
Genetic Deletion of HLJ1 Does Not Affect Blood Coagulation in Mice

 and
and 
Abstract
1. Introduction
2. Results
2.1. HLJ1 Is Expressed in the Bone Marrow and Plasma of Mice
2.2. The Role of HLJ1 in the Bleeding Time and Blood Loss in Mice
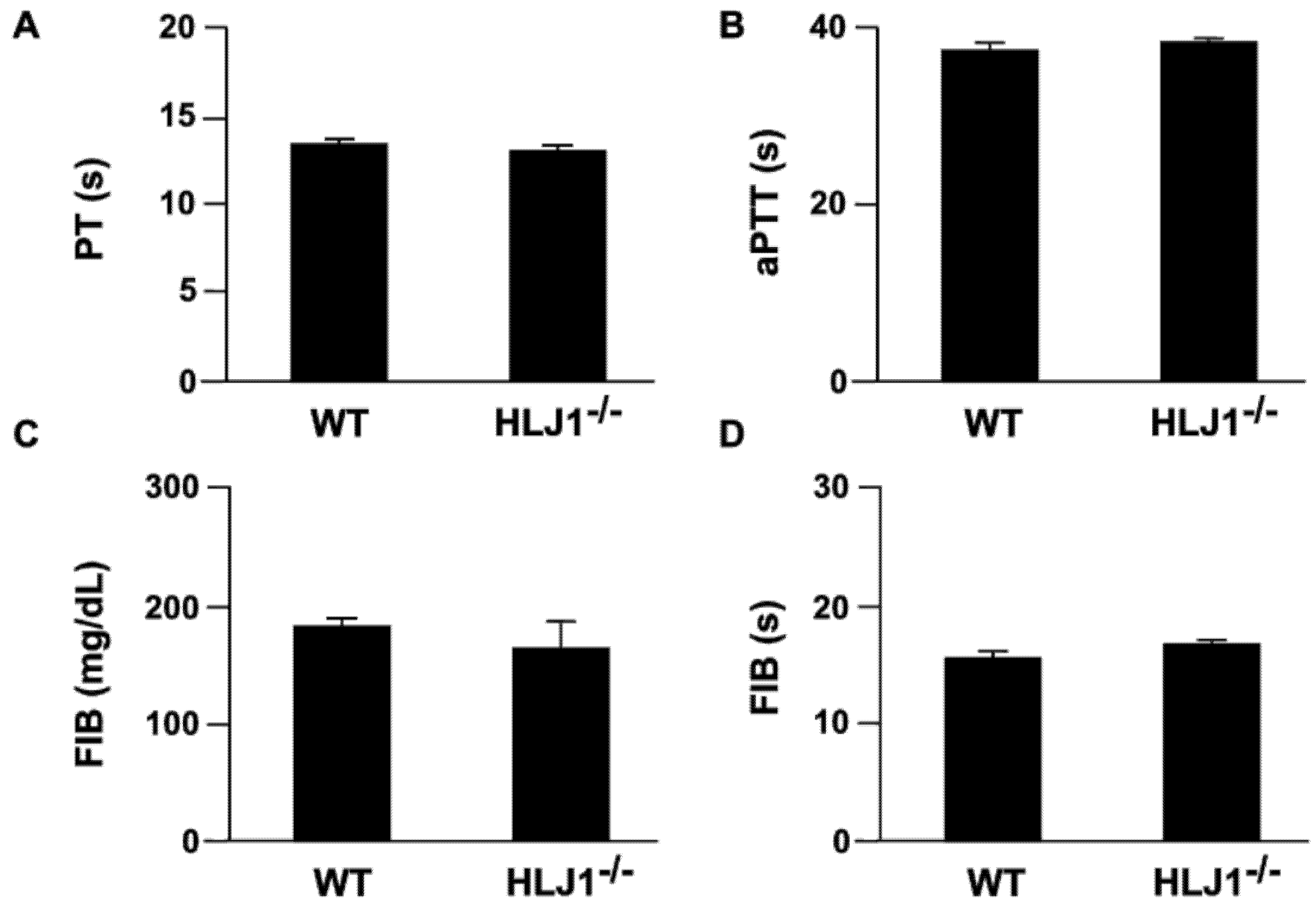
2.3. The Role of HLJ1 in the Activity of Blood Coagulation in Mice
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Animal
4.3. Immunohistochemistry
4.4. Western Blot Analysis
4.5. Tail Bleeding Assay
4.6. Coagulation Test
4.7. Thromboelastography (TEG)
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Balestra, D.; Branchini, A. Molecular mechanisms and determinants of innovative correction approaches in coagulation factor deficiencies. Int. J. Mol. Sci. 2019, 20, 3036. [Google Scholar] [CrossRef] [PubMed]
- Periayah, M.H.; Halim, A.S.; Mat Saad, A.Z. Mechanism action of platelets and crucial blood coagulation pathways in hemostasis. Int. J. Hematol. Oncol. Stem Cell Res. 2017, 11, 319–327. [Google Scholar] [PubMed]
- Palta, S.; Saroa, R.; Palta, A. Overview of the coagulation system. Indian J. Anaesth. 2014, 8, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Mackman, N.; Tilley, R.E.; Key, N.S. Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1687–1693. [Google Scholar] [CrossRef]
- Mertens, K.; Bertina, R.M. Activation of human coagulation factor VIII by activated factor X, the common product of the intrinsic and the extrinsic pathway of blood coagulation. Thromb. Haemost. 1982, 47, 96–100. [Google Scholar] [CrossRef]
- Maas, C.; Govers-Riemslag, J.W.; Bouma, B.; Schiks, B.; Hazenberg, B.P.; Lokhorst, H.M.; Hammarström, P.; ten Cate, H.; de Groot, P.G.; Bouma, B.N.; et al. Misfolded proteins activate factor XII in humans, leading to kallikrein formation without initiating coagulation. J. Clin. Investig. 2008, 118, 3208–3218. [Google Scholar] [CrossRef]
- Zamolodchikov, D.; Renné, T.; Strickland, S. The Alzheimer’s disease peptide β-amyloid promotes thrombin generation through activation of coagulation factor XII. J. Thromb. Haemost. 2016, 14, 995–1007. [Google Scholar] [CrossRef]
- Saibil, H. Chaperone machines for protein folding, unfolding and disaggregation. Nat. Rev. Mol. Cell Biol. 2013, 14, 630–642. [Google Scholar] [CrossRef]
- Foit, L.; George, J.S.; Zhang, B.W.; Brooks, C.L.; Bardwell, J.C. Chaperone activation by unfolding. Proc. Natl. Acad. Sci. USA 2013, 110, E1254–E1262. [Google Scholar] [CrossRef]
- Poothong, J.; Pottekat, A.; Siirin, M.; Campos, A.R.; Paton, A.W.; Paton, J.C.; Lagunas-Acosta, J.; Chen, Z.; Swift, M.; Volkmann, N.; et al. Factor VIII exhibits chaperone-dependent and glucose-regulated reversible amyloid formation in the endoplasmic reticulum. Blood 2020, 135, 1899–1911. [Google Scholar] [CrossRef]
