
Fusarium graminearum Infection Strategy in Wheat Involves a Highly Conserved Genetic Program That Controls the Expression of a Core Effectome

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. In Planta Expression Signature of the F. graminearum Gene Set Coding Secreted Proteins
2.2. Characterization of Differential Expression of SP Gene Sets at the Early Stages of Infection
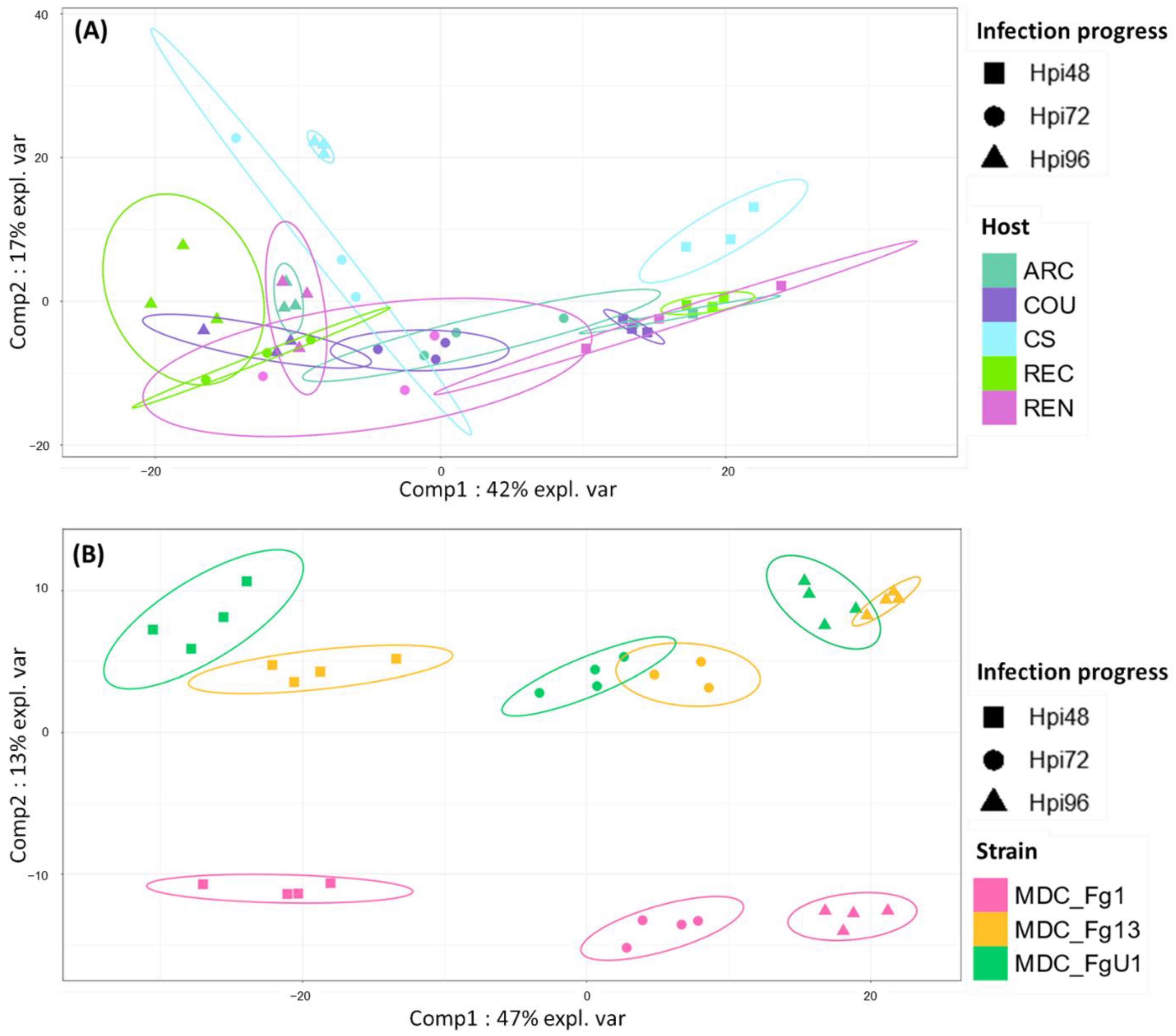
2.2.1. Fungal Gene Expression in Different Host Cultivars
2.2.2. Fungal Strain Specificities
2.3. Parsing Secretome Gene Sets towards Effectome Gene Sets That Are Regulated along the Infection Progress
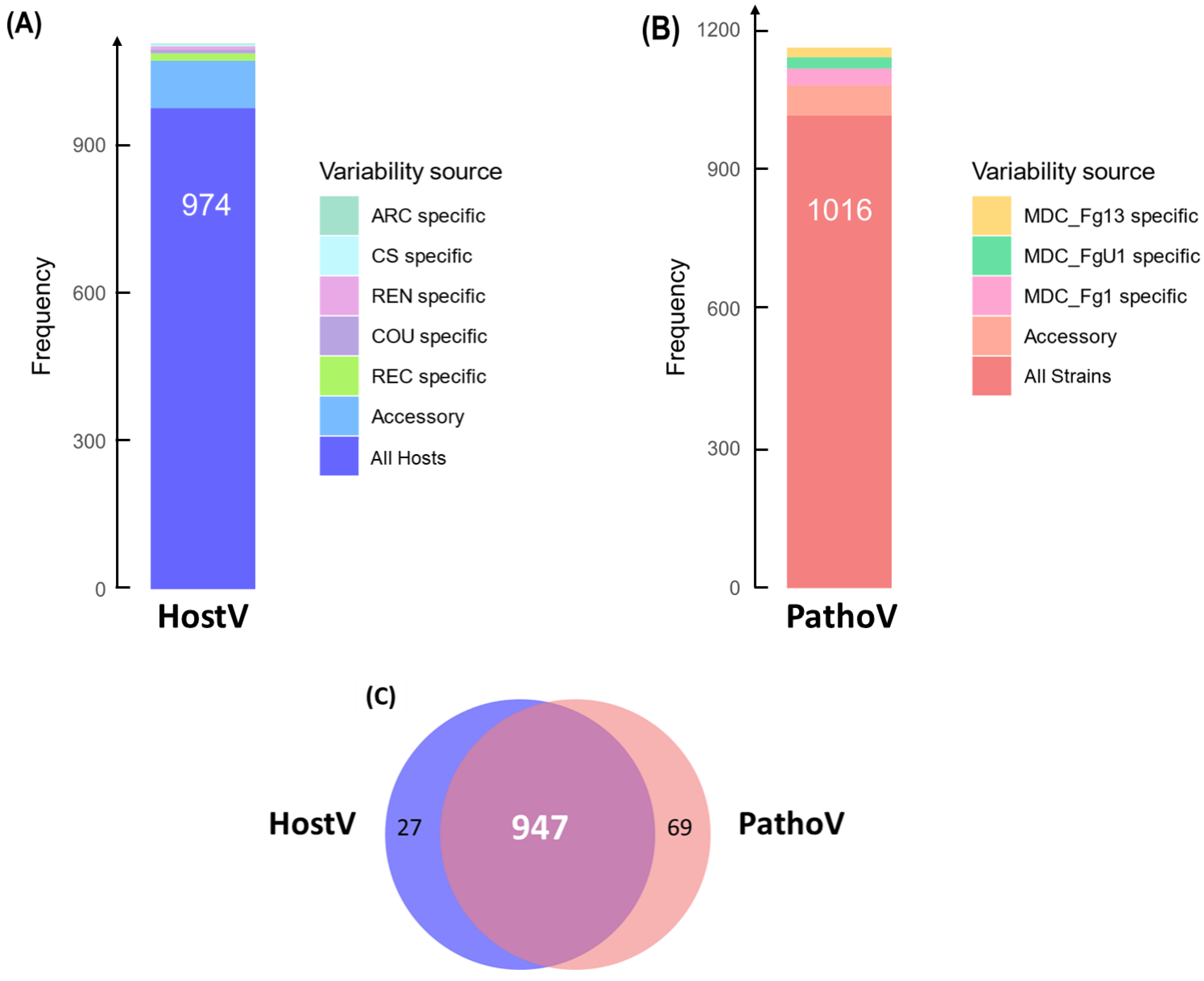
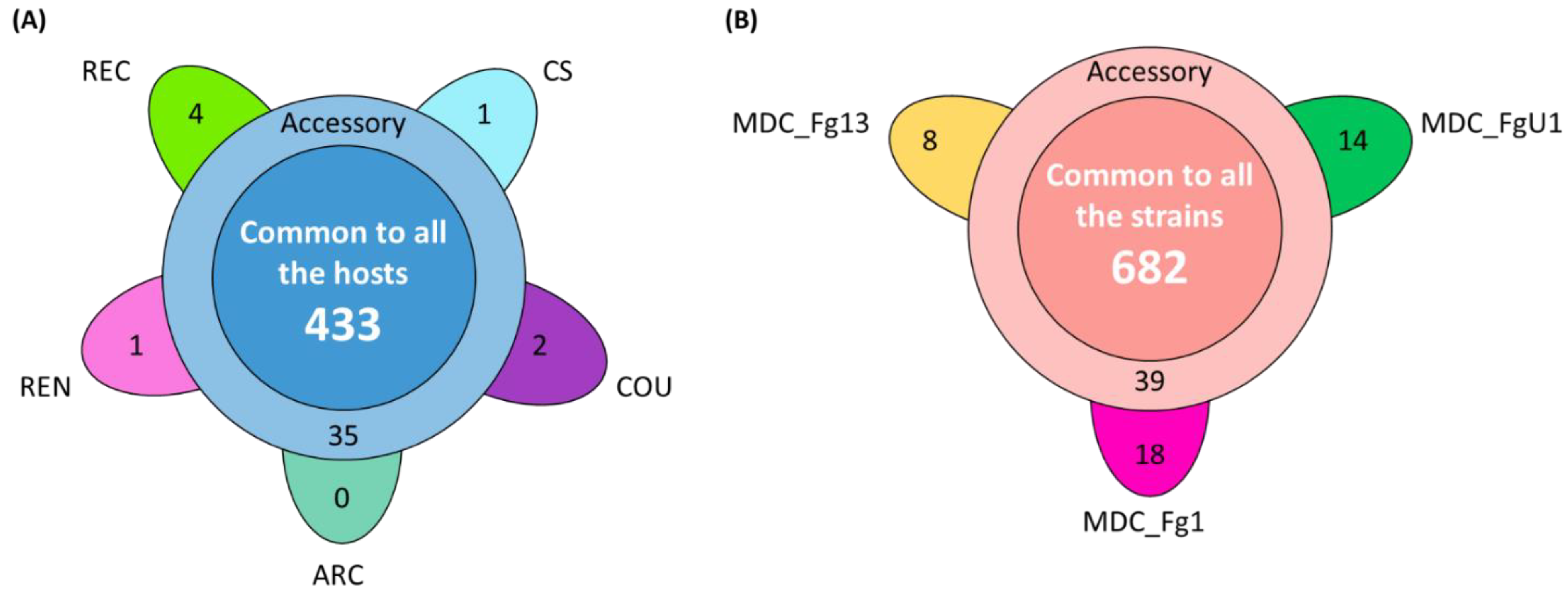
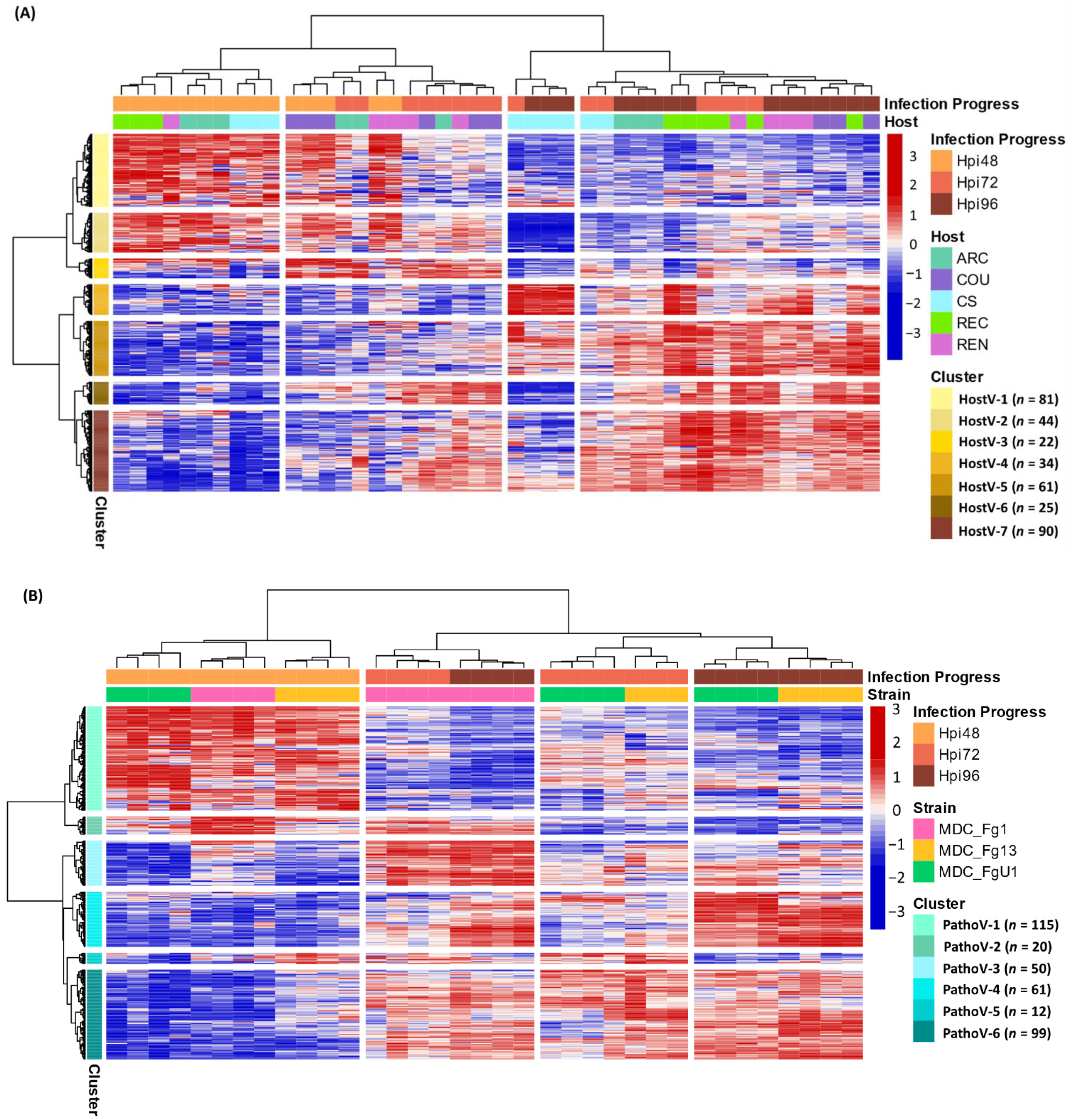
2.4. Different Wheat and F. graminearum Genetic Backgrounds Evidenced a Relevant Core Effectome Gene Set Expressed at Specific Infection Stages
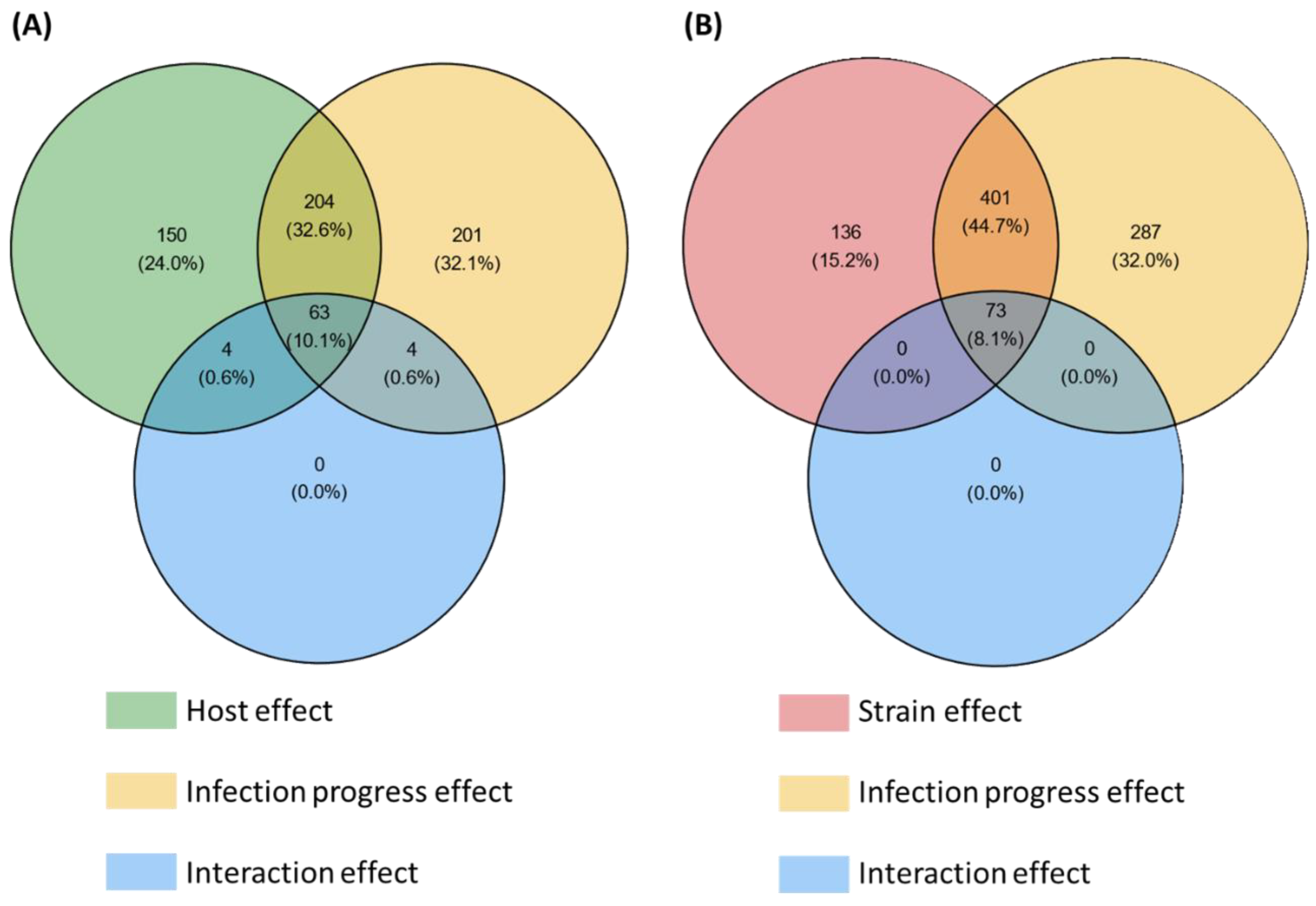
2.5. Host and Strain Driven Regulations of the Core Effectome Gene Set
2.5.1. Host Cultivar Effects
2.5.2. Fungal Strain Effects
2.6. Fusarium graminearum Putative Effectome Displayed Several Targets in Wheat Spikes at Different Infection Stages
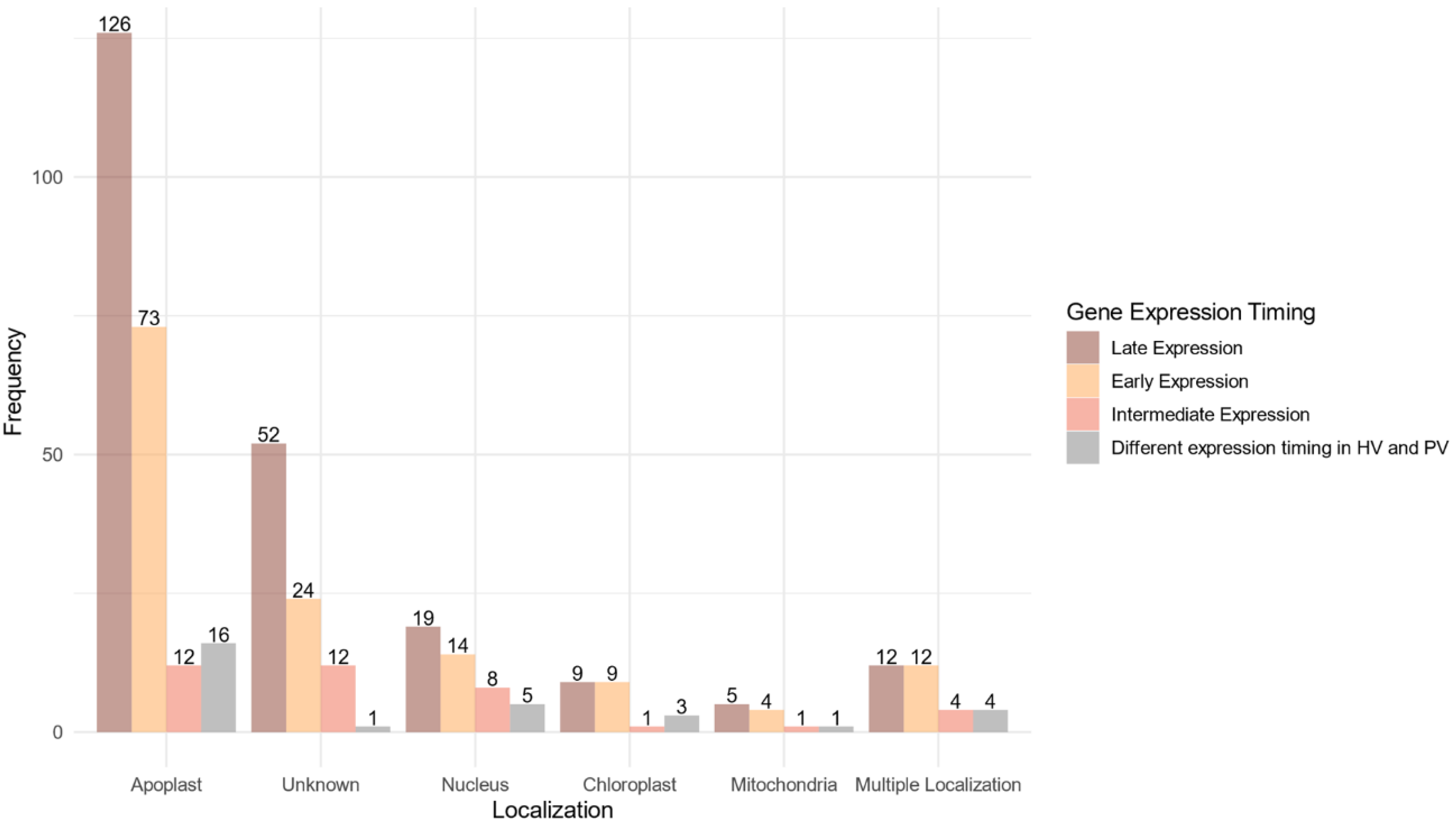
2.7. Identification of Additional Effector Features
2.7.1. In Silico Prediction
2.7.2. Localization in the Fast-Evolving Subgenome
2.8. Predicted Functions of the Core Effectome Are Highly Diverse
3. Discussion
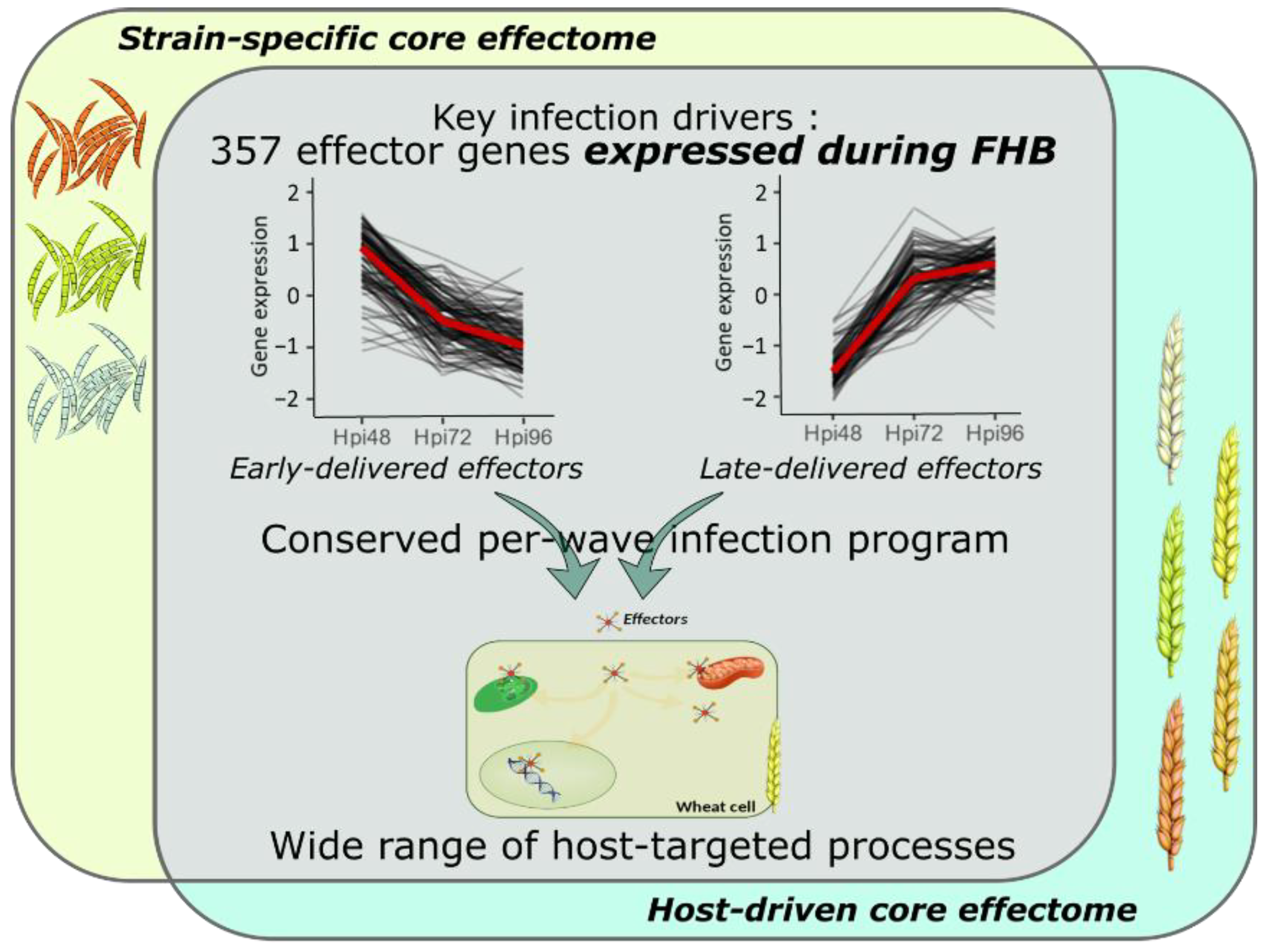
3.1. Fusarium graminearum Infection Involves a Highly Conserved Effectome
3.2. F. graminearum Core Effectors Are Delivered in a Conservative Per-Wave Expression
3.3. F. graminearum Infection Strategy Involves Integrative Host Cellular Processes
4. Materials and Methods
4.1. Experiments and Biological Material
4.1.1. Preparation of the Fusarium graminearum Inoculum
4.1.2. Plant Growth Conditions
4.1.3. Experimental Procedures
4.1.4. RNA Extraction and Sequencing
4.2. Fusarium graminearum Pangenome Construction and Characterization
4.3. RNA-Seq Bioinformatic Analysis
4.3.1. Data Cleaning Step
4.3.2. Mapping and Assignation
4.4. Statistical Analysis of F. graminearum Expression Data
4.4.1. Gene Filtering and Normalization
4.4.2. Differential Expression Analysis
4.4.3. Expression Pattern Characterization
4.4.4. Functional Enrichment Analysis
4.5. Genomic Localization of Effectome Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Parry, D.W.; Jenkinson, P.; McLeod, L. Fusarium Ear Blight (Scab) in Small Grain Cereals—A Review. Plant Pathol. 1995, 44, 207–238. [Google Scholar] [CrossRef]
- Goswami, R.S.; Kistler, H.C. Heading for Disaster: Fusarium Graminearum on Cereal Crops. Mol. Plant Pathol. 2004, 5, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Boyacioǧlu, D.; Hettiarachchy, N.S. Changes in Some Biochemical Components of Wheat Grain That Was Infected with Fusarium Graminearum. J. Cereal Sci. 1995, 21, 57–62. [Google Scholar] [CrossRef]
- Argyris, J.; Sanford, D.; TeKrony, D. Fusarium Graminearum Infection during Wheat Seed Development and Its Effect on Seed Quality. Crop Sci. 2003, 43, 1782–1788. [Google Scholar] [CrossRef]
- Chen, Y.; Kistler, H.C.; Ma, Z. Fusarium Graminearum Trichothecene Mycotoxins: Biosynthesis, Regulation, and Management. Annu. Rev. Phytopathol. 2019, 57, 15–39. [Google Scholar] [CrossRef] [Green Version]
