
Why Monoamine Oxidase B Preferably Metabolizes N-Methylhistamine over Histamine: Evidence from the Multiscale Simulation of the Rate-Limiting Step




Abstract
:
1. Introduction
2. Results and Discussion
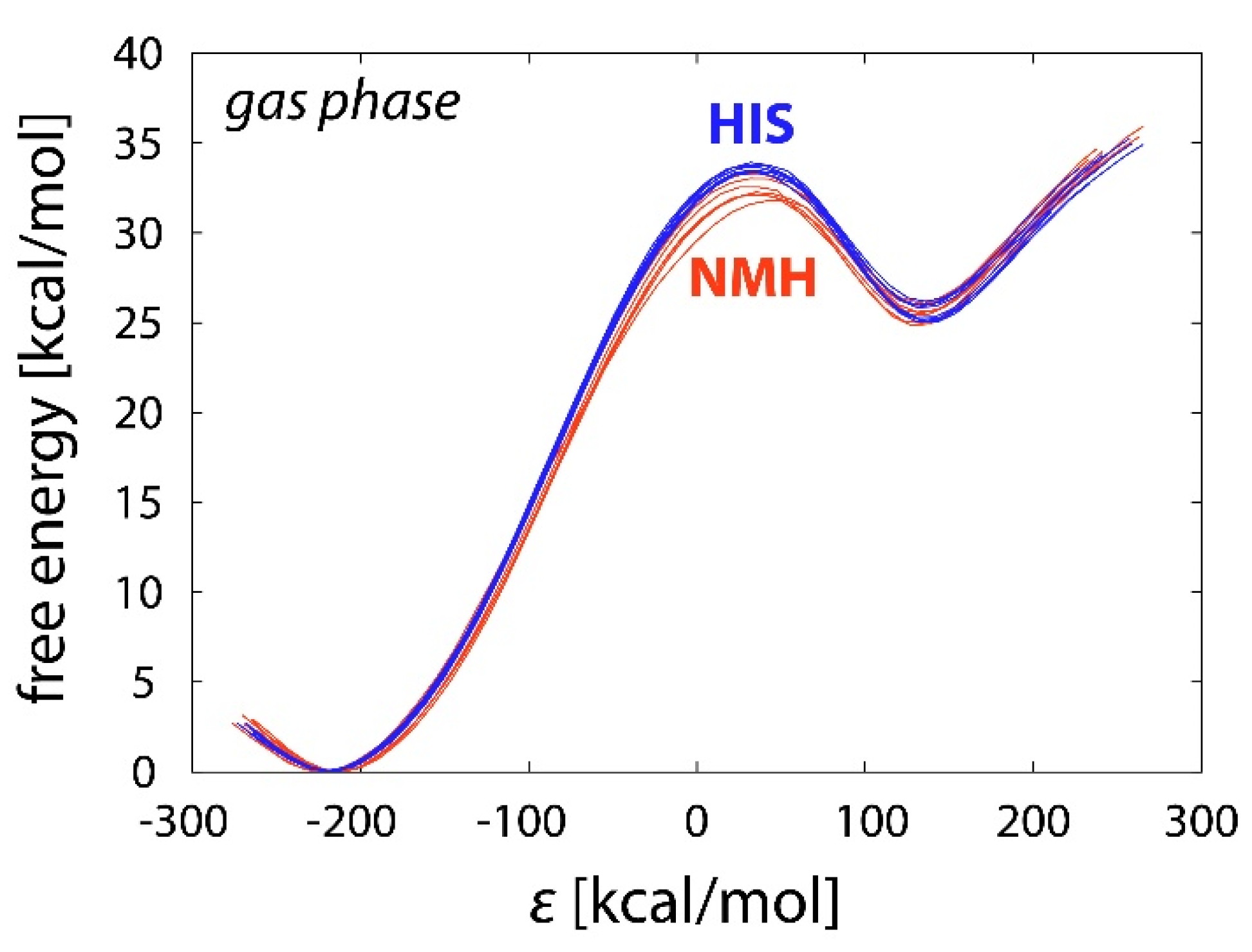
2.1. Reference Reactions in the Gas Phase
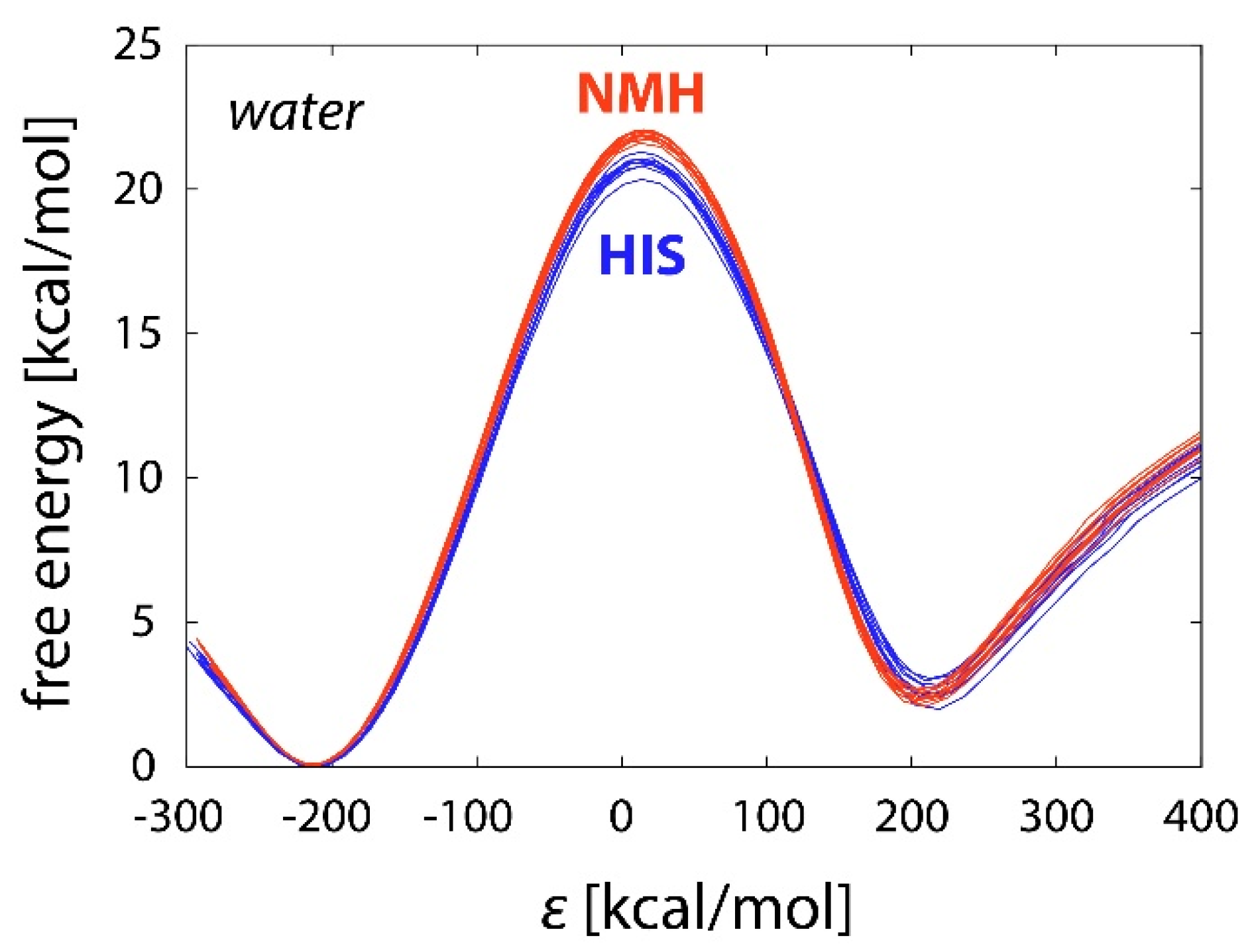
2.2. Reactions in Water
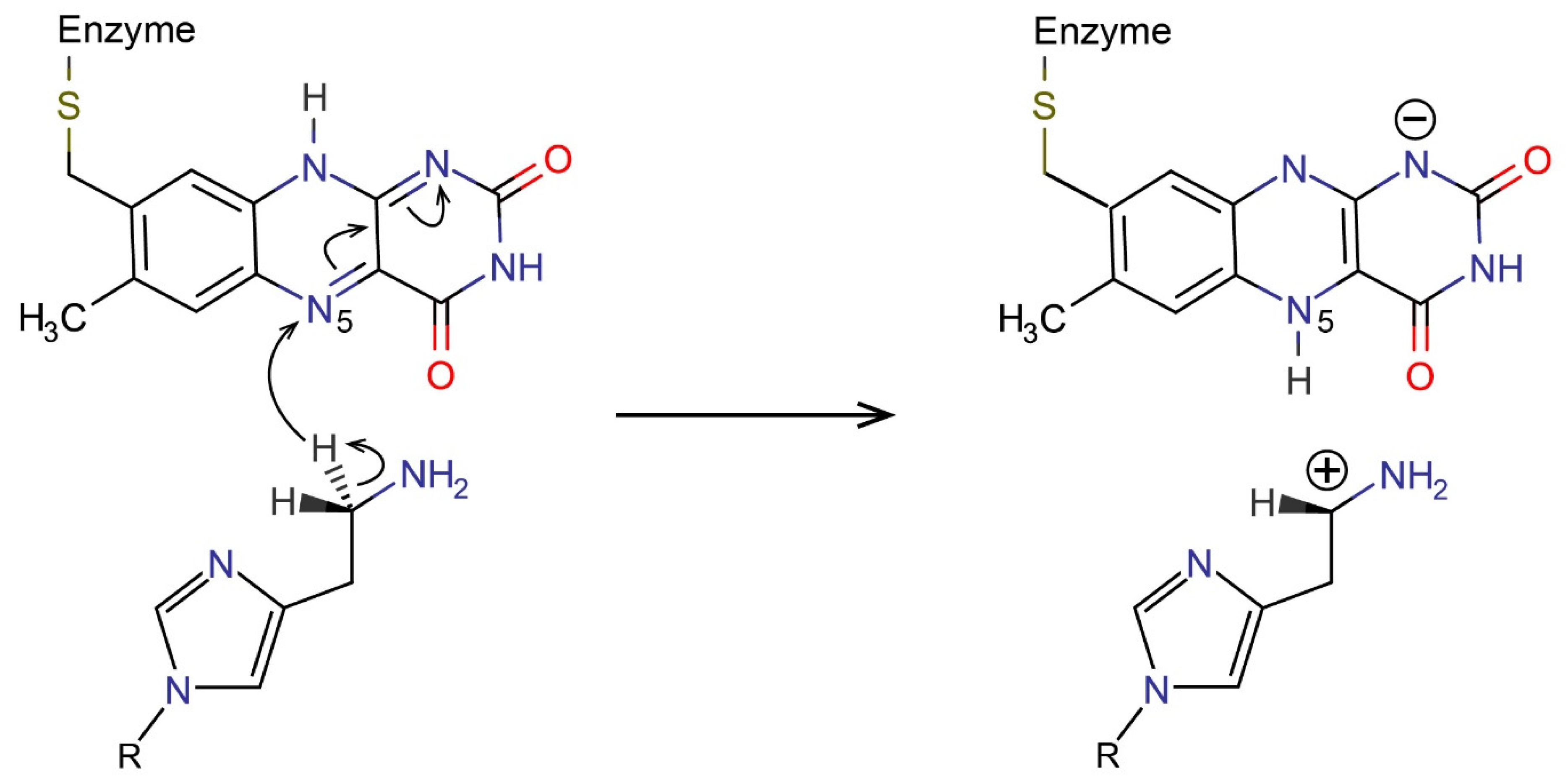
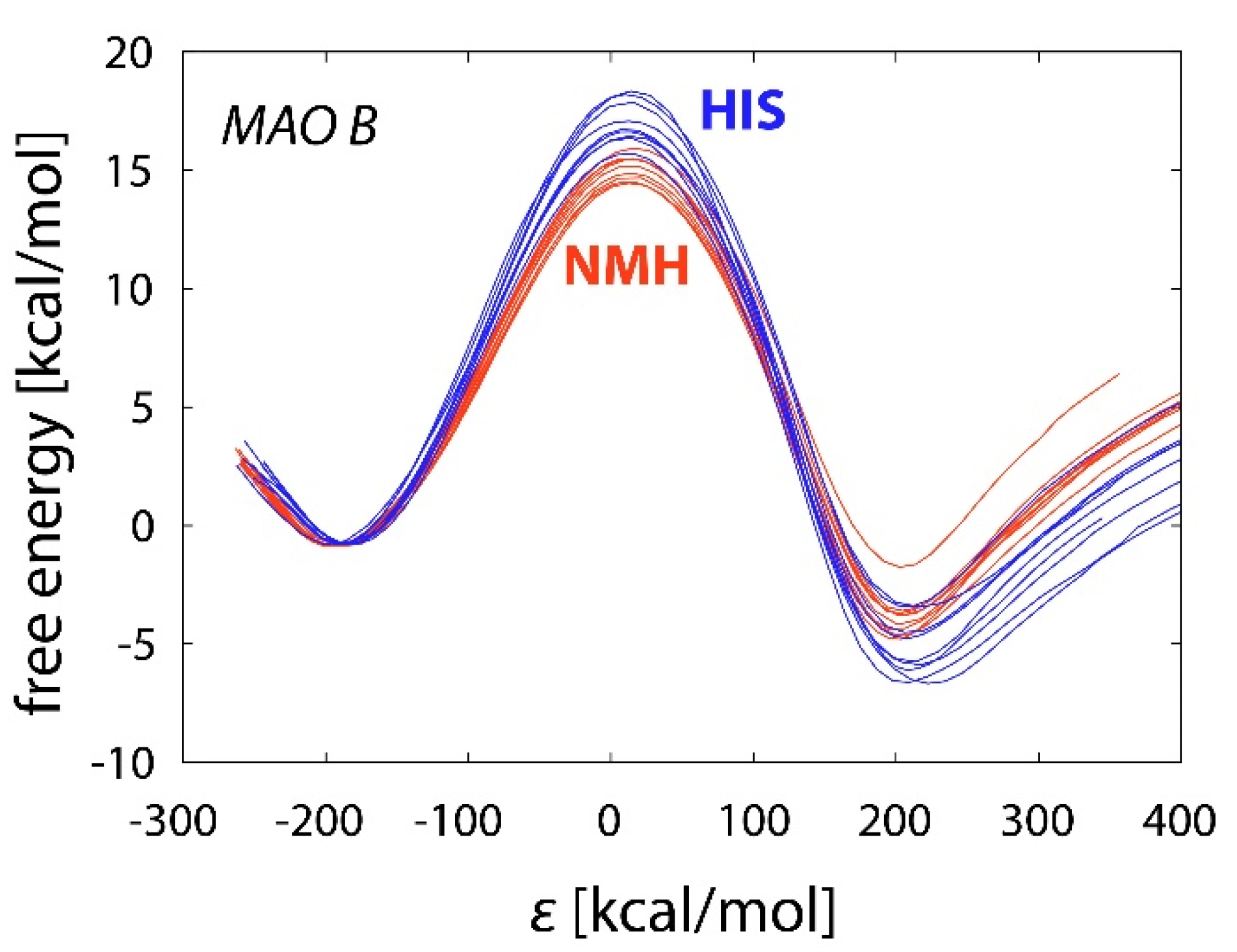
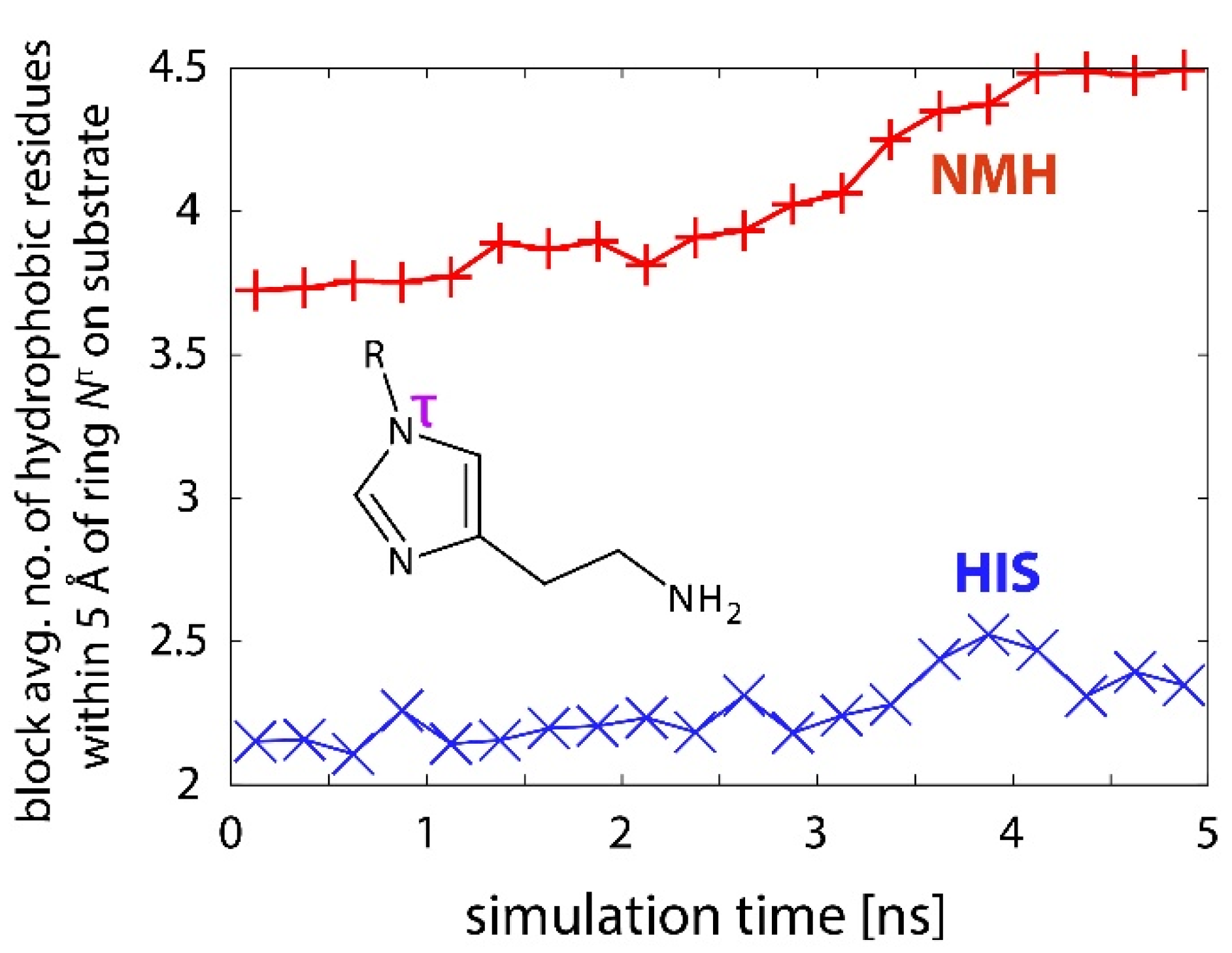
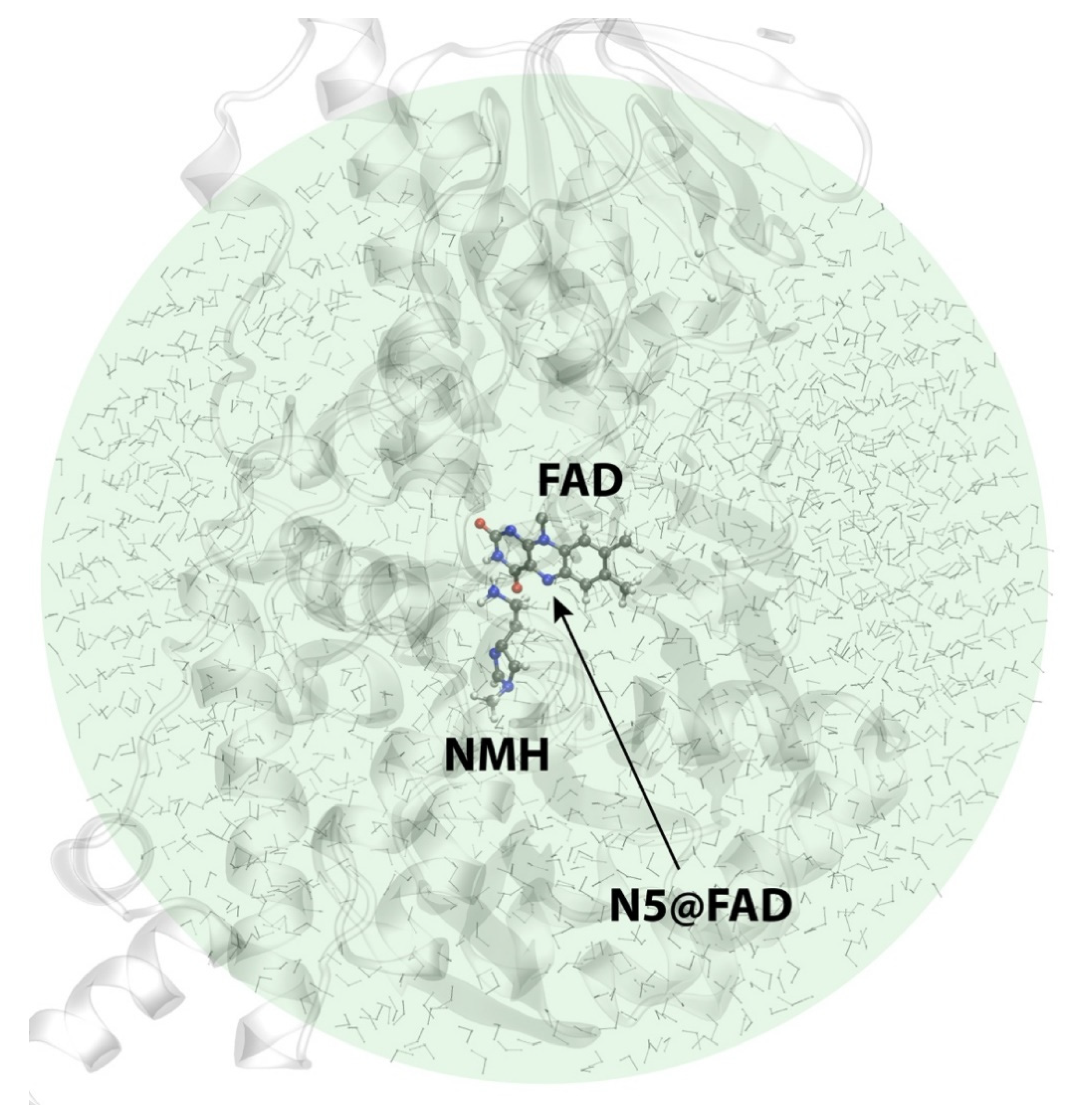
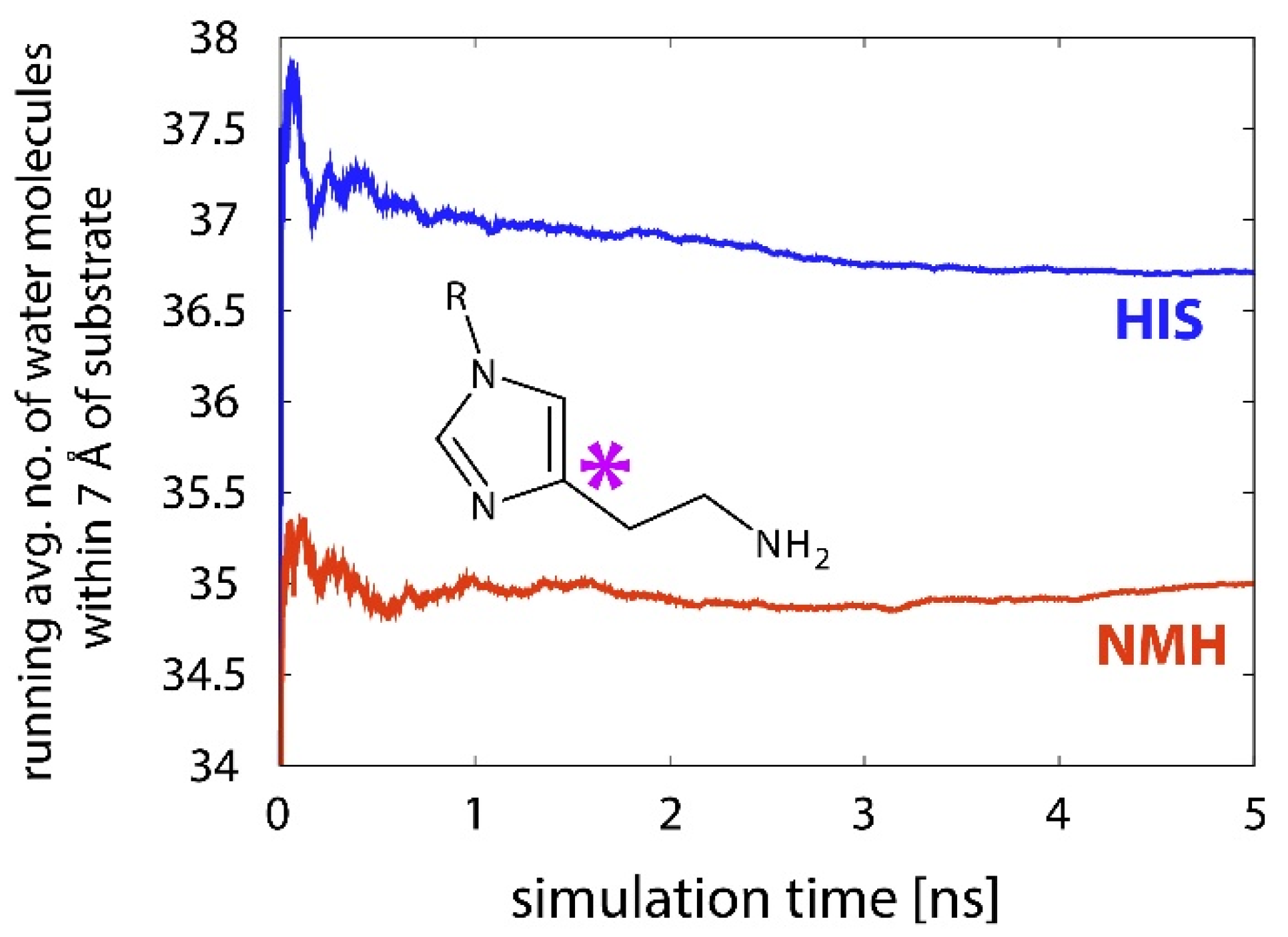
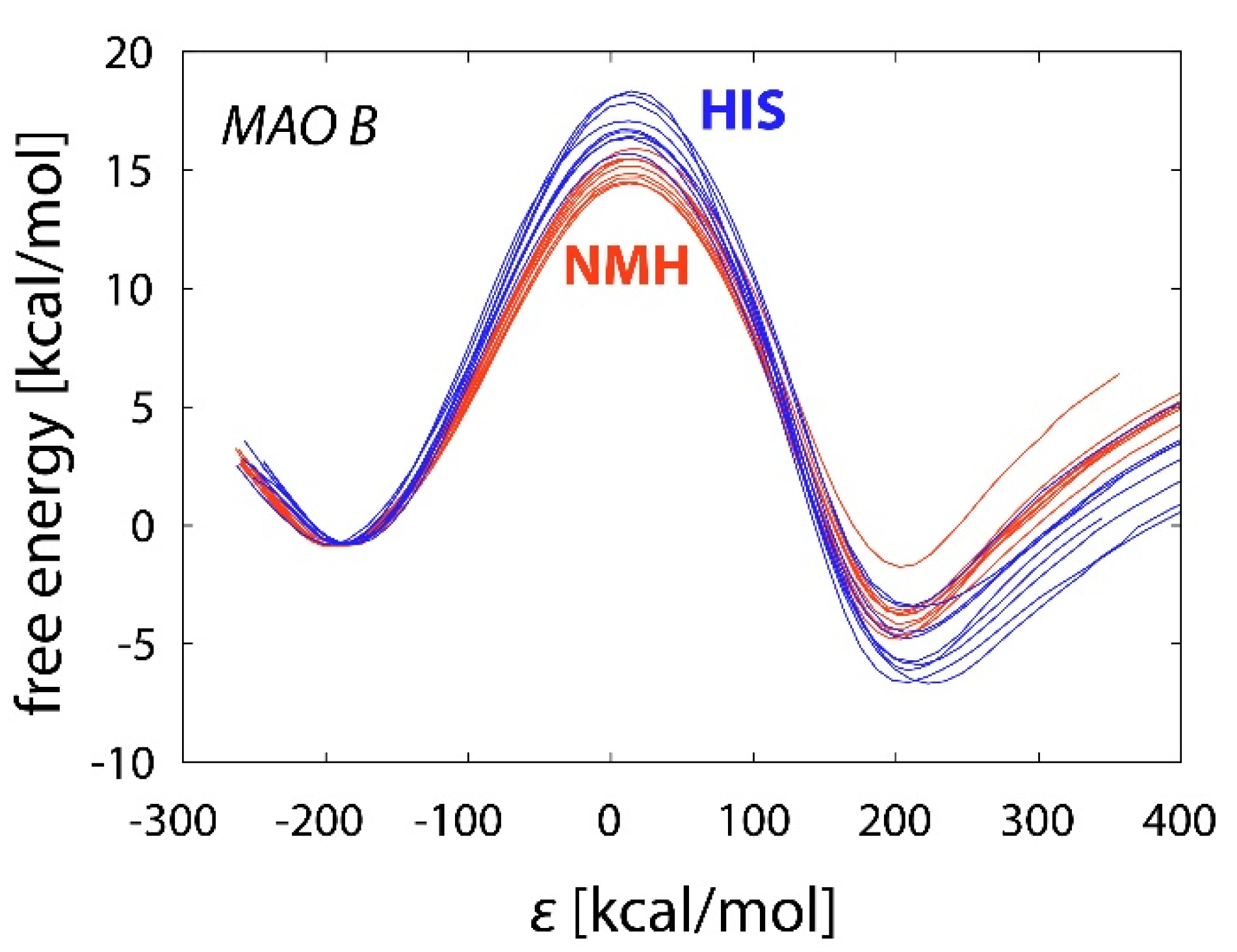
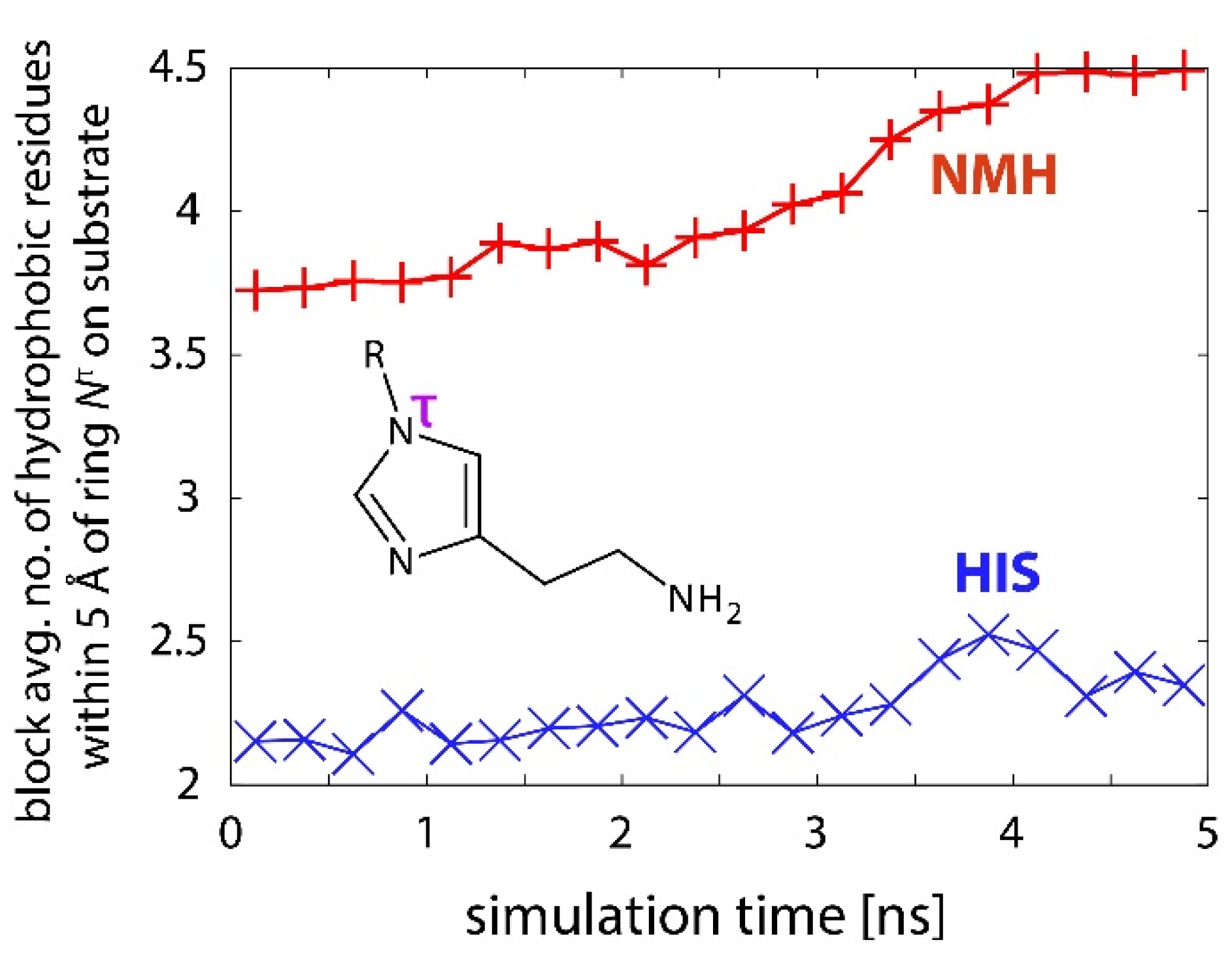
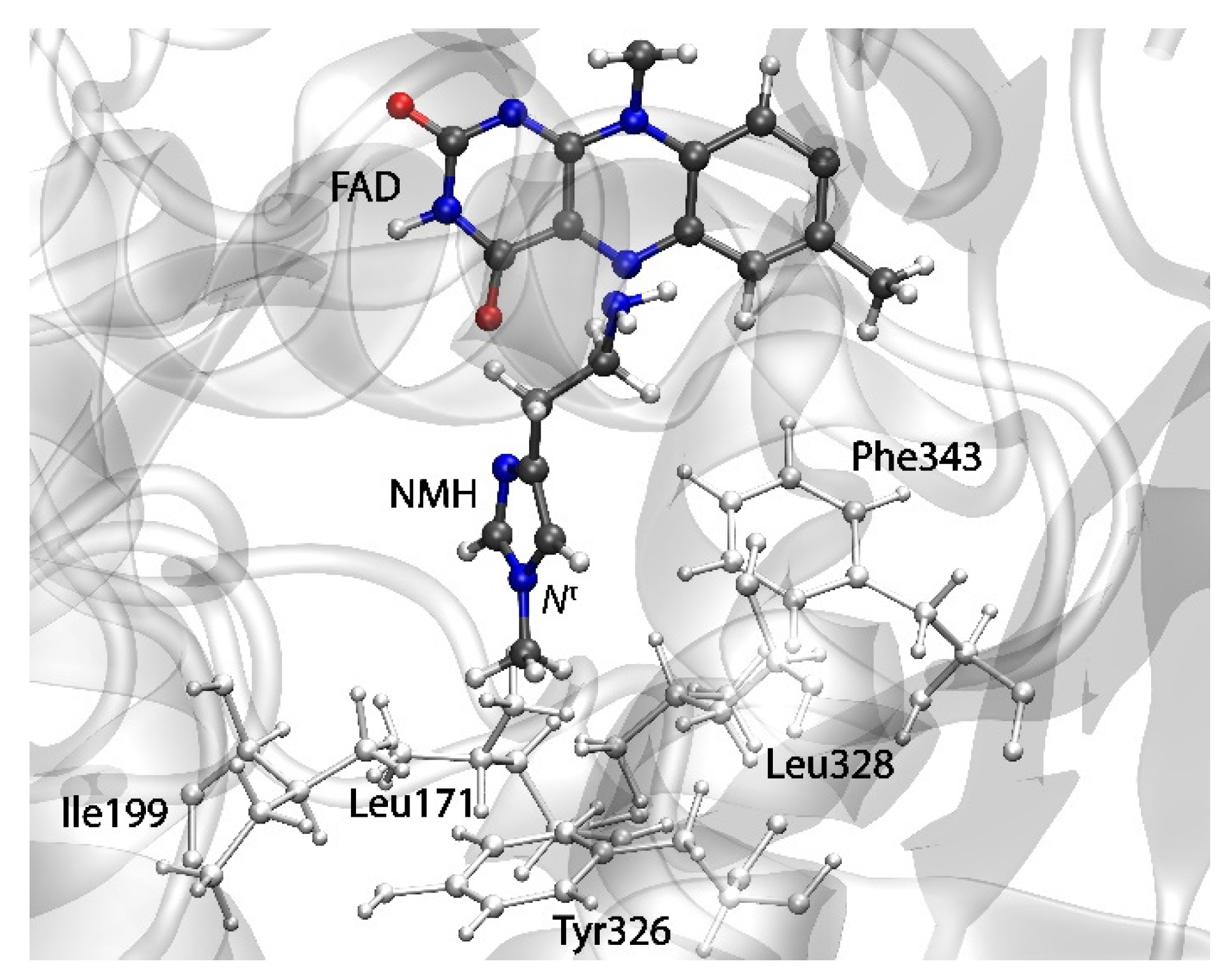
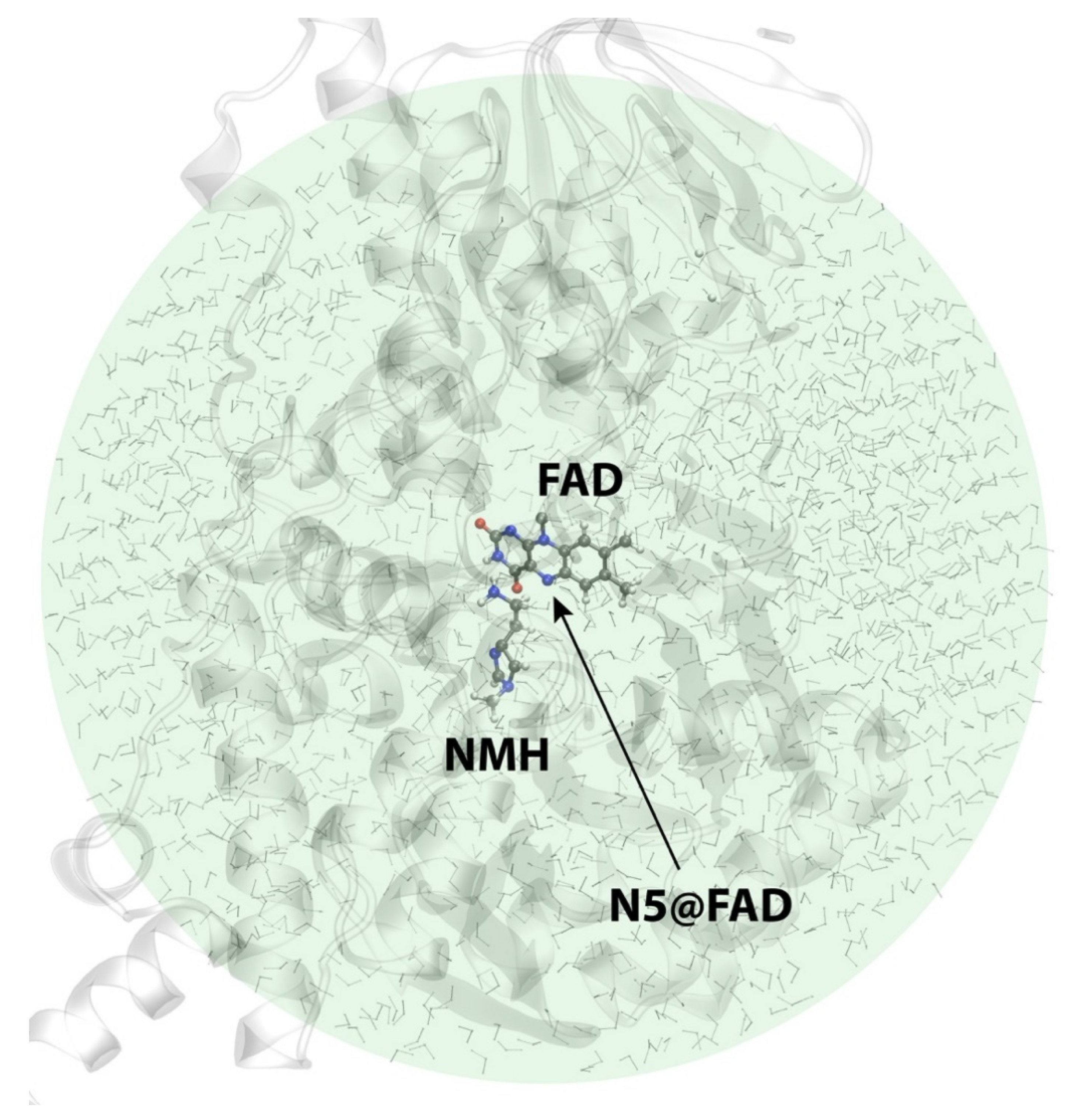
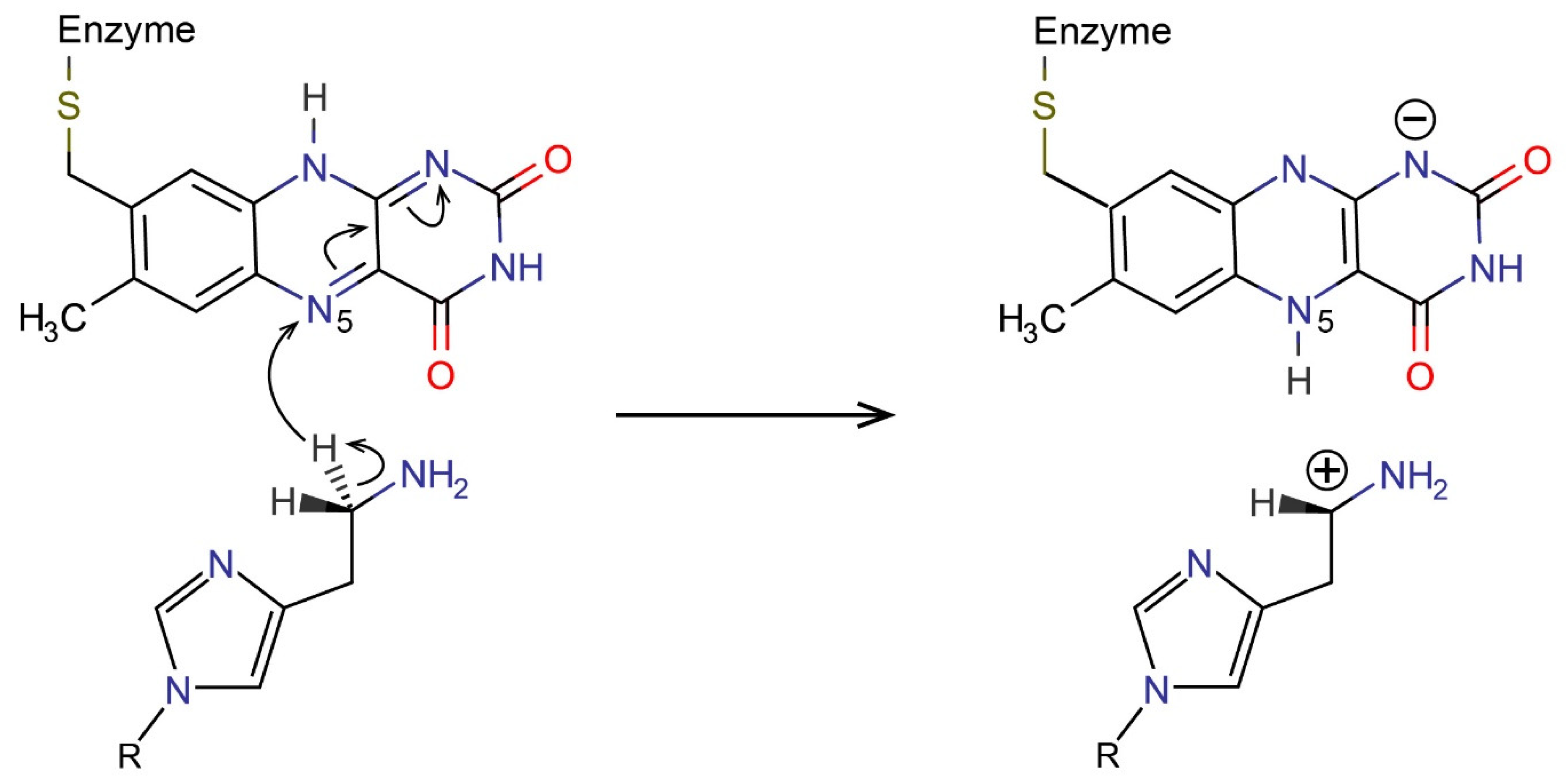
2.3. Reactions in the Enzyme
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Parsons, M.E.; Ganellin, C.R. Histamine and its receptors. Brit. J. Pharmacol. 2006, 147, S127–S135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, L.; Liu, J.Y.; Chen, Z. The Histaminergic System in Neuropsychiatric Disorders. Biomolecules 2021, 11, 1345. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.; Miszta, P.; Filipek, S. Molecular Modeling of Histamine Receptors-Recent Advances in Drug Discovery. Molecules 2021, 26, 1776. [Google Scholar] [CrossRef] [PubMed]
- Carthy, E.; Ellender, T. Histamine, Neuroinflammation and Neurodevelopment: A Review. Front. Neurosci. 2021, 15, 870. [Google Scholar] [CrossRef] [PubMed]
- Shulpekova, Y.O.; Nechaev, V.M.; Popova, I.R.; Deeva, T.A.; Kopylov, A.T.; Malsagova, K.A.; Kaysheva, A.L.; Ivashkin, V.T. Food Intolerance: The Role of Histamine. Nutrients 2021, 13, 3207. [Google Scholar] [CrossRef]
- Bieganski, T.; Kusche, J.; Feussner, K.D.; Hesterberg, R.; Richter, H.; Lorenz, W. The Importance of Human Intestinal Diamine Oxidase in the Oxidation of Histamine and-or Putrescine. Arch. Immunol. Et Ther. Exp. 1980, 28, 901–906. [Google Scholar]
- Bieganski, T.; Kusche, J.; Feussner, K.D.; Hesterberg, R.; Richter, H.; Lorenz, W. Human Intestinal Diamine Oxidase–Substrate-Specificity and Comparative Inhibitor Study. Agents Actions 1980, 10, 108–110. [Google Scholar] [CrossRef]
- Schwelberger, H.G.; Ahrens, F.; Fogel, W.A.; Sánchez-Jiménez, F. Chapter 3 Histamine Metabolism. In Histamine H4 Receptor; Holger, S., Ed.; De Gruyter Open Poland: Warsaw, Poland, 2013; pp. 63–102. [Google Scholar] [CrossRef] [Green Version]