- Pignani, S.; Todaro, A.; Ferrarese, M.; Marchi, S.; Lombardi, S.; Balestra, D.; Pinton, P.; Bernardi, F.; Pinotti, M.; Branchini, A. The chaperone-like sodium phenylbutyrate improves factor IX intracellular trafficking and activity impaired by the frequent p.R294Q mutation. J. Thromb. Haemost. 2018, 16, 2035–2043. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhou, J.; Zhang, C.; Fu, W.; Xiao, X.; Ruan, S.; Zhang, Y.; Luo, X.; Tang, M. HLJ1 is a novel biomarker for colorectal carcinoma progression and overall patient survival. Int. J. Clin. Exp. Pathol. 2014, 7, 969–977. [Google Scholar] [PubMed]
- Zhang, L.; Cai, X.; Chen, K.; Wang, Z.; Wang, L.; Ren, M.; Huang, A.; Tang, H. Hepatitis B virus protein up-regulated HLJ1 expression via the transcription factor YY1 in human hepatocarcinoma cells. Virus Res. 2011, 157, 76. [Google Scholar] [CrossRef] [PubMed]
- Simões-Correia, J.; Silva, D.I.; Melo, S.; Figueiredo, J.; Caldeira, J.; Pinto, M.T.; Girão, H.; Pereira, P.; Seruca, R. DNAJB4 molecular chaperone distinguishes WT from mutant E-cadherin, determining their fate in vitro and in vivo. Hum. Mol. Genet. 2014, 23, 2094–2105. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.F.; Wang, C.C.; Chang, G.C.; Chen, C.Y.; Chen, H.Y.; Cheng, C.L.; Yang, Y.P.; Wu, C.Y.; Shih, F.Y.; Liu, C.C.; et al. A new tumor suppressor DnaJ-like heat shock protein, HLJ1, and survival of patients with non-small-cell lung carcinoma. J. Natl. Cancer Inst. 2006, 98, 825–838. [Google Scholar] [CrossRef]
- Chen, C.H.; Chang, W.H.; Su, K.Y.; Ku, W.H.; Chang, G.C.; Hong, Q.S.; Hsiao, Y.J.; Chen, H.C.; Chen, H.Y.; Wu, R.; et al. HLJ1 is an endogenous Src inhibitor suppressing cancer progression through dual mechanisms. Oncogene 2016, 35, 5674–5685. [Google Scholar] [CrossRef]
- Vabulas, R.M.; Raychaudhuri, S.; Hayer-Hartl, M.; Hartl, F.U. Protein folding in the cytoplasm and the heat shock response. Cold Spring Harb. Perspect. Biol. 2010, 2, a004390. [Google Scholar] [CrossRef]
- Khalil, A.A.; Kabapy, N.F.; Deraz, S.F.; Smith, C. Heat shock proteins in oncology: Diagnostic biomarkers or therapeutic targets? Biochim. Biophys. Acta 2011, 1816, 89–104. [Google Scholar] [CrossRef]
- Moura, C.S.; Lollo, P.C.B.; Morato, P.N.; Amaya-Farfan, J. Dietary nutrients and bioactive substances modulate heat shock protein (HSP) expression: A review. Nutrients 2018, 10, 683. [Google Scholar] [CrossRef]
- Feidantsis, K.; Giantsis, I.A.; Vratsistas, A.; Makri, S.; Pappa, A.Z.; Drosopoulou, E.; Anestis, A.; Mavridou, E.; Exadactylos, A.; Vafidis, D.; et al. Correlation between intermediary metabolism, Hsp gene expression, and oxidative stress-related proteins in long-term thermal-stressed Mytilus galloprovincialis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2020, 319, R264–R281. [Google Scholar] [CrossRef]
