebeyer-Masotta M, Eichhorn T, Weiss R, Semak V, Lauková L, Fischer MB, Weber V. Heparin-Functionalized Adsorbents Eliminate Central Effectors of Immunothrombosis, including Platelet Factor 4, High-Mobility Group Box 1 Protein and Histones. International Journal of Molecular Sciences. 2022; 23(3):1823. https://doi.org/10.3390/ijms23031823
Chicago/Turabian StyleEbeyer-Masotta, Marie, Tanja Eichhorn, René Weiss, Vladislav Semak, Lucia Lauková, Michael B. Fischer, and Viktoria Weber. 2022. "Heparin-Functionalized Adsorbents Eliminate Central Effectors of Immunothrombosis, including Platelet Factor 4, High-Mobility Group Box 1 Protein and Histones" International Journal of Molecular Sciences 23, no. 3: 1823. https://doi.org/10.3390/ijms23031823
APA StyleEbeyer-Masotta, M., Eichhorn, T., Weiss, R., Semak, V., Lauková, L., Fischer, M. B., & Weber, V. (2022). Heparin-Functionalized Adsorbents Eliminate Central Effectors of Immunothrombosis, including Platelet Factor 4, High-Mobility Group Box 1 Protein and Histones. International Journal of Molecular Sciences, 23(3), 1823. https://doi.org/10.3390/ijms23031823