- Schayer, R.W.; Karjala, S.A. Ring N methylation; a major route of histamine metabolism. J. Biol. Chem. 1956, 221, 307–313. [Google Scholar] [CrossRef]
- Elsworth, J.D.; Glover, V.; Sandler, M. Tele-Methylhistamine Is a Specific Mao B Substrate in Man. Psychopharmacology 1980, 69, 287–290. [Google Scholar] [CrossRef]
- Chajkowski-Scarry, S.; Rimoldi, J.M. Monoamine oxidase A and B substrates: Probing the pathway for drug development. Future Med. Chem. 2014, 6, 697–717. [Google Scholar] [CrossRef]
- Ramsay, R.R.; Albreht, A. Questions in the Chemical Enzymology of MAO. Chemistry 2021, 3, 69. [Google Scholar] [CrossRef]
- Edmondson, D.E.; Binda, C.; Wang, J.; Upadhyay, A.K.; Mattevi, A. Molecular and Mechanistic Properties of the Membrane-Bound Mitochondrial Monoamine Oxidases. Biochemistry 2009, 48, 4220–4230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hough, L.B. Dynamics of Histamine in the Brain. In Basic Neurochemistry: Molecular, Cellular and Medical Aspects, 6th ed.; Siegel, G.J., Agranoff, B.W., Albers, R.W., Fisher, S.K., Uhler, M.D., Eds.; Lippincott-Raven: Philadelphia, Pennsylvania, 1999. [Google Scholar]
- Schwartz, J.C.; Arrang, J.M.; Garbarg, M.; Pollard, H.; Ruat, M. Histaminergic Transmission in the Mammalian Brain. Physiol. Rev. 1991, 71, 1–51. [Google Scholar] [CrossRef] [PubMed]
- Thomas, B.; Prell, G.D. Imidazoleacetic Acid, a Gamma-Aminobutyric-Acid Receptor Agonist, Can Be Formed in Rat-Brain by Oxidation of Histamine. J. Neurochem. 1995, 65, 818–826. [Google Scholar] [CrossRef] [PubMed]
- Elmore, B.O.; Bollinger, J.A.; Dooley, D.M. Human kidney diamine oxidase: Heterologous expression, purification, and characterization. J. Biol. Inorg. Chem. 2002, 7, 565–579. [Google Scholar] [CrossRef]
- De Deurwaerdere, P.; Ramsay, R.R.; Di Giovanni, G. Neurobiology and neuropharmacology of monoaminergic systems. Prog. Neurobiol. 2017, 151, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Marsavelski, A.; Vianello, R. What a Difference a Methyl Group Makes: The Selectivity of Monoamine Oxidase B Towards Histamine and N-Methylhistamine. Chem. Eur. J. 2017, 23, 2915–2925. [Google Scholar] [CrossRef]
- Warshel, A.; Weiss, R.M. An Empirical Valence Bond Approach for Comparing Reactions in Solutions and in Enzymes. J. Am. Chem. Soc. 1980, 102, 6218–6226. [Google Scholar] [CrossRef]
- Aqvist, J.; Warshel, A. Simulation of Enzyme-Reactions Using Valence-Bond Force-Fields and Other Hybrid Quantum-Classical Approaches. Chem. Rev. 1993, 93, 2523–2544. [Google Scholar] [CrossRef]