- Dill-Macky, R. Chapter1—Fusarium Head Blight: Recent Epidemics and Research Efforts in the Upper Midwest of the United States. In Fusarium Head Scab: Global Status and Future Prospects; Dubin, H.J., Gilchrist, L., Reeves, J., McNab, A., Eds.; CIMMYT: El Batán, Mexico, 1997. [Google Scholar]
- Windels, C.E. Economic and Social Impacts of Fusarium Head Blight: Changing Farms and Rural Communities in the Northern Great Plains. Phytopathology 2000, 90, 17–21. [Google Scholar] [CrossRef] [Green Version]
- McMullen, M.; Bergstrom, G.; De Wolf, E.; Dill-Macky, R.; Hershman, D.; Shaner, G.; Van Sanford, D. A Unified Effort to Fight an Enemy of Wheat and Barley: Fusarium Head Blight. Plant Dis. 2012, 96, 1712–1728. [Google Scholar] [CrossRef] [Green Version]
- Dahl, B.; Wilson, W.W. Risk Premiums Due to Fusarium Head Blight (FHB) in Wheat and Barley. Agric. Syst. 2018, 162, 145–153. [Google Scholar] [CrossRef]
- Wilson, W.; Dahl, B.; Nganje, W. Economic Costs of Fusarium Head Blight, Scab and Deoxynivalenol. World Mycotoxin J. 2018, 11, 291–302. [Google Scholar] [CrossRef]
- Vaughan, M.; Backhouse, D.; Ponte, E.M.D. Climate Change Impacts on the Ecology of Fusarium Graminearum Species Complex and Susceptibility of Wheat to Fusarium Head Blight: A Review. World Mycotoxin J. 2016, 9, 685–700. [Google Scholar] [CrossRef] [Green Version]
- Mylonas, I.; Stavrakoudis, D.; Katsantonis, D.; Korpetis, E. Chapter 1—Better farming practices to combat climate change. In Climate Change and Food Security with Emphasis on Wheat, 1st ed.; Ozturk, M., Gul, A., Eds.; Academic Press: Cambridge, MA, USA, 2020; pp. 1–29. [Google Scholar]
- Xia, R.; Schaafsma, A.W.; Wu, F.; Hooker, D.C. Impact of the Improvements in Fusarium Head Blight and Agronomic Management on Economics of Winter Wheat. World Mycotoxin J. 2020, 13, 423–439. [Google Scholar] [CrossRef]
- Venske, E.; dos Santos, R.S.; Farias, D.d.R.; Rother, V.; da Maia, L.C.; Pegoraro, C.; Costa de Oliveira, A. Meta-Analysis of the QTLome of Fusarium Head Blight Resistance in Bread Wheat: Refining the Current Puzzle. Front. Plant Sci. 2019, 10, 727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, T.; Hua, C.; Li, L.; Sun, Z.; Yuan, M.; Bai, G.; Humphreys, G.; Li, T. Integration of Meta-QTL Discovery with Omics: Towards a Molecular Breeding Platform for Improving Wheat Resistance to Fusarium Head Blight. Crop J. 2021, 9, 739–749. [Google Scholar] [CrossRef]
- Pavan, S.; Jacobsen, E.; Visser, R.G.F.; Bai, Y. Loss of Susceptibility as a Novel Breeding Strategy for Durable and Broad-Spectrum Resistance. Mol. Breed. 2010, 25, 1–12. [Google Scholar] [CrossRef] [Green Version]
- van Schie, C.C.N.; Takken, F.L.W. Susceptibility Genes 101: How to Be a Good Host. Annu. Rev. Phytopathol. 2014, 52, 551–581. [Google Scholar] [CrossRef]
- Ma, H.-X.; Bai, G.-H.; Gill, B.S.; Hart, L.P. Deletion of a Chromosome Arm Altered Wheat Resistance to Fusarium Head Blight and Deoxynivalenol Accumulation in Chinese Spring. Plant Dis. 2006, 90, 1545–1549. [Google Scholar] [CrossRef]
- Fabre, F.; Vignassa, M.; Urbach, S.; Langin, T.; Bonhomme, L. Time-resolved Dissection of the Molecular Crosstalk Driving Fusarium Head Blight in Wheat Provides New Insights into Host Susceptibility Determinism. Plant Cell Environ. 2019, 42, 2291–2308. [Google Scholar] [CrossRef]
- Fabre, F.; Bormann, J.; Urbach, S.; Roche, S.; Langin, T.; Bonhomme, L. Unbalanced Roles of Fungal Aggressiveness and Host Cultivars in the Establishment of the Fusarium Head Blight in Bread Wheat. Front. Microbiol. 2019, 10, 2857. [Google Scholar] [CrossRef]