- Balasubramaniam, B.; Vinitha, T.; Deepika, S.; JebaMercy, G.; VenkataKrishna, L.M.; Balamurugan, K. Analysis of Caenorhabditis elegans phosphoproteome reveals the involvement of a molecular chaperone, HSP-90 protein during Salmonella enterica Serovar Typhi infection. Int. J. Biol. Macromol. 2019, 37, 620–646. [Google Scholar] [CrossRef] [PubMed]
- Polanowska-Grabowska, R.; Gear, A.R. Heat-shock proteins and platelet function. Platelets 2000, 11, 6–22. [Google Scholar] [CrossRef] [PubMed]
- İn, E.; Deveci, F.; Kaman, D. Assessment of heat shock proteins and endothelial dysfunction in acute pulmonary embolism. Blood Coagul. Fibrinolysis 2016, 27, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Kosuge, Y.; Kobayashi, K.; Kurosaki, Y.; Ishii, N.; Aoyama, N.; Ishihara, K.; Ichikawa, T. Heat-shock protein 72 promotes platelet aggregation induced by various platelet activators in rats. Biomed. Res. 2017, 38, 175–182. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, F.; He, M.; Yang, M.; Fan, Y.; Chen, Y.; Xia, X.; Xie, Y.; Deng, D. Alteration of heat shock protein 20 expression in preeclamptic patients and its effect in vascular and coagulation function. Front. Med. 2018, 12, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Bakogiannis, C.; Sachse, M.; Stamatelopoulos, K.; Stellos, K. Platelet-derived chemokines in inflammation and atherosclerosis. Cytokine 2019, 122, 154–157. [Google Scholar] [CrossRef] [PubMed]
- Golebiewska, E.M.; Poole, A.W. Platelet secretion: From haemostasis to wound healing and beyond. Blood Rev. 2015, 29, 153–162. [Google Scholar] [CrossRef]
- Zhu, Y.; O’Neill, S.; Saklatvala, J.; Tassi, L.; Mendelsohn, M.E. Phosphorylated HSP27 associates with the activation-dependent cytoskeleton in human platelets. Blood 1994, 84, 3715–3723. [Google Scholar] [CrossRef]
- Rigg, R.A.; Healy, L.D.; Nowak, M.S.; Mallet, J.; Thierheimer, M.L.; Pang, J.; McCarty, O.J.; Aslan, J.E. Heat shock protein 70 regulates platelet integrin activation, granule secretion and aggregation. Am. J. Physiol. Cell Physiol. 2016, 310, C568–C575. [Google Scholar] [CrossRef]
- Jackson, J.W.; Rivera-Marquez, G.M.; Beebe, K.; Tran, A.D.; Trepel, J.B.; Gestwicki, J.E.; Blagg, B.S.J.; Ohkubo, S.; Neckers, L.M. Pharmacologic dissection of the overlapping impact of heat shock protein family members on platelet function. J. Thromb. Haemost. 2020, 18, 1197–1209. [Google Scholar] [CrossRef]
- Kumarapeli, A.R.; Su, H.; Huang, W.; Tang, M.; Zheng, H.; Horak, K.M.; Li, M.; Wang, X. Alpha B-crystallin suppresses pressure overload cardiac hypertrophy. Circ. Res. 2008, 103, 1473–1482. [Google Scholar] [CrossRef] [PubMed]
- Kumarapeli, A.R.; Horak, K.; Wang, X. Protein quality control in protection against systolic overload cardiomyopathy: The longterm role of small heat shock proteins. Am. J. Transl. Res. 2010, 2, 390–401. [Google Scholar] [PubMed]