- Kamerlin, S.C.L.; Warshel, A. The empirical valence bond model: Theory and applications. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 30–45. [Google Scholar] [CrossRef]
- Pisliakov, A.V.; Cao, J.; Kamerlin, S.C.L.; Warshel, A. Enzyme millisecond conformational dynamics do not catalyze the chemical step. Proc. Natl. Acad. Sci. USA 2009, 106, 17359–17364. [Google Scholar] [CrossRef] [Green Version]
- Olsson, M.H.M.; Mavri, J.; Warshel, A. Transition state theory can be used in studies of enzyme catalysis: Lessons from simulations of tunnelling and dynamical effects in lipoxygenase and other systems. Philos. Trans. R. Soc. B 2006, 361, 1417–1432. [Google Scholar] [CrossRef] [Green Version]
- Warshel, A.; Sharma, P.K.; Kato, M.; Xiang, Y.; Liu, H.B.; Olsson, M.H.M. Electrostatic basis for enzyme catalysis. Chem. Rev. 2006, 106, 3210–3235. [Google Scholar] [CrossRef]
- Adamczyk, A.J.; Cao, J.; Kamerlin, S.C.L.; Warshel, A. Catalysis by dihydrofolate reductase and other enzymes arises from electrostatic preorganization, not conformational motions. Proc. Natl. Acad. Sci. USA 2011, 108, 14115–14120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feierberg, I.; Luzhkov, V.; Aqvist, J. Computer simulation of primary kinetic isotope effects in the proposed rate-limiting step of the glyoxalase I catalyzed reaction. J. Biol. Chem. 2000, 275, 22657–22662. [Google Scholar] [CrossRef] [Green Version]
- Purg, M.; Pabis, A.; Baier, F.; Tokuriki, N.; Jackson, C.; Kamerlin, S.C.L. Probing the mechanisms for the selectivity and promiscuity of methyl parathion hydrolase. Philos. Trans. R. Soc. A 2016, 374. [Google Scholar] [CrossRef]
- Serrano-Hervás, E.; Garcia-Borràs, M.; Osuna, S. Exploring the origins of selectivity in soluble epoxide hydrolase from Bacillus megaterium. Org. Biomol. Chem. 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florian, J.; Goodman, M.F.; Warshel, A. Computer simulation of the chemical catalysis of DNA polymerases: Discriminating between alternative nucleotide insertion mechanisms for T7 DNA polymerase. J. Am. Chem. Soc. 2003, 125, 8163–8177. [Google Scholar] [CrossRef] [PubMed]
- Warshel, A.; Bora, R.P. Perspective: Defining and quantifying the role of dynamics in enzyme catalysis. J. Chem. Phys. 2016, 144. [Google Scholar] [CrossRef] [PubMed]