- Su, Z.; Bernardo, A.; Tian, B.; Chen, H.; Wang, S.; Ma, H.; Cai, S.; Liu, D.; Zhang, D.; Li, T.; et al. A Deletion Mutation in TaHRC Confers Fhb1 Resistance to Fusarium Head Blight in Wheat. Nat. Genet. 2019, 51, 1099–1105. [Google Scholar] [CrossRef]
- Hales, B.; Steed, A.; Giovannelli, V.; Burt, C.; Lemmens, M.; Molnár-Láng, M.; Nicholson, P. Type II Fusarium Head Blight Susceptibility Conferred by a Region on Wheat Chromosome 4D. J. Exp. Bot. 2020, 71, 4703–4714. [Google Scholar] [CrossRef]
- Fabre, F.; Urbach, S.; Roche, S.; Langin, T.; Bonhomme, L. Proteomics-Based Data Integration of Wheat Cultivars Facing Fusarium Graminearum Strains Revealed a Core-Responsive Pattern Controlling Fusarium Head Blight. Front. Plant Sci. 2021, 12, 644810. [Google Scholar] [CrossRef]
- Zaidi, S.S.-A.; Mukhtar, M.S.; Mansoor, S. Genome Editing: Targeting Susceptibility Genes for Plant Disease Resistance. Trends Biotechnol. 2018, 36, 898–906. [Google Scholar] [CrossRef] [PubMed]
- Fabre, F.; Rocher, F.; Alouane, T.; Langin, T.; Bonhomme, L. Searching for FHB Resistances in Bread Wheat: Susceptibility at the Crossroad. Front. Plant Sci. 2020, 11, 731. [Google Scholar] [CrossRef]
- Gorash, A.; Armonienė, R.; Kazan, K. Can Effectoromics and Loss-of-Susceptibility Be Exploited for Improving Fusarium Head Blight Resistance in Wheat? Crop J. 2021, 9, 1–16. [Google Scholar] [CrossRef]
- O’Connell, R.J.; Panstruga, R. Tête à Tête inside a Plant Cell: Establishing Compatibility between Plants and Biotrophic Fungi and Oomycetes. New Phytol. 2006, 171, 699–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selin, C.; de Kievit, T.R.; Belmonte, M.F.; Fernando, W.G.D. Elucidating the Role of Effectors in Plant-Fungal Interactions: Progress and Challenges. Front. Microbiol. 2016, 7, 600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeilinger, S.; Gupta, V.K.; Dahms, T.E.S.; Silva, R.N.; Singh, H.B.; Upadhyay, R.S.; Gomes, E.V.; Tsui, C.K.-M.; Nayak, S.C. Friends or Foes? Emerging Insights from Fungal Interactions with Plants. FEMS Microbiol. Rev. 2016, 40, 182–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.; Seto, D.; Subramaniam, R.; Desveaux, D. Oh, the Places They’ll Go! A Survey of Phytopathogen Effectors and Their Host Targets. Plant J. 2018, 93, 651–663. [Google Scholar] [CrossRef]
- He, Q.; McLellan, H.; Boevink, P.C.; Birch, P.R.J. All Roads Lead to Susceptibility: The Many Modes of Action of Fungal and Oomycete Intracellular Effectors. Plant Commun. 2020, 1, 100050. [Google Scholar] [CrossRef]
- Jaswal, R.; Kiran, K.; Rajarammohan, S.; Dubey, H.; Singh, P.K.; Sharma, Y.; Deshmukh, R.; Sonah, H.; Gupta, N.; Sharma, T.R. Effector Biology of Biotrophic Plant Fungal Pathogens: Current Advances and Future Prospects. Microbiol. Res. 2020, 241, 126567. [Google Scholar] [CrossRef]
- Brown, N.A.; Hammond-Kosack, K.E. Chapter 19—Secreted biomolecules in fungal plant pathogenesis. In Fungal Biomolecules: Sources, Applications and Recent Developments; Gupta, V.K., Mach, R.L., Sreenivasaprasad, S., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2015; pp. 263–310. [Google Scholar]
- Lo Presti, L.; Lanver, D.; Schweizer, G.; Tanaka, S.; Liang, L.; Tollot, M.; Zuccaro, A.; Reissmann, S.; Kahmann, R. Fungal Effectors and Plant Susceptibility. Annu. Rev. Plant Biol. 2015, 66, 513–545. [Google Scholar] [CrossRef] [PubMed]