- Xu, Q.; Li, D.G.; Holbrook, N.J.; Udelsman, R. Acute hypertension induces heat-shock protein 70 gene expression in rat aorta. Circulation 1995, 92, 1223–1229. [Google Scholar] [CrossRef] [PubMed]
- Patton, W.F.; Erdjument-Bromage, H.; Marks, A.R.; Tempst, P.; Taubman, M.B. Components of the protein synthesis and folding machinery are induced in vascular smooth muscle cells by hypertrophic and hyperplastic agents. Identification by comparative protein phenotyping and microsequencing. J. Biol. Chem. 1995, 270, 21404–21410. [Google Scholar] [CrossRef] [PubMed]
- Di Naso, F.C.; Porto, R.R.; Fillmann, H.S.; Maggioni, L.; Padoin, A.V.; Ramos, R.J.; Mottin, C.C.; Bittencourt, A.; Marroni, N.A.; de Bittencourt, P.I., Jr. Obesity depresses the anti-inflammatory HSP70 pathway, contributing to NAFLD progression. Obesity 2015, 23, 120–129. [Google Scholar] [CrossRef]
- Chung, J.; Nguyen, A.K.; Henstridge, D.C.; Holmes, A.G.; Chan, M.H.; Mesa, J.L.; Lancaster, G.I.; Southgate, R.J.; Bruce, C.R.; Duffy, S.J.; et al. HSP72 protects against obesity-induced insulin resistance. Proc. Natl. Acad. Sci. USA 2008, 105, 1739–1744. [Google Scholar] [CrossRef] [PubMed]
- Konstantinova, E.V.; Chipigina, N.S.; Shurdumova, M.H.; Kovalenko, E.I.; Sapozhnikov, A.M. Heat shock protein 70 kDa as a target for diagnostics and therapy of cardiovascular and cerebrovascular diseases. Curr. Pharm. Des. 2019, 25, 710–714. [Google Scholar] [CrossRef]
- Gungor, B.; Vanharanta, L.; Hölttä-Vuori, M.; Pirhonen, J.; Petersen, N.H.T.; Gramolelli, S.; Ojala, P.M.; Kirkegaard, T.; Ikonen, E. HSP70 induces liver X receptor pathway activation and cholesterol reduction in vitro and in vivo. Mol. Metab. 2019, 28, 135–143. [Google Scholar] [CrossRef]
- Grundtman, C.; Kreutmayer, S.B.; Almanzar, G.; Wick, M.C.; Wick, G. Heat shock protein 60 and immune inflammatory responses in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 960–968. [Google Scholar] [CrossRef]
- Habich, C.; Sell, H. Heat shock proteins in obesity: Links to cardiovascular disease. Horm. Mol. Biol. Clin. Investig. 2015, 21, 117–124. [Google Scholar] [CrossRef]
- Archer, A.E.; Von Schulze, A.T.; Geiger, P.C. Exercise, heat shock proteins and insulin resistance. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20160529. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Iturbe, B.; Lanaspa, M.A.; Johnson, R.J. The role of autoimmune reactivity induced by heat shock protein 70 in the pathogenesis of essential hypertension. Br. J. Pharmacol. 2019, 176, 1829–1838. [Google Scholar] [CrossRef] [PubMed]
- Bozaykut, P.; Ozer, N.K.; Karademir, B. Regulation of protein turnover by heat shock proteins. Free Radic. Biol. Med. 2014, 77, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Burns, T.F. Targeting heat shock proteins in cancer: A promising therapeutic approach. Int. J. Mol. Sci. 2017, 18, 1978. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organ Weight | WT (n = 9) | HLJ1-/- (n = 9) | p-Value |