- Morgenstern, A.; Jaszai, M.; Eberhart, M.E.; Alexandrova, A.N. Quantified electrostatic preorganization in enzymes using the geometry of the electron charge density. Chem. Sci. 2017, 8, 5010–5018. [Google Scholar] [CrossRef] [Green Version]
- Hennefarth, M.R.; Alexandrova, A.N. Direct Look at the Electric Field in Ketosteroid Isomerase and Its Variants. Acs. Catal. 2020, 10, 9915–9924. [Google Scholar] [CrossRef]
- Fried, S.D.; Bagchi, S.; Boxer, S.G. Extreme electric fields power catalysis in the active site of ketosteroid isomerase. Science 2014, 346, 1510–1514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prah, A.; Franciskovic, E.; Mavri, J.; Stare, J. Electrostatics as the Driving Force Behind the Catalytic Function of the Monoamine Oxidase A Enzyme Confirmed by Quantum Computations. Acs Catal. 2019, 9, 1231–1240. [Google Scholar] [CrossRef]
- Repič, M.; Vianello, R.; Purg, M.; Duarte, F.; Bauer, P.; Kamerlin, S.C.L.; Mavri, J. Empirical valence bond simulations of the hydride transfer step in the monoamine oxidase B catalyzed metabolism of dopamine. Proteins 2014, 82, 3347–3355. [Google Scholar] [CrossRef] [Green Version]
- Vianello, R.; Repic, M.; Mavri, J. How are Biogenic Amines Metabolized by Monoamine Oxidases? Eur. J. Org. Chem. 2012, 36, 7057–7065. [Google Scholar] [CrossRef]
- Oanca, G.; Purg, M.; Mavri, J.; Shih, J.C.; Stare, J. Insights into enzyme point mutation effect by molecular simulation: Phenylethylamine oxidation catalyzed by monoamine oxidase A. Phys. Chem. Chem. Phys. 2016, 18, 13346–13356. [Google Scholar] [CrossRef] [Green Version]
- Geha, R.M.; Rebrin, I.; Chen, K.; Shih, J.C. Substrate and inhibitor specificities for human monoamine oxidase A and B are influenced by a single amino acid. J. Biol. Chem. 2001, 276, 9877–9882. [Google Scholar] [CrossRef] [Green Version]
- Oanca, G.; Stare, J.; Vianello, R.; Mavri, J. Multiscale simulation of monoamine oxidase catalyzed decomposition of phenylethylamine analogs. Eur. J. Pharmacol. 2017, 817, 46–50. [Google Scholar] [CrossRef]
- Poberznik, M.; Purg, M.; Repic, M.; Mavri, J.; Vianello, R. Empirical valence bond simulations of the hydride-transfer step in the monoamine oxidase A catalyzed metabolism of noradrenaline. J. Phys. Chem. B 2016, 120, 11419–11427. [Google Scholar] [CrossRef]
- Oanca, G.; Stare, J.; Mavri, J. How Fast Monoamine Oxidases Decompose Adrenaline? Kinetics of Isoenzymes A and B Evaluated by Empirical Valence Bond Simulation. Proteins 2017, 85, 2170–2178. [Google Scholar] [CrossRef]