- Sperschneider, J.; Dodds, P.N.; Gardiner, D.M.; Manners, J.M.; Singh, K.B.; Taylor, J.M. Advances and Challenges in Computational Prediction of Effectors from Plant Pathogenic Fungi. PLoS Pathog. 2015, 11, e1004806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nejat, N.; Rookes, J.; Mantri, N.L.; Cahill, D.M. Plant–Pathogen Interactions: Toward Development of next-Generation Disease-Resistant Plants. Crit. Rev. Biotechnol. 2017, 37, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Prasad, P.; Savadi, S.; Bhardwaj, S.C.; Gangwar, O.P.; Kumar, S. Rust Pathogen Effectors: Perspectives in Resistance Breeding. Planta 2019, 250, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Uhse, S.; Djamei, A. Effectors of Plant-Colonizing Fungi and Beyond. PLoS Pathog. 2018, 14, e1006992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorrain, C.; Gonçalves dos Santos, K.C.; Germain, H.; Hecker, A.; Duplessis, S. Advances in Understanding Obligate Biotrophy in Rust Fungi. New Phytol. 2019, 222, 1190–1206. [Google Scholar] [CrossRef] [Green Version]
- Depotter, J.R.L.; Doehlemann, G. Target the Core: Durable Plant Resistance against Filamentous Plant Pathogens through Effector Recognition. Pest Manag. Sci. 2020, 76, 426–431. [Google Scholar] [CrossRef] [Green Version]
- Arroyo-Velez, N.; González-Fuente, M.; Peeters, N.; Lauber, E.; Noël, L.D. From Effectors to Effectomes: Are Functional Studies of Individual Effectors Enough to Decipher Plant Pathogen Infectious Strategies? PLoS Pathog. 2020, 16, e1009059. [Google Scholar] [CrossRef]
- Badet, T.; Croll, D. The Rise and Fall of Genes: Origins and Functions of Plant Pathogen Pangenomes. Curr. Opin. Plant Biol. 2020, 56, 65–73. [Google Scholar] [CrossRef]
- Petre, B.; Lorrain, C.; Stukenbrock, E.H.; Duplessis, S. Host-Specialized Transcriptome of Plant-Associated Organisms. Curr. Opin. Plant Biol. 2020, 56, 81–88. [Google Scholar] [CrossRef]
- Bruce, M.; Neugebauer, K.A.; Joly, D.L.; Migeon, P.; Cuomo, C.A.; Wang, S.; Akhunov, E.; Bakkeren, G.; Kolmer, J.A.; Fellers, J.P. Using Transcription of Six Puccinia Triticina Races to Identify the Effective Secretome during Infection of Wheat. Front. Plant Sci. 2014, 4, 520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamel, L.; Tang, N.; Malbreil, M.; San Clemente, H.; Le Marquer, M.; Roux, C.; Frei dit Frey, N. The Comparison of Expressed Candidate Secreted Proteins from Two Arbuscular Mycorrhizal Fungi Unravels Common and Specific Molecular Tools to Invade Different Host Plants. Front. Plant Sci. 2017, 8, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutter, W.B.; Salcedo, A.; Akhunova, A.; He, F.; Wang, S.; Liang, H.; Bowden, R.L.; Akhunov, E. Divergent and Convergent Modes of Interaction between Wheat and Puccinia Graminis f. Sp. Tritici Isolates Revealed by the Comparative Gene Co-Expression Network and Genome Analyses. BMC Genom. 2017, 18, 291. [Google Scholar] [CrossRef] [Green Version]
- Haueisen, J.; Möller, M.; Eschenbrenner, C.J.; Grandaubert, J.; Seybold, H.; Adamiak, H.; Stukenbrock, E.H. Highly Flexible Infection Programs in a Specialized Wheat Pathogen. Ecol. Evol. 2019, 9, 275–294. [Google Scholar] [CrossRef] [PubMed]
- Human, M.P.; Berger, D.K.; Crampton, B.G. Time-Course RNAseq Reveals Exserohilum Turcicum Effectors and Pathogenicity Determinants. Front. Microbiol. 2020, 11, 360. [Google Scholar] [CrossRef] [Green Version]
- Alouane, T.; Rimbert, H.; Bormann, J.; González-Montiel, G.A.; Loesgen, S.; Schäfer, W.; Freitag, M.; Langin, T.; Bonhomme, L. Comparative Genomics of Eight Fusarium graminearum Strains with Contrasting Aggressiveness Reveals an Expanded Open Pangenome and Extended Effector Content Signatures. Int. J. Mol. Sci. 2021, 22, 6257. [Google Scholar] [CrossRef]