|---|---|---|---|
| Body weight | 22.61 ± 0.765 | 24.68 ± 0.635 * | 0.0236 * |
| Heart | 0.17 ± 0.003 | 0.20 ± 0.002 * | 0.0033 * |
| Liver | 1.24 ± 0.072 | 1.29 ± 0.095 | 0.453 |
| Spleen | 0.06 ± 0.001 | 0.06 ± 0.009 | 0.822 |
| Lung | 0.13 ± 0.005 | 0.12 ± 0.004 | 0.247 |
| Kidney | 0.30 ± 0.014 | 0.35 ± 0.038 | 0.375 |
| Brown adipose tissue | 0.04 ± 0.004 | 0.05 ± 0.006 | 0.315 |
| White adipose tissue | 0.48 ± 0.054 | 0.64 ± 0.036 * | 0.004 * |
| Gastrocnemius muscle | 0.25 ± 0.015 | 0.24 ± 0.011 | 0.871 |
| Brain | 0.45 ± 0.03 | 0.45 ± 0.004 | 0.470 |
| The ratio of organ weight to body weight | |||
| Heart (g/BW) | 0.0075 ± 0.000144 | 0.0081 ± 0.000112 * | 0.0253 * |
| Liver (g/BW) | 0.0548 ± 0.00255 | 0.0522 ± 0.00409 | 0.199 |
| Spleen (g/BW) | 0.0024 ± 0.0000422 | 0.0024 ± 0.0000881 | 0.273 |
| Lung (g/BW) | 0.0056 ± 0.00021 | 0.0055 ± 0.000298 | 0.803 |
| Kidney (g/BW) | 0.0132 ± 0.000480 | 0.0141 ± 0.00121 | 0.453 |
| Brown adipose tissue (g/BW) | 0.0017 ± 0.000164 | 0.0019 ± 0.0004 | 0.977 |
| White adipose tissue (g/BW) | 0.0212 ± 0.00008 | 0.0259 ± 0.00028 * | 0.0359 * |
| Gastrocnemius muscle (g/BW) | 0.0110 ± 0.000508 | 0.0097 ± 0.000683 | 0.309 |
| Brain (g/BW) | 0.0199 ± 0.000232 | 0.0189 ± 0.000482 | 0.15 |
| Peripheral Blood Counts | WT (n = 5) | HLJ1-/- (n = 5) | p-Value |
|---|---|---|---|
| RBC (×106/mL) | 10.82 ± 0.21 | 10.49 ± 0.088 | 0.236 |
| PLT (×103/mL) | 922.00 ± 42.02 | 1074.33 ± 83.73 | 0.176 |
| WBC (×103/mL) | 6.58 ± 0.71 | 5.29 ± 0.60 | 0.296 |
| NEUT (×103/mL) | 0.47 ± 0.11 | 0.41 ± 0.10 | 0.703 |
| LYMPH (×103/mL) | 6.00 ± 0.58 | 4.81 ± 0.49 | 0.474 |
| MONO (×103/mL) | 0.04 ± 0.002 | 0.03 ± 0.008 | 0.338 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, M.-C.; Luo, W.-J.; Guo, B.-C.; Chen, C.-H.; Hu, P.-A.; Tsai, Y.-H.; Su, K.-Y.; Lee, T.-S. Genetic Deletion of HLJ1 Does Not Affect Blood Coagulation in Mice. Int. J. Mol. Sci. 2022, 23, 2064. https://doi.org/10.3390/ijms23042064
Hsu M-C, Luo W-J, Guo B-C, Chen C-H, Hu P-A, Tsai Y-H, Su K-Y, Lee T-S. Genetic Deletion of HLJ1 Does Not Affect Blood Coagulation in Mice. International Journal of Molecular Sciences. 2022; 23(4):2064. https://doi.org/10.3390/ijms23042064
Chicago/Turabian StyleHsu, Man-Chen, Wei-Jia Luo, Bei-Chia Guo, Chia-Hui Chen, Po-An Hu, Yi-Hsuan Tsai, Kang-Yi Su, and Tzong-Shyuan Lee. 2022. "Genetic Deletion of HLJ1 Does Not Affect Blood Coagulation in Mice" International Journal of Molecular Sciences 23, no. 4: 2064. https://doi.org/10.3390/ijms23042064
APA StyleHsu, M.-C., Luo, W.-J., Guo, B.-C., Chen, C.-H., Hu, P.-A., Tsai, Y.-H., Su, K.-Y., & Lee, T.-S. (2022). Genetic Deletion of HLJ1 Does Not Affect Blood Coagulation in Mice. International Journal of Molecular Sciences, 23(4), 2064. https://doi.org/10.3390/ijms23042064