- Prah, A.; Purg, M.; Stare, J.; Vianello, R.; Mavri, J. How Monoamine Oxidase A Decomposes Serotonin: An Empirical Valence Bond Simulation of the Reactive Step. J. Phys. Chem. B 2020, 124, 8259–8265. [Google Scholar] [CrossRef]
- Marsavelski, A.; Petrovic, D.; Bauer, P.; Vianello, R.; Kamerlin, S.C.L. Empirical Valence Bond Simulations Suggest a Direct Hydride Transfer Mechanism for Human Diamine Oxidase. ACS Omega 2018, 3, 3665–3674. [Google Scholar] [CrossRef]
- Abad, E.; Zenn, R.K.; Kastner, J. Reaction Mechanism of Monoamine Oxidase from QM/MM Calculations. J. Phys. Chem. B 2013, 117, 14238–14246. [Google Scholar] [CrossRef] [PubMed]
- Zapata-Torres, G.; Fierro, A.; Barriga-Gonzalez, G.; Salgado, J.C.; Celis-Barros, C. Revealing Monoamine Oxidase B Catalytic Mechanisms by Means of the Quantum Chemical Cluster Approach. J. Chem. Inf. Model. 2015, 55, 1349–1360. [Google Scholar] [CrossRef] [Green Version]
- Fierro, A.; Edmondson, D.E.; Celis-Barros, C.; Rebolledo-Fuentes, M.; Zapata-Torres, G. Why p-OMe- and p-Cl-beta-Methylphenethylamines Display Distinct Activities uponMAO-B Binding. PLoS ONE 2016, 11, 0154989. [Google Scholar] [CrossRef] [Green Version]
- Tandaric, T.; Vianello, R. Computational Insight into the Mechanism of the Irreversible Inhibition of Monoamine Oxidase Enzymes by the Antiparkinsonian Propargylamine Inhibitors Rasagiline and Selegiline. Acs Chem. Neurosci. 2019, 10, 3532–3542. [Google Scholar] [CrossRef]
- Tandaric, T.; Prah, A.; Stare, J.; Mavri, J.; Vianello, R. Hydride Abstraction as the Rate-Limiting Step of the Irreversible Inhibition of Monoamine Oxidase B by Rasagiline and Selegiline: A Computational Empirical Valence Bond Study. Int. J. Mol. Sci. 2020, 21, 6151. [Google Scholar] [CrossRef]
- Youdim, M.B.H.; Edmondson, D.; Tipton, K.F. The therapeutic potential of monoamine oxidase inhibitors. Nat. Rev. Neurosci. 2006, 7, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, R.R. Inhibitor Design for Monoamine Oxidases. Curr. Pharm. Des. 2013, 19, 2529–2539. [Google Scholar] [CrossRef] [PubMed]
- Prah, A.; Mavri, J.; Stare, J. An electrostatic duel: Subtle differences in catalytic performance of monoamine oxidase A and B isoenzymes elucidated at a residue level by quantum computations. Phys. Chem. Chem. Phys. 2021. [Google Scholar] [CrossRef]