- Brown, N.A.; Antoniw, J.; Hammond-Kosack, K.E. The Predicted Secretome of the Plant Pathogenic Fungus Fusarium graminearum: A Refined Comparative Analysis. PLoS ONE 2012, 7, e33731. [Google Scholar] [CrossRef] [Green Version]
- Lysøe, E.; Seong, K.-Y.; Kistler, H.C. The Transcriptome of Fusarium graminearum During the Infection of Wheat. Mol. Plant Microbe Interact. 2011, 24, 995–1000. [Google Scholar] [CrossRef] [Green Version]
- Brown, N.A.; Evans, J.; Mead, A.; Hammond-Kosack, K.E. A Spatial Temporal Analysis of the Fusarium graminearum Transcriptome during Symptomless and Symptomatic Wheat Infection: Transcriptome of Symptomless Fusarium Infection. Mol. Plant Pathol. 2017, 18, 1295–1312. [Google Scholar] [CrossRef] [Green Version]
- Sperschneider, J.; Dodds, P.N.; Gardiner, D.M.; Singh, K.B.; Taylor, J.M. Improved Prediction of Fungal Effector Proteins from Secretomes with EffectorP 2.0. Mol. Plant Pathol. 2018, 19, 2094–2110. [Google Scholar] [CrossRef] [Green Version]
- Laurent, B.; Moinard, M.; Spataro, C.; Ponts, N.; Barreau, C.; Foulongne-Oriol, M. Landscape of Genomic Diversity and Host Adaptation in Fusarium graminearum. BMC Genom. 2017, 18, 203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Jiang, C.; Wang, C.; Chen, C.; Xu, J.-R.; Liu, H. Characterization of the Two-Speed Subgenomes of Fusarium graminearum Reveals the Fast-Speed Subgenome Specialized for Adaption and Infection. Front. Plant Sci. 2017, 8, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.; Liu, Z.; Rocheleau, H.; Fauteux, F.; Wang, Y.; McCartney, C.; Ouellet, T. Transcriptome Dynamics Associated with Resistance and Susceptibility against Fusarium Head Blight in Four Wheat Genotypes. BMC Genom. 2018, 19, 642. [Google Scholar] [CrossRef] [PubMed]
- Li, H.B.; Xie, G.Q.; Ma, J.; Liu, G.R.; Wen, S.M.; Ban, T.; Chakraborty, S.; Liu, C.J. Genetic Relationships between Resistances to Fusarium Head Blight and Crown Rot in Bread Wheat (Triticum Aestivum L.). Theor. Appl. Genet. 2010, 121, 941–950. [Google Scholar] [CrossRef] [PubMed]
- Woolhouse, M.E.J.; Webster, J.P.; Domingo, E.; Charlesworth, B.; Levin, B.R. Biological and Biomedical Implications of the Co-Evolution of Pathogens and Their Hosts. Nat. Genet. 2002, 32, 569–577. [Google Scholar] [CrossRef] [Green Version]
- Newman, T.E.; Derbyshire, M.C. The Evolutionary and Molecular Features of Broad Host-Range Necrotrophy in Plant Pathogenic Fungi. Front. Plant Sci. 2020, 11, 591733. [Google Scholar] [CrossRef]
- Alouane, T.; Rimbert, H.; Fabre, F.; Cambon, F.; Langin, T.; Bonhomme, L. Genome Sequence of Fusarium graminearum Strain MDC_Fg1, Isolated from Bread Wheat Grown in France. Microbiol. Resour. Announc. 2018, 7, e01260-18. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wang, Y. Trick or Treat: Microbial Pathogens Evolved Apoplastic Effectors Modulating Plant Susceptibility to Infection. Mol. Plant Microbe Interact. 2017, 31, 6–12. [Google Scholar] [CrossRef] [Green Version]
- Jashni, M.K.; Dols, I.H.M.; Iida, Y.; Boeren, S.; Beenen, H.G.; Mehrabi, R.; Collemare, J.; de Wit, P.J.G.M. Synergistic Action of a Metalloprotease and a Serine Protease from Fusarium oxysporum f. sp. lycopersici Cleaves Chitin-Binding Tomato Chitinases, Reduces Their Antifungal Activity, and Enhances Fungal Virulence. Mol. Plant Microbe Interact. 2015, 28, 996–1008. [Google Scholar] [CrossRef] [Green Version]
- Jashni, M.K.; Mehrabi, R.; Collemare, J.; Mesarich, C.H.; de Wit, P.J.G.M. The Battle in the Apoplast: Further Insights into the Roles of Proteases and Their Inhibitors in Plant–Pathogen Interactions. Front. Plant Sci. 2015, 6, 584. [Google Scholar] [CrossRef] [Green Version]
- Muszewska, A.; Stepniewska-Dziubinska, M.M.; Steczkiewicz, K.; Pawlowska, J.; Dziedzic, A.; Ginalski, K. Fungal Lifestyle Reflected in Serine Protease Repertoire. Sci. Rep. 2017, 7, 9147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jashni, M.K.; van der Burgt, A.; Battaglia, E.; Mehrabi, R.; Collemare, J.; de Wit, P.J.G.M. Transcriptome and Proteome Analyses of Proteases in Biotroph Fungal Pathogen Cladosporium fulvum. J. Plant Pathol. 2020, 102, 377–386. [Google Scholar] [CrossRef]
- Rocafort, M.; Fudal, I.; Mesarich, C.H. Apoplastic Effector Proteins of Plant-Associated Fungi and Oomycetes. Curr. Opin. Plant Biol. 2020, 56, 9–19. [Google Scholar] [CrossRef]
- Lu, S.; Faris, J.D. Fusarium graminearum KP4-like Proteins Possess Root Growth-Inhibiting Activity against Wheat and Potentially Contribute to Fungal Virulence in Seedling Rot. Fungal Genet. Biol. 2019, 123, 1–13. [Google Scholar] [CrossRef]
- Lu, S.; Edwards, M.C. Molecular Characterization and Functional Analysis of PR-1-Like Proteins Identified from the Wheat Head Blight Fungus Fusarium graminearum. Phytopathology 2018, 108, 510–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, Y.; Liu, Z.; Yin, Y.; Jiang, J.; Chen, Y.; Xu, J.-R.; Ma, Z. Functional Analysis of the Fusarium graminearum Phosphatome. New Phytol. 2015, 207, 119–134. [Google Scholar] [CrossRef]
- Zhang, Y.; He, J.; Jia, L.-J.; Yuan, T.-L.; Zhang, D.; Guo, Y.; Wang, Y.; Tang, W.-H. Cellular Tracking and Gene Profiling of Fusarium graminearum during Maize Stalk Rot Disease Development Elucidates Its Strategies in Confronting Phosphorus Limitation in the Host Apoplast. PLoS Pathog. 2016, 12, e1005485. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-G.; Stork, W.; Mudgett, M.B. Xanthomonas Type III Effector XopD Desumoylates Tomato Transcription Factor SlERF4 to Suppress Ethylene Responses and Promote Pathogen Growth. Cell Host Microbe 2013, 13, 143–154. [Google Scholar] [CrossRef] [Green Version]
- Gimenez-Ibanez, S.; Boter, M.; Fernández-Barbero, G.; Chini, A.; Rathjen, J.P.; Solano, R. The Bacterial Effector HopX1 Targets JAZ Transcriptional Repressors to Activate Jasmonate Signaling and Promote Infection in Arabidopsis. PLoS Biol. 2014, 12, e1001792. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, M.B.; dos Santos, K.C.G.; Sanchez, I.B.; Petre, B.; Lorrain, C.; Plourde, M.B.; Duplessis, S.; Desgagné-Penix, I.; Germain, H. A Rust Fungal Effector Binds Plant DNA and Modulates Transcription. Sci. Rep. 2018, 8, 14718. [Google Scholar] [CrossRef]
- Lu, Y.; Yao, J. Chloroplasts at the Crossroad of Photosynthesis, Pathogen Infection and Plant Defense. Int. J. Mol. Sci. 2018, 19, 3900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Aken, O. Mitochondrial Redox Systems as Central Hubs in Plant Metabolism and Signaling. Plant Physiol. 2021, 186, 36–52. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Tang, C.; Wang, X.; Sun, S.; Zhao, J.; Kang, Z.; Wang, X. An Effector Protein of the Wheat Stripe Rust Fungus Targets Chloroplasts and Suppresses Chloroplast Function. Nat. Commun. 2019, 10, 5571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, G.; Zhong, X.; Shi, Y.; Liu, Z.; Jiang, N.; Liu, J.; Ding, B.; Li, Z.; Kang, H.; Ning, Y.; et al. A Fungal Effector Targets a Heat Shock–Dynamin Protein Complex to Modulate Mitochondrial Dynamics and Reduce Plant Immunity. Sci. Adv. 2020, 6, eabb7719. [Google Scholar] [CrossRef] [PubMed]