- Dale, M.M.; Rang, H.P.; Dale, M.M. Rang & Dale’s Pharmacology; Churchill Livingstone: Edinburgh, Scotland, 2007. [Google Scholar]
- Hong, G.Y.; Rosta, E.; Warshel, A. Using the constrained DFT approach in generating diabatic surfaces and off diagonal empirical valence bond terms for modeling reactions in condensed phases. J. Phys. Chem. B 2006, 110, 19570–19574. [Google Scholar] [CrossRef]
- Muller, P. Glossary of Terms Used in Physical Organic-Chemistry. Pure Appl. Chem. 1994, 66, 1077–1184. [Google Scholar] [CrossRef] [Green Version]
- Repič, M.; Purg, M.; Vianello, R.; Mavri, J. Examining electrostatic preorganization in monoamine oxidases A and B by structural comparison and pKa calculations. J. Phys. Chem. B 2014, 118, 4326–4332. [Google Scholar] [CrossRef]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Felix, E.; Magarinos, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef] [PubMed]
- Perrin, D.D.; Dempsey, B.; Serjeant, E.P. pKa Prediction for Organic Acids and Bases; Chapman and Hall: London, NY, USA, 1981; p. 146. [Google Scholar]
- Borštnar, R.; Repič, M.; Kamerlin, S.C.L.; Vianello, R.; Mavri, J. Computational study of the pKa values of potential catalytic residues in the active site of monoamine oxidase B. J. Chem. Theory Comput. 2012, 8, 3864–3870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sham, Y.Y.; Chu, Z.T.; Warshel, A. Consistent calculations of pKa’s of ionizable residues in proteins: Semi-microscopic and microscopic approaches. J. Phys. Chem. B 1997, 101, 4458–4472. [Google Scholar] [CrossRef]
- Simonson, T.; Carlsson, J.; Case, D.A. Proton binding to proteins: pKa calculations with explicit and implicit solvent models. J. Am. Chem. Soc. 2004, 126, 4167–4180. [Google Scholar] [CrossRef] [PubMed]
- Stare, J. Complete sampling of an enzyme reaction pathway: A lesson from gas phase simulations. RSC Adv. 2017, 2017, 8740–8754. [Google Scholar] [CrossRef] [Green Version]
- Dapprich, S.; Komaromi, I.; Byun, K.S.; Morokuma, K.; Frisch, M.J. A new ONIOM implementation in Gaussian98. Part I. The calculation of energies, gradients, vibrational frequencies and electric field derivatives. J. Mol. Struc. Theochem. 1999, 461, 1–21. [Google Scholar] [CrossRef]