- Petre, B.; Saunders, D.G.O.; Sklenar, J.; Lorrain, C.; Win, J.; Duplessis, S.; Kamoun, S. Candidate Effector Proteins of the Rust Pathogen Melampsora larici-populina Target Diverse Plant Cell Compartments. Mol. Plant Microbe Interact. 2015, 28, 689–700. [Google Scholar] [CrossRef] [Green Version]
- Petre, B.; Lorrain, C.; Saunders, D.G.O.; Win, J.; Sklenar, J.; Duplessis, S.; Kamoun, S. Rust Fungal Effectors Mimic Host Transit Peptides to Translocate into Chloroplasts. Cell. Microbiol. 2016, 18, 453–465. [Google Scholar] [CrossRef] [Green Version]
- Pennington, H.G.; Gheorghe, D.M.; Damerum, A.; Pliego, C.; Spanu, P.D.; Cramer, R.; Bindschedler, L.V. Interactions between the Powdery Mildew Effector BEC1054 and Barley Proteins Identify Candidate Host Targets. J. Proteome Res. 2016, 15, 826–839. [Google Scholar] [CrossRef] [Green Version]
- CEA Genoscope–Centre National de Séquençage. Available online: https://jacob.cea.fr/drf/ifrancoisjacob/Pages/Departements/Genoscope.aspx (accessed on 30 August 2021).
- GeT—Génomique & Transcriptomique. Available online: https://get.genotoul.fr/ (accessed on 30 August 2021).
- Gardiner, D.M.; Stiller, J.; Kazan, K. Genome Sequence of Fusarium graminearum Isolate CS3005. Genome Announc. 2014, 2, e00227-14. [Google Scholar] [CrossRef] [Green Version]
- Walkowiak, S.; Rowland, O.; Rodrigue, N.; Subramaniam, R. Whole Genome Sequencing and Comparative Genomics of Closely Related Fusarium Head Blight Fungi: Fusarium graminearum, F meridionale and F. asiaticum. BMC Genom. 2016, 17, 1014. [Google Scholar] [CrossRef] [Green Version]
- King, R.; Urban, M.; Hammond-Kosack, M.C.U.; Hassani-Pak, K.; Hammond-Kosack, K.E. The Completed Genome Sequence of the Pathogenic Ascomycete Fungus Fusarium graminearum. BMC Genom. 2015, 16, 544. [Google Scholar] [CrossRef] [Green Version]
- King, R.; Urban, M.; Hammond-Kosack, K.E. Annotation of Fusarium graminearum (PH-1) Version 5.0. Genome Announc. 2017, 5, e01479-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, C.-S.; Alexander, D.H.; Marks, P.; Klammer, A.A.; Drake, J.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.E.; et al. Nonhybrid, Finished Microbial Genome Assemblies from Long-Read SMRT Sequencing Data. Nat. Methods 2013, 10, 563–569. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-Hit: A Fast Program for Clustering and Comparing Large Sets of Protein or Nucleotide Sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [Green Version]
- Holt, C.; Yandell, M. MAKER2: An Annotation Pipeline and Genome-Database Management Tool for Second-Generation Genome Projects. BMC Bioinform. 2011, 12, 491. [Google Scholar] [CrossRef] [Green Version]
- Korf, I. Gene Finding in Novel Genomes. BMC Bioinform. 2004, 5, 59. [Google Scholar] [CrossRef] [Green Version]
- Stanke, M.; Keller, O.; Gunduz, I.; Hayes, A.; Waack, S.; Morgenstern, B. AUGUSTUS: Ab Initio Prediction of Alternative Transcripts. Nucleic Acids Res. 2006, 34, W435–W439. [Google Scholar] [CrossRef] [Green Version]
- Jurka, J. Repeats in Genomic DNA: Mining and Meaning. Curr. Opin. Struct. Biol. 1998, 8, 333–337. [Google Scholar] [CrossRef]
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker. Available online: http://repeatmasker.org (accessed on 13 January 2021).