- Atalay, V.E.; Erdem, S.S. A comparative computational investigation on the proton and hydride transfer mechanisms of monoamine oxidase using model molecules. Comput. Biol. Chem. 2013, 47, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Akyuz, M.A.; Erdem, S.S. Computational modeling of the direct hydride transfer mechanism for the MAO catalyzed oxidation of phenethylamine and benzylamine: ONIOM (QM/QM) calculations. J. Neural. Transm. 2013, 120, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Marelius, J.; Kolmodin, K.; Feierberg, I.; Aqvist, J. Q: A molecular dynamics program for free energy calculations and empirical valence bond simulations in biomolecular systems. J. Mol. Graph. Model. 1998, 16, 213. [Google Scholar] [CrossRef]
- Bonivento, D.; Milczek, E.M.; McDonald, G.R.; Binda, C.; Holt, A.; Edmondson, D.E.; Mattevi, A. Potentiation of Ligand Binding through Cooperative Effects in Monoamine Oxidase B. J. Biol. Chem. 2010, 285, 36849–36856. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Maxwell, D.S.; TiradoRives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Tirado-Rives, J. The OPLS Potential Functions for Proteins–Energy Minimizations for Crystals of Cyclic-Peptides and Crambin. J. Am. Chem. Soc. 1988, 110, 1657–1666. [Google Scholar] [CrossRef]
- Wang, J.M.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Berryman, J.T.; Betz, R.M.; Cerutti, D.S.; T.E. Cheatham, I.; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; et al. AMBER 2015; University of California: San Francisco, CA, USA.
- Jug, U.; Pregeljc, D.; Mavri, J.; Vianello, R.; Stare, J. Elementary S(N)2 reaction revisited. Effects of solvent and alkyl chain length on kinetics of halogen exchange in haloalkanes elucidated by Empirical Valence Bond simulation. Comput. Theor. Chem. 2017, 1116, 96–101. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Jones, T.Z.E.; Balsa, D.; Unzeta, M.; Ramsay, R.R. Variations in activity and inhibition with pH: The protonated amine is the substrate for monoamine oxidase, but uncharged inhibitors bind better. J. Neural. Transm. 2007, 114, 707–712. [Google Scholar] [CrossRef]
- Dunn, R.V.; Marshall, K.R.; Munro, A.W.; Scrutton, N.S. The pH dependence of kinetic isotope effects in monoamine oxidase A indicates stabilization of the neutral amine in the enzyme-substrate complex. Febs. J. 2008, 275, 3850–3858. [Google Scholar] [CrossRef]
- Tan, A.K.; Ramsay, R.R. Substrate-Specific Enhancement of the Oxidative Half-Reaction of Monoamine-Oxidase. Biochemistry 1993, 32, 2137–2143. [Google Scholar] [CrossRef] [PubMed]
- Roberts, E.; Simonsen, D.G. A hypnotic and possible analgesic effect of imidazoleacetic acid in mice. Biochem. Pharmacol. 1966, 15, 1875–1877. [Google Scholar] [CrossRef]