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 Improves Signal Peptide Predictions Using Deep Neural Networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef]
- Sperschneider, J.; Dodds, P.N.; Singh, K.B.; Taylor, J.M. ApoplastP: Prediction of Effectors and Plant Proteins in the Apoplast Using Machine Learning. New Phytol. 2018, 217, 1764–1778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sperschneider, J.; Catanzariti, A.-M.; DeBoer, K.; Petre, B.; Gardiner, D.M.; Singh, K.B.; Dodds, P.N.; Taylor, J.M. LOCALIZER: Subcellular Localization Prediction of Both Plant and Effector Proteins in the Plant Cell. Sci. Rep. 2017, 7, 44598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-Scale Protein Function Classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Winnenburg, R.; Baldwin, T.K.; Urban, M.; Rawlings, C.; Köhler, J.; Hammond-Kosack, K.E. PHI-Base: A New Database for Pathogen Host Interactions. Nucleic Acids Res. 2006, 34, D459–D464. [Google Scholar] [CrossRef]
- Urban, M.; Cuzick, A.; Seager, J.; Wood, V.; Rutherford, K.; Venkatesh, S.Y.; De Silva, N.; Martinez, M.C.; Pedro, H.; Yates, A.D.; et al. PHI-Base: The Pathogen–Host Interactions Database. Nucleic Acids Res. 2020, 48, D613–D620. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Barrett, A.J.; Thomas, P.D.; Huang, X.; Bateman, A.; Finn, R.D. The MEROPS Database of Proteolytic Enzymes, Their Substrates and Inhibitors in 2017 and a Comparison with Peptidases in the PANTHER Database. Nucleic Acids Res. 2018, 46, D624–D632. [Google Scholar] [CrossRef]
- HMMER. HMMER: Biosequence Analysis Using Profile Hidden Markov Models. Available online: http://hmmer.org/ (accessed on 1 September 2021).
- Yin, Y.; Mao, X.; Yang, J.; Chen, X.; Mao, F.; Xu, Y. DbCAN: A Web Resource for Automated Carbohydrate-Active Enzyme Annotation. Nucleic Acids Res. 2012, 40, W445–W451. [Google Scholar] [CrossRef]
- UCA. Mésocentre. Available online: https://mesocentre.uca.fr/accueil-mesocentre (accessed on 1 September 2021).
- CEA. Très Grand Centre de Calcul du CEA. Available online: http://www-hpc.cea.fr/fr/complexe/tgcc.htm (accessed on 1 September 2021).
- Babraham Institute. Babraham Bioinformatics—Trim Galore! Available online: https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (accessed on 30 August 2021).
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- NCBI. RefSeq: NCBI Reference Sequence Database. Available online: https://www.ncbi.nlm.nih.gov/refseq/ (accessed on 13 January 2022).
- Kopylova, E.; Noé, L.; Touzet, H. SortMeRNA: Fast and Accurate Filtering of Ribosomal RNAs in Metatranscriptomic Data. Bioinformatics 2012, 28, 3211–3217. [Google Scholar] [CrossRef]
- EnsemblPlants. Triticum_aestivum (IWGSC)—Ensembl Genomes. Available online: https://plants.ensembl.org/Triticum_aestivum/Info/Index (accessed on 13 January 2022).
- IWGSC. IWGSC RefSeq v1.1 Reference Genome and Annotation. Available online: https://urgi.versailles.inra.fr/download/iwgsc/IWGSC_RefSeq_Annotations/v1.1/ (accessed on 31 August 2021).
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The R Foundation. The R Project for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 31 August 2021).
- Robinson, M.D.; Oshlack, A. A Scaling Normalization Method for Differential Expression Analysis of RNA-Seq Data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, I.; Paysant-Le Roux, C.; Colella, S.; Martin-Magniette, M.-L. DiCoExpress: A Tool to Process Multifactorial RNAseq Experiments from Quality Controls to Co-Expression Analysis through Differential Analysis Based on Contrasts inside GLM Models. Plant Methods 2020, 16, 68. [Google Scholar] [CrossRef]
- RDocumentation. Hypergeometric: The Hypergeometric Distribution. Available online: https://www.rdocumentation.org/packages/stats/versions/3.6.2/topics/Hypergeometric (accessed on 4 January 2022).
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohart, F.; Gautier, B.; Singh, A.; Cao, K.-A.L. MixOmics: An R Package for ‘omics Feature Selection and Multiple Data Integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef] [Green Version]
- Kolde, R. Pheatmap. Available online: https://github.com/raivokolde/pheatmap (accessed on 1 September 2021).
- Murtagh, F.; Legendre, P. Ward’s Hierarchical Agglomerative Clustering Method: Which Algorithms Implement Ward’s Criterion? J. Classif. 2014, 31, 274–295. [Google Scholar] [CrossRef] [Green Version]
- Gel, B.; Serra, E. KaryoploteR: An R/Bioconductor Package to Plot Customizable Genomes Displaying Arbitrary Data. Bioinformatics 2017, 33, 3088–3090. [Google Scholar] [CrossRef]
- Wang, Q. Wangqinhu/Fgreseq. Available online: https://github.com/wangqinhu/fgreseq (accessed on 20 October 2021).







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rocher, F.; Alouane, T.; Philippe, G.; Martin, M.-L.; Label, P.; Langin, T.; Bonhomme, L. Fusarium graminearum Infection Strategy in Wheat Involves a Highly Conserved Genetic Program That Controls the Expression of a Core Effectome. Int. J. Mol. Sci. 2022, 23, 1914. https://doi.org/10.3390/ijms23031914
Rocher F, Alouane T, Philippe G, Martin M-L, Label P, Langin T, Bonhomme L. Fusarium graminearum Infection Strategy in Wheat Involves a Highly Conserved Genetic Program That Controls the Expression of a Core Effectome. International Journal of Molecular Sciences. 2022; 23(3):1914. https://doi.org/10.3390/ijms23031914
Chicago/Turabian StyleRocher, Florian, Tarek Alouane, Géraldine Philippe, Marie-Laure Martin, Philippe Label, Thierry Langin, and Ludovic Bonhomme. 2022. "Fusarium graminearum Infection Strategy in Wheat Involves a Highly Conserved Genetic Program That Controls the Expression of a Core Effectome" International Journal of Molecular Sciences 23, no. 3: 1914. https://doi.org/10.3390/ijms23031914
APA StyleRocher, F., Alouane, T., Philippe, G., Martin, M.-L., Label, P., Langin, T., & Bonhomme, L. (2022). Fusarium graminearum Infection Strategy in Wheat Involves a Highly Conserved Genetic Program That Controls the Expression of a Core Effectome. International Journal of Molecular Sciences, 23(3), 1914. https://doi.org/10.3390/ijms23031914