- Marcus, R.J.; Winters, W.D.; Roberts, E.; Simonsen, D.G. Neuropharmacological studies of imidazole-4-acetic acid actions in the mouse and rat. Neuropharmacology 1971, 10, 203–215. [Google Scholar] [CrossRef]
- Tunnicliff, G. Pharmacology and Function of Imidazole 4-Acetic Acid in Brain. Gen. Pharmacol. Vasc. Syst. 1998, 31, 503–509. [Google Scholar] [CrossRef]
- Prell, G.D.; Martinelli, G.P.; Holstein, G.R.; Matulić-Adamić, J.; Watanabe, K.A.; Chan, S.L.F.; Morgan, N.G.; Haxhiu, M.A.; Ernsberger, P. Imidazoleacetic acid-ribotide: An endogenous ligand that stimulates imidazol(in)e receptors. Proc. Natl. Acad. Sci. USA 2004, 101, 13677. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate | DFT Calculations | Calibrated EVB Parameters | ||
|---|---|---|---|---|
| ΔG‡ [kcal/mol] | ΔGR [kcal/mol] | H12 [kcal/mol] | α20 [kcal/mol] | |
| HIS | 33.52 | 25.84 | 72.81 | 88.08 |
| NMH | 32.46 | 26.30 | 74.94 | 98.50 |
| Substrate | Gas Phase | Water | MAO-B | Exp. |
|---|---|---|---|---|
| HIS | 33.52 (0.06) | 24.12 (0.08) | 21.04 (0.29) | 19.26 |
| NMH | 32.46 (0.17) | 24.75 (0.05) | 18.95 (0.16) | 17.89 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maršavelski, A.; Mavri, J.; Vianello, R.; Stare, J. Why Monoamine Oxidase B Preferably Metabolizes N-Methylhistamine over Histamine: Evidence from the Multiscale Simulation of the Rate-Limiting Step. Int. J. Mol. Sci. 2022, 23, 1910. https://doi.org/10.3390/ijms23031910
Maršavelski A, Mavri J, Vianello R, Stare J. Why Monoamine Oxidase B Preferably Metabolizes N-Methylhistamine over Histamine: Evidence from the Multiscale Simulation of the Rate-Limiting Step. International Journal of Molecular Sciences. 2022; 23(3):1910. https://doi.org/10.3390/ijms23031910
Chicago/Turabian StyleMaršavelski, Aleksandra, Janez Mavri, Robert Vianello, and Jernej Stare. 2022. "Why Monoamine Oxidase B Preferably Metabolizes N-Methylhistamine over Histamine: Evidence from the Multiscale Simulation of the Rate-Limiting Step" International Journal of Molecular Sciences 23, no. 3: 1910. https://doi.org/10.3390/ijms23031910
APA StyleMaršavelski, A., Mavri, J., Vianello, R., & Stare, J. (2022). Why Monoamine Oxidase B Preferably Metabolizes N-Methylhistamine over Histamine: Evidence from the Multiscale Simulation of the Rate-Limiting Step. International Journal of Molecular Sciences, 23(3), 1910. https://doi.org/10.3390/ijms